

Abstract
Objectives:
To perform a comprehensive population genetic study of PARK2. PARK2 mutations are associated with juvenile parkinsonism, Alzheimer disease, cancer, leprosy, and diabetes mellitus, yet ironically, there has been no comprehensive study of PARK2 in control subjects; and to resolve controversial association of PARK2 heterozygous mutations with Parkinson disease (PD) in a well-powered study.
Methods:
We studied 1,686 control subjects (mean age 66.1 ± 13.1 years) and 2,091 patients with PD (mean onset age 58.3 ± 12.1 years). We tested for PARK2 deletions/multiplications/copy number variations (CNV) using semiquantitative PCR and multiplex ligation-dependent probe amplification, and validated the mutations by real-time quantitative PCR. Subjects were tested for point mutations previously. Association with PD was tested as PARK2 main effect, and in combination with known PD risk factors: SNCA, MAPT, APOE, smoking, and coffee intake.
Results:
A total of 0.95% of control subjects and 0.86% of patients carried a heterozygous CNV mutation. CNV mutations found in 16 control subjects were all in exons 1–4, sparing exons that encode functionally critical protein domains. Thirteen patients had 2 CNV mutations, 5 had 1 CNV and 1 point mutation, and 18 had 1 CNV mutation. Mutations found in patients spanned exons 2–9. In whites, having 1 CNV was not associated with increased risk (odds ratio 1.05, p = 0.89) or earlier onset of PD (64.7 ± 8.6 heterozygous vs 58.5 ± 11.8 normal).
Conclusions:
This comprehensive population genetic study in control subjects fills the void for a PARK2 reference dataset. There is no compelling evidence for association of heterozygous PARK2 mutations, by themselves or in combination with known risk factors, with PD.
GLOSSARY
- ARJP
= autosomal recessive juvenile parkinsonism;
- CI
= confidence interval;
- CNV
= copy number variation;
- MAP
= moving average plots;
- MLPA
= multiplex ligation-dependent probe amplification;
- NGRC
= NeuroGenetics Research Consortium;
- OR
= odds ratio;
- PD
= Parkinson disease.
PARK2, a large gene in a fragile site on chromosome 6q25.2-q27,1 encodes Parkin, an E3 ubiquitin-protein ligase. PARK2 is also a tumor suppressor gene.2,3 Originally discovered as a cause of autosomal recessive juvenile parkinsonism (ARJP),4 PARK2 has subsequently been linked to cancer,2,3 leprosy,5 autism,6 type 2 diabetes mellitus,7 and Alzheimer disease.8 Despite increasing appreciation for the potential involvement of PARK2 in common disorders of diverse origins, there has not been a systematic large study of PARK2 in control subjects. Our first aim was to conduct a population genetic study of PARK2 and establish a reference dataset for the increasing number of PARK2-disease associations that are emerging from genome-wide association studies.
PARK2 is a molecular diagnostic test for parkinsonism. The majority of positive results are heterozygous and difficult for clinicians to interpret because whether having one mutation can cause, increase risk, or accelerate onset of Parkinson disease (PD) is unknown.9–21 The second aim of our study was to determine, conclusively, if heterozygous mutations are associated with PD. We designed this study specifically to address this question, and to that end, amassed a large sample size to ensure analytic power, analyzed the coding regions for all types of variations, and importantly, used the same rigorous mutation analysis and validation methods in control subjects as in patients. In the first phase, we established that heterozygous point mutations are as frequent in control subjects as in patients with PD.22 In this final phase, we present a comprehensive analysis of deletions/multiplications/copy number variations (CNV).
METHODS
Subjects included 3,777 genetically unrelated individuals (1,686 control subjects and 2,091 patients with PD) from the NeuroGenetics Research Consortium (NGRC). Control subjects consisted of 1,644 white, 13 black, 11 Hispanic, 5 Asian, 4 Native American, and 9 other race/ethnicity, ages 25–99 years at blood draw (mean 66.1 ± 13.1 years), and 43.8% were men. Patients with PD consisted of 1,968 white, 18 black, 31 Hispanic, 29 Asian, 9 Native American, 2 Pacific Islander, and 34 other/unknown. They were diagnosed by a movement disorder neurologist according to the modified UK PD Brain Bank criteria23 at one of 8 NGRC clinics in Oregon, Washington, New York, and Georgia. Patients were 8–93 years old at disease onset (58.3 ± 12.1) and 21–96 years old at blood draw (mean 67.3 ± 10.7 years), 67.8% were men, and 22.7% had a positive family history (first-degree or second-degree relative with PD).
Standard protocol approvals, registrations, and patient consents.
The study was approved by the Institutional Review Boards of participating institutions. Informed consent was obtained from all subjects.
Molecular analysis.
CNV genotyping was completed for 2,091 patients and 1,686 controls using high-molecular-weight genomic DNA from blood. Genotyping was performed in phases over 7 years (details in appendix e-1 on the Neurology® Web site at www.neurology.org). In 2004, we published on 39 patients with young-onset PD.24 While performing the large-scale sequencing (2004–2007), we identified 40 subjects with rare sequence variants and genotyped them for CNVs.22 The large-scale CNV analysis started with a modified semiquantitative PCR protocol25 that was used for 252 patients and 299 control subjects. With the advent of PD-specific multiplex ligation-dependent probe amplification (MLPA),26,27 we switched to the SALSA MLPA kit P052B from MRC-Holland (Amsterdam, the Netherlands) for 1,956 patients and 1,477 control subjects. We used more conservative measures than recommended by the MLPA manufacturer and were able to establish false-positive and false-negative rates (appendix e-1). We used real-time qPCR to verify all abnormal and equivocal findings from MLPA and semiquantitative PCR. CNV carriers were sequenced for all exons to detect point mutations. Finally, using DNA from relatives, we established phase unequivocally for 39 of 52 CNV carriers.
Data analysis.
Logistic regression, χ2 tests, and Fisher exact tests were used to test frequencies and estimate odds ratios (ORs) and corresponding 95% confidence intervals (95% CI). Age at onset was tested using Kaplan-Meier survival analysis, log-rank statistics, and analysis of variance. Data were adjusted for recruitment site, gender, age at blood draw, cigarette smoking,28 caffeinated coffee consumption,28 and PD susceptibility genotypes at SNCA REP1,29 SNCA 5′ promoter polymorphism rs2619364,30 MAPT H1/H2 haplotypes,31 and the APOE polymorphism.32 We tested for PARK2 × environment (smoking and coffee) and PARK2 × genotype (SNCA, MAPT, APOE) interaction using likelihood ratio test statistics. We selected the most significant factors using backward variable selection criteria. The model goodness-of-fit was tested using Hosmer and Lemeshow test statistics. Frequencies of CNVs in patients and control subjects were visualized as a function of age using moving average plots (MAP).33
RESULTS
Reference data.
Reference data are detailed in table e-1 and figure 1. Sixteen of 1,686 control subjects had a CNV, yielding a heterozygous CNV carrier frequency of 0.95% (95% CI 0.48–1.42). None of the carriers was homozygous or compound heterozygous. The mutations included deletions and multiplications occurring predominantly in exons 2, 3, and 4. One control subject had a mutation in exon 1. No CNVs were detected in exons 5–12.

Figure 1 Location of PARK2 copy number variations
Deletions (hatched arrows) and multiplications (open arrows) detected among 2,091 patients (top) and 1,686 control subjects (bottom). The PARK2 coding region is shown in gray; the 2 ring domains are shown in black. Introns not drawn to scale. E = exon.
Parkinson study.
The Parkinson study is described in table e-2 and figure 1. Among the 2,091 patients with PD, 13 had 2 CNVs, 5 had 1 CNV and 1 point mutation, and 18 had only 1 CNV. The mutations included deletions and multiplications in exons 2–9. No mutations were found in exons 1 or 10–12. All subjects with 2 PARK2 mutations had developed PD prior to age 55, which is consistent with autosomal recessive mutations causing young-onset PD. Among 171 patients with onset ≤40 years, 14 had 2 PARK2 mutations (Parkin disease) and none of the other 296 was heterozygous.
The question that we aimed to resolve was whether heterozygous mutations increase the risk or accelerate onset age of PD. Heterozygous CNV carrier frequency in patients was 0.86% (95% CI 0.46–1.26) as compared to 0.95% (95% CI 0.48–1.42) in control subjects. For the following analyses, we excluded subjects with mutations in LRRK2, SNCA, or SCA2 and nonwhites; the sample included 1,937 patients and 1,642 control subjects.
A striking feature of recessive PARK2 disease is the very early onset. We therefore questioned if having one mutation might accelerate onset age in common forms of PD. Mean age at onset was not earlier in PARK2 heterozygotes (64.7 ± 8.6 years) than in patients lacking a PARK2 CNV mutation (58.5 ± 11.8; table 1). Kaplan-Meier–generated age at onset distributions revealed a highly significant age at onset effect (p = 1 × 10−40), which stemmed from ∼30 years earlier onset for patients with 2 mutations (figure 2). The Kaplan-Meier age at onset distribution for heterozygotes was not significantly different from nonmutation carriers; in fact, heterozygotes appeared to have slightly delayed onset age (also reflected in mean onset ages), which is contrary to the hypothesis that PARK2 accelerates PD onset.
Table 1 Association of PARK2 CNV heterozygous mutations with PD in white individuals
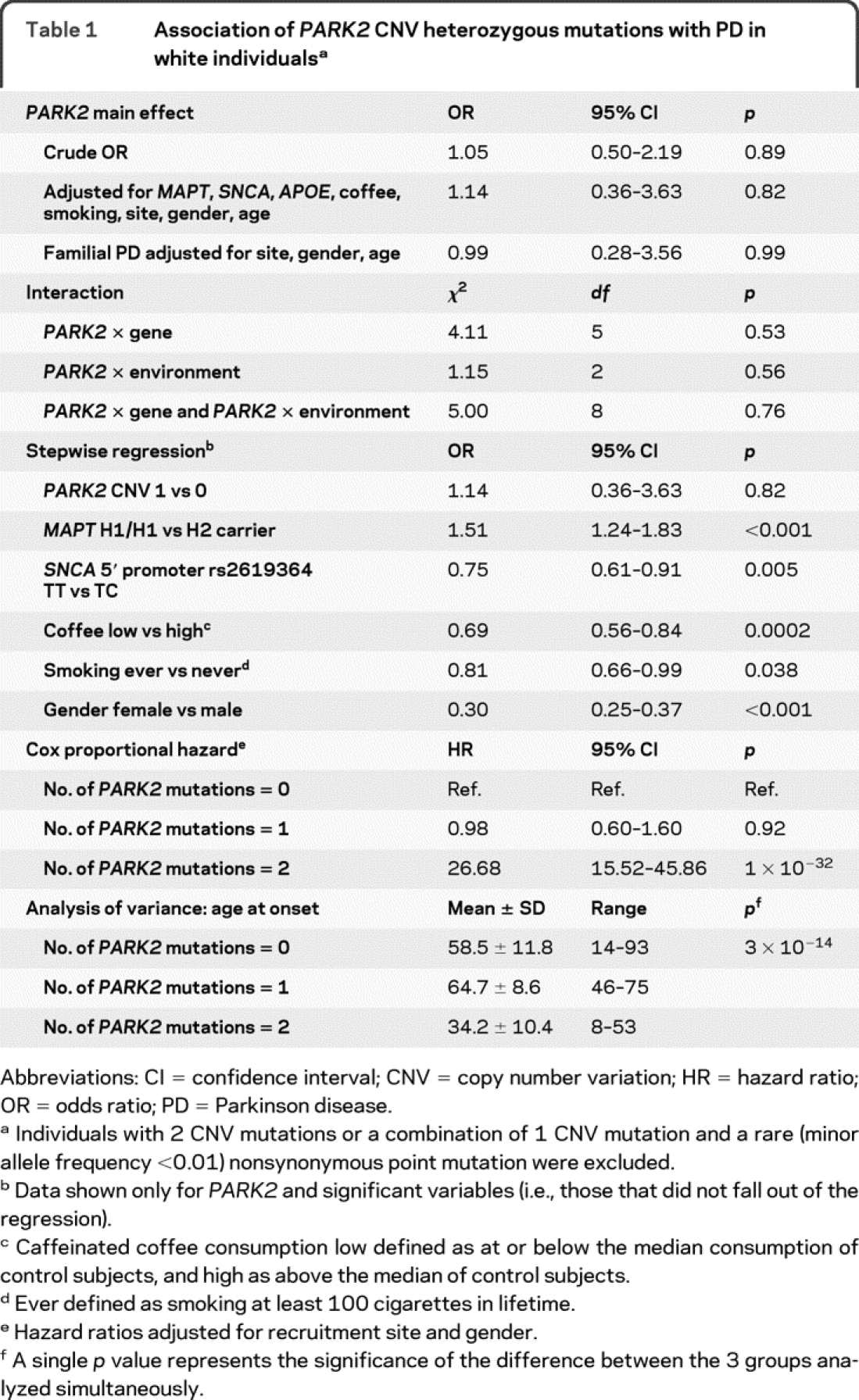
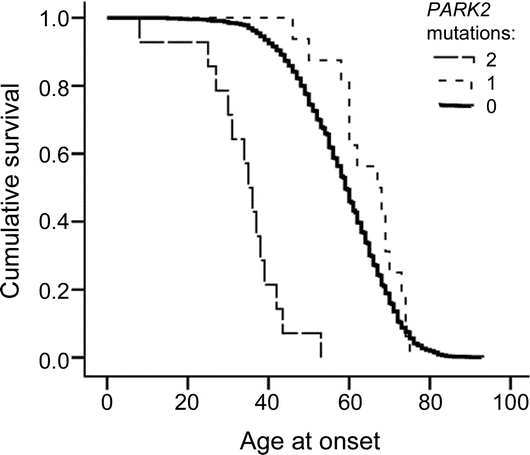
Figure 2 Age at onset of Parkinson disease as a function of number of PARK2 mutations
Patients with 2 (long dashed line), 1 (short dashed line), and 0 (solid line) PARK2 mutations. Rare nonsynonymous sequence variants were considered to be a mutation only when found in an individual carrying a copy number variation (CNV). Age at onset in heterozygous PARK2 patients is not earlier than in patients lacking a PARK2 CNV mutation, as opposed to patients with compound mutations who have substantially earlier onset. The graph was generated using Kaplan-Meier survival analysis and the differences were tested using log-rank statistics. Overall, p = 1 × 10−40, which is driven by compound mutation carriers in recessive young-onset disease.
To assess PARK2 heterozygosity in relation to risk of PD, we included all patients and control subjects, regardless of age at onset or family history, and compared those with one mutation to those without. Fourteen subjects with compound PARK2 mutations were excluded. The sample included 1,923 unrelated white patients and 1,642 unrelated white control subjects. We found no evidence (p = 0.89) for association of PARK2 heterozygosity with PD risk in the overall sample (OR 1.05, 95% CI 0.50–2.19; table 1) or in patients with positive family history (n = 423, OR 0.99, 95% CI 0.28–3.56; table 1).
We hypothesized that PARK2 heterozygosity might increase PD risk if patients also had a high-risk genotype at MAPT, SNCA, or APOE, did not smoke, did not consume large amounts of caffeinated beverages, were male, or were at advanced age. Adjusting for these factors in the model did not change the outcome (OR 1.14, p = 0.82; table 1). We tested for interaction between PARK2 and the aforementioned genes and exposures, and found none (p = 0.53–0.76). We performed stepwise and backward regression, which yielded similar results. We detected significant effects on PD risk for every factor known to be associated with PD, but not for PARK2 heterozygosity. We performed numerous iterative subanalyses to search for evidence that might be hidden in the type of mutation (deletion vs multiplication), exon location (exon 2 vs 3 vs 4, for example), and mutation phase, where we assigned and analyzed seemingly contiguous deletions/multiplications once as heterozygous and once as compound (data not shown). We did not find any compelling evidence for association with PD.
The MAP (figure 3) captures the results at a glance: mutation frequency starts very high in young-onset PD, declines sharply with increasing age at onset until it meets the control frequency at around age 45, and from age 45 until 90, stays completely superimposed on control subjects.

Figure 3 Moving average plots of PARK2 mutation frequency as a function of age
Moving average frequency of PARK2 mutations was estimated as a function of age at onset in patients (red triangles) vs age at blood draw in controls (blue circles) with 95% central posterior probabilities (red and blue lines). Plots show that PARK2 mutation frequency is very high in young-onset Parkinson disease (PD), declines with increasing age at onset, and by age 45 and thereafter, the mutation frequency in PD and control subjects are completely superimposed. Gray bars at the bottom of the graph show the statistical significance of the difference between patients and control subjects, calculated in the moving window (light gray ≥95% probability, dark gray ≥99% probability). Due to few numbers at the extremes, subjects with ages ≤20 or ≥90 years were merged.
DISCUSSION
Currently, there exists no population genetic study of PARK2 in the literature. The present study fills this void. We demonstrated that PARK2 deletions, multiplications, and CNVs (this report), and rare sequence variants including point mutations (reported previously),22 are not exclusive to disease populations. We found CNVs in ∼1% and point mutations in ∼3% of control subjects. Heterozygous CNV mutations in exons 2–4 are common and well-tolerated. However, we did not find any CNVs in exons 5–12 in control subjects, which include the coding region for the highly conserved functional domains of Parkin. Mutations that affect these regions may be deleterious and hence rare or absent in healthy individuals. Alterations in these exons have been observed in ARJP as recessive germline mutations, and in malignancies as heterozygous somatic mutations. None of the 1,686 control subjects was homozygous or compound heterozygous, which implies biallelic mutations are pathogenic. This is certainly the case for ARJP. We do not know, however, if recessive genotypes can cause disorders other than ARJP.
The second component of the study, the role of PARK2 in common PD, addressed a much-debated and extensively published controversy.9–21 Studies have reported heterozygous mutations in patients that were absent in controls, suggesting PARK2 heterozygosity is a risk factor for PD.10–14,16,17 However, not all studies found a higher frequency in patients than in controls.20 Although the sampling of patients varied across studies (early-onset, late-onset, familial PD), the frequency of PARK2 mutations in our patients with PD, when subgrouped to match each study, are generally in line with most prior studies. The main difference between our data and most other published datasets is in the controls. The frequency of PARK2 mutation in our controls is higher than most, but is in line with Lincoln et al.20 and Bruggemann et al.,21 who, like us, genotyped controls comprehensively. Most other studies performed detailed genotyping in patients and screened control subjects only for the mutations found in patients; thus, we suspect, missed the mutations that might have been present in controls but not in patients. The 3 studies that genotyped controls comprehensively had sample sizes of 192,20 356,21 and 1,686 (this study), and all 3 report 3%–4% of controls carrying heterozygous PARK2 mutations. A study of familial PD, however, found no CNV mutations in 263 control subjects, which was significant compared to their patient population.14 Our study had 99% power to detect the effect size reported for familial PD,14 and ∼90% power to detect a minimum OR of 1.5 overall; yet our OR and p values were ∼1. The varied results of published data may be consolidated as follows when 2 key variables are considered: age and genotyping method. 1) PARK2 mutation frequency is high in young-onset PD and drops sharply with increasing onset age, as clearly shown in figure 3. 2) Having PARK2 mutations on both chromosomes causes disease, which is well-established. 3) Having one mutation occurs at a low frequency (<5%) in both patients and control subjects, and is detectable only when the sample size is large and rigorous genotyping methods are used.
There are caveats to our study. Results cannot be generalized to nonwhite populations, nor do they speak to subclinical disease that may be present in heterozygotes.18 Interaction tests had low power. It is possible that a single PARK2 mutation is a “silent” etiologic factor that manifests clinically in combination with as yet unknown PD triggers.
AUTHOR CONTRIBUTIONS
Statistical analysis was conducted by Dr. D.M. Kay and Dr. T.H. Hamza.
ACKNOWLEDGMENT
The authors thank the patients, their family members, and the volunteers who participated in this study. MLPA genotyping was performed by the Applied Genomics Technologies Core facility of the Wadsworth Center, New York State Department of Health.
DISCLOSURE
Dr. Kay, Dr. Stevens, Dr. Hamza, and J.S. Montimurro report no disclosures. Dr. Zabetian has received speaker honoraria from the Swedish Medical Center, the American Parkinson Disease Association, and the Portland Veterans Affairs Research Foundation; receives research support from the NIH (NINDS R01 NS065070-01 [PI], NINDS P50 NS062684-01 [PI Project 3], NINDSR01 NS036960-09 [coinvestigator], NINDS R01 NS057567-01 [coinvestigator], and NIA R01 AG033398-01 [coinvestigator]), the US Department of Veterans Affairs, the Parkinson's Disease Foundation, and the American Parkinson Disease Association. Dr. Factor serves as a Section Editor for Current Treatment Options in Neurology; receives royalties from the publication of Parkinson's Disease Diagnosis and Clinical Management (Demos, 2008) and Drug Induced Movement Disorders (Blackwell Futura, 2005); serves as a consultant for Allergan, Inc., UCB, and Lundbeck Inc.; receives research support from Teva Pharmaceutical Industries Ltd., Ipsen, UCB, and Schering-Plough Corp.; and has served as an expert witness on behalf of Boehringer Ingelheim. Dr. Samii has received funding for travel and speaker honoraria from Teva Pharmaceutical Industries Ltd., Boehringer Ingelheim, and Ipsen. Dr. Griffith has served on a scientific advisory board for and received speaker honoraria from Teva Pharmaceutical Industries Ltd.; and serves on speakers' bureaus for Teva Pharmaceutical Industries Ltd. and Novartis. Dr. Roberts has received speaker honoraria from Teva Pharmaceutical Industries Ltd. Dr. Molho has served on scientific advisory boards for Allergan, Inc., Ipsen, and Merz Pharmaceuticals, LLC; has served on speakers' bureaus for Allergan, Inc., Boehringer Ingelheim, and Teva Pharmaceutical Industries Ltd.; and receives research support from Teva Pharmaceutical Industries Ltd., IMPAX Laboratories, Inc., Allergan, Inc., the NIH [NINDS 1 UO1 NS50324-01A1] [site investigator]), and Molecular Biometrics, Inc. Dr. Higgins has served on speakers' bureaus for GlaxoSmithKline and Boehringer Ingelheim and has received research support from Acadia Pharmaceuticals and Schwarz Pharma. Dr. Gancher, L. Moses, and Dr. Zareparsi report no disclosures. Dr. Poorkaj has received research support from the NIH (NIA 5K01AG024329-02 [PI]) and the University of Washington. Dr. Bird serves on scientific advisory boards for the Association for Frontotemporal Dementia and Charcot-Marie-Tooth Association; serves on the speakers' bureau for and has received funding for travel and a speaker honoraria from Athena Diagnostics, Inc.; serves on the editorial boards of Brain and Neurology Today; has received license fee payments from Athena Diagnostics, Inc. for patents re: Mutations In PKCγ: are the cause for spinocerebellar ataxia, and Mutations associated with a human demyelinating neuropathy (Charcot-Marie-Tooth Disease Type 1C); receives royalties from the publication of Human Genetics: Principles and Approaches, 4th ed (Springer-Verlag GmbH Biomedical Sciences, 2010); and receives research support from the US Department of Veterans Affairs. Dr. Nutt has received funding for travel from Novartis and Teva Pharmaceutical Industries Ltd.; has received speaker honoraria from Novartis and Teva; has served as a consultant for XenoPort Inc., IMPAX Laboratories, Inc., Neurogen Inc., Synosia Therapeutics, NeuroDerm, Ltd., Merck, Lilly-Medtronics, Elan and Lundbeck; and has received research support from Schering-Plough Corp, the NIH (NINDS R01 NS 21062 [PI] and UL1-RR024140 [PI]), the Veterans Administration (PADRECC [Co-PI]), and the National Parkinson Foundation. Dr. Schellenberg serves on a scientific advisory board and receives honoraria from the American Health Assistance Foundation; has served as a consultant for and received funding for travel from IntegraGen; serves on the editorial boards of the American Journal of Alzheimer's Disease and Other Dementias, Neurodegenerative Diseases, Current Alzheimer Research, and Pathology and Laboratory Medicine International; holds/has filed patents re: Chromosome 14 and familial Alzheimer's disease genetic markers and assays, Chromosome 1 gene and gene products related to Alzheimer's disease, and Genetic basis of Alzheimer's disease and diagnosis and treatment thereof; and receives/has received research support from the NIH (NIA R01 AG 11762 [PI], NIA R01 AG 21544 [PI], NHGRI ADD GRANT NUMBER [co-PI], NIA UO1AG032984 [PI], NIA RC2AG036528 [PI], and NIMH R01 MH089004 [PI]), the Autism Genome Project, Autism Speaks, the PSP Genetics Consortium, and CurePSP. Dr. Payami receives research support from the NIH (NINDS NS R01-36960 [PI]) and the New York Attorney General's Office.
Supplementary Material
Address correspondence and reprint requests to Dr. Haydeh Payami, Genomics Institute, New York State Department of Health Wadsworth Center, PO Box 22002 Albany, NY 12201-2002 hpayami@wadsworth.org
Supplemental data at www.neurology.org
Study funding: Supported by National Institutes of Health grant NS R01-36960 from the National Institutes of Neurologic Disease and Stroke (NINDS; H.P.); Veterans Affairs Research Funds (T.B., C.P.Z.); and the Riley Family Chair in Parkinson's Disease (E.S.M.). A subset of control subjects were acquired through the Indiana University National Cell Repository for Alzheimer's Disease, and NIH National Institutes of Aging grant AG 08017 (Jeffrey Kaye). The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Disclosure: Author disclosures are provided at the end of the article.
Received February 2, 2010. Accepted in final form June 8, 2010.
REFERENCES
- 1.Asakawa S, Tsunematsu K, Takayanagi A, et al. The genomic structure and promoter region of the human parkin gene. Biochem Biophys Res Commun 2001;286:863–868. [DOI] [PubMed] [Google Scholar]
- 2.Cesari R, Martin ES, Calin GA, et al. Parkin, a gene implicated in autosomal recessive juvenile parkinsonism, is a candidate tumor suppressor gene on chromosome 6q25-q27. Proc Natl Acad Sci USA 2003;100:5956–5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Veeriah S, Taylor BS, Meng S, et al. Somatic mutations of the Parkinson's disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat Genet 2010;42:77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998;392:605–608. [DOI] [PubMed] [Google Scholar]
- 5.Mira MT, Alcais A, Nguyen VT, et al. Susceptibility to leprosy is associated with PARK2 and PACRG. Nature 2004;427:636–640. [DOI] [PubMed] [Google Scholar]
- 6.Glessner JT, Wang K, Cai G, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 2009;459:569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wongseree W, Assawamakin A, Piroonratana T, Sinsomros S, Limwongse C, Chaiyaratana N. Detecting purely epistatic multi-locus interactions by an omnibus permutation test on ensembles of two-locus analyses. BMC Bioinformatics 2009;10:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burns MP, Zhang L, Rebeck GW, Querfurth HW, Moussa CE. Parkin promotes intracellular Abeta1–42 clearance. Hum Mol Genet 2009;18:3206–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chien HF, Rohe CF, Costa MD, et al. Early-onset Parkinson's disease caused by a novel parkin mutation in a genetic isolate from north-eastern Brazil. Neurogenetics 2006;7:13–19. [DOI] [PubMed] [Google Scholar]
- 10.Hedrich K, Marder K, Harris J, et al. Evaluation of 50 probands with early-onset Parkinson's disease for Parkin mutations. Neurology 2002;58:1239–1246. [DOI] [PubMed] [Google Scholar]
- 11.Lesage S, Lohmann E, Tison F, Durif F, Durr A, Brice A. Rare heterozygous parkin variants in French early-onset Parkinson disease patients and controls. J Med Genet 2008;45:43–46. [DOI] [PubMed] [Google Scholar]
- 12.Clark LN, Afridi S, Karlins E, et al. Case-control study of the parkin gene in early-onset Parkinson disease. Arch Neurol 2006;63:548–552. [DOI] [PubMed] [Google Scholar]
- 13.Foroud T, Uniacke SK, Liu L, et al. Heterozygosity for a mutation in the parkin gene leads to later onset Parkinson disease. Neurology 2003;60:796–801. [DOI] [PubMed] [Google Scholar]
- 14.Pankratz N, Kissell DK, Pauciulo MW, et al. Parkin dosage mutations have greater pathogenicity in familial PD than simple sequence mutations. Neurology 2009;73:279–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun M, Latourelle JC, Wooten GF, et al. Influence of heterozygosity for parkin mutation on onset age in familial Parkinson disease: the GenePD study. Arch Neurol 2006;63:826–832. [DOI] [PubMed] [Google Scholar]
- 16.Oliveira SA, Scott WK, Martin ER, et al. Parkin mutations and susceptibility alleles in late-onset Parkinson's disease. Ann Neurol 2003;53:624–629. [DOI] [PubMed] [Google Scholar]
- 17.Schlitter AM, Kurz M, Larsen JP, et al. Parkin gene variations in late-onset Parkinson's disease: comparison between Norwegian and German cohorts. Acta Neurol Scand 2006;113:9–13. [DOI] [PubMed] [Google Scholar]
- 18.Khan NL, Scherfler C, Graham E, et al. Dopaminergic dysfunction in unrelated, asymptomatic carriers of a single parkin mutation. Neurology 2005;64:134–136. [DOI] [PubMed] [Google Scholar]
- 19.Mortiboys H, Thomas KJ, Koopman WJ, et al. Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann Neurol 2008;64:555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lincoln SJ, Maraganore DM, Lesnick TG, et al. Parkin variants in North American Parkinson's disease: cases and controls. Mov Disord 2003;18:1306–1311. [DOI] [PubMed] [Google Scholar]
- 21.Bruggemann N, Mitterer M, Lanthaler AJ, et al. Frequency of heterozygous Parkin mutations in healthy subjects: need for careful prospective follow-up examination of mutation carriers. Parkinsonism Relat Disord 2009;15:425–429. [DOI] [PubMed] [Google Scholar]
- 22.Kay DM, Moran D, Moses L, et al. Heterozygous parkin point mutations are as common in control subjects as in Parkinson's patients. Ann Neurol 2007;61:47–54. [DOI] [PubMed] [Google Scholar]
- 23.Gibb W, Lees A. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson's disease. J Neurol Neurosurg Psychiatry 1988;51:745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poorkaj P, Nutt JG, James D, et al. Parkin mutation analysis in clinic patients with early-onset Parkinson's disease. Am J Med Genet A 2004;129:44–50. [DOI] [PubMed] [Google Scholar]
- 25.Lucking CB, Durr A, Bonifati V, et al. Association between early-onset Parkinson's disease and mutations in the parkin gene: French Parkinson's Disease Genetics Study Group. N Engl J Med 2000;342:1560–1567. [DOI] [PubMed] [Google Scholar]
- 26.Djarmati A, Guzvic M, Grunewald A, et al. Rapid and reliable detection of exon rearrangements in various movement disorders genes by multiplex ligation-dependent probe amplification. Mov Disord 2007;22:1708–1714. [DOI] [PubMed] [Google Scholar]
- 27.Scarciolla O, Brancati F, Valente EM, et al. Multiplex ligation-dependent probe amplification assay for simultaneous detection of Parkinson's disease gene rearrangements. Mov Disord 2007;22:2274–2278. [DOI] [PubMed] [Google Scholar]
- 28.Powers K, Kay D, Factor S, et al. Combined effects of smoking, coffee and NSAIDs on Parkinson's disease risk. Mov Disord 2008;23:88–95. [DOI] [PubMed] [Google Scholar]
- 29.Kay DM, Factor SA, Samii A, et al. Genetic association between alpha-synuclein and idiopathic Parkinson's disease. Am J Med Genet B Neuropsychiatr Genet 2008;147B:1222–1230. [DOI] [PubMed] [Google Scholar]
- 30.Mata I, Samii A, Factor S, et al. Variation in the 3′ region of the alpha-synuclein gene modifies risk for Parkinson's disease. Mov Disord 2009;24:S145. [Google Scholar]
- 31.Zabetian CP, Hutter CM, Factor SA, et al. Association analysis of MAPT H1 haplotype and subhaplotypes in Parkinson's disease. Ann Neurol 2007;62:137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Available at: http://www.pdgene.org/. Accessed November 29, 2009.
- 33.Payami H, Kay DM, Zabetian CP, Schellenberg GD, Factor SA, McCulloch CC. Visualizing disease associations: graphic analysis of frequency distributions as a function of age using moving average plots (MAP) with application to Alzheimer's and Parkinson's disease. Genet Epidemiol 2009;34:92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.