


Abstract
Objectives:
Observational and experimental studies suggest that nonsteroidal anti-inflammatory drugs (NSAIDs) may protect against Alzheimer disease (AD); however, clinical trials and other observational studies, including the Adult Changes in Thought (ACT) study, show no protection or promotion of AD. The objective of this study is to determine the relationship between common dementia-associated pathologies and mid- to late-life NSAID exposure.
Methods:
We examined the association of mid- to late-life NSAID use with neuropathologic findings on 257 autopsies from ACT, a population-based study of brain aging and incident dementia. Cumulative standard daily doses (SDD) of nonselective NSAIDs were determined from ≥10 years of computerized pharmacy dispensing data. Analyses were adjusted for selection bias to broaden generalizability of results to 3,026 eligible participants in the ACT cohort. Seven pathologic indices were evaluated: intermediate or frequent score for neuritic plaques, Braak stages V or VI for neurofibrillary tangles, >2 cerebral microinfarcts, the presence of any neocortical Lewy bodies, any macroscopic infarcts, any amyloid angiopathy, and moderate or severe atherosclerosis.
Results:
Of the neuropathologic indices evaluated, only neuritic plaque score was significantly increased in participants with greater use of nonselective NSAIDs (p = 0.065), specifically in those with high levels of cumulative use: 1,000–2,000 SDD (adjusted relative risk [RR] 2.16, 95% confidence interval [CI] 1.02–4.25, compared to light/nonuse [<60 SDD]) and >2,000 SDD (adjusted RR 2.37, 95% CI 1.24–4.67).
Conclusions:
Increased neuritic plaque accumulation may explain the association between heavy use of nonselective NSAIDs and increased risk of dementia among ACT participants.
GLOSSARY
- AA
= amyloid angiopathy;
- ACT
= Adult Changes in Thought;
- AD
= Alzheimer disease;
- CERAD
= Consortium to Establish a Registry for Alzheimer's Disease;
- CI
= confidence interval;
- CMI
= cerebral microinfarct;
- DSM-IV
= Diagnostic and Statistical Manual of Mental Disorders, 4th edition;
- GHC
= Group Health Cooperative;
- LBD
= Lewy body disease;
- NFT
= neurofibrillary tangle;
- NP
= neuritic plaque;
- NSAID
= nonsteroidal anti-inflammatory drug;
- OTC
= over-the-counter;
- RR
= relative risk;
- SDD
= standard daily dose;
- VBI
= vascular brain injury.
Several observational studies have concluded that the use of nonselective nonsteroidal anti-inflammatory drugs (NSAIDs) is associated with reduced risk of clinically diagnosed dementia and Alzheimer disease (AD) dementia. These associations appear to be particularly strong in those with protracted NSAID use,1 and perhaps in individuals with an ε4 allele of APOE.2,3 Results from these studies are reinforced by experiments in which transgenic mice treated with NSAIDs accumulate less cerebral Aβ-containing plaques and less hyperphosphorylated tau, both features of AD, along with improved behavior.4–6 However, not all studies have found an apparent protective effect of nonselective NSAIDs.1 In particular, the Adult Changes in Thought (ACT) study observed increased incidence of all-cause dementia and AD dementia in individuals with heavy nonselective NSAID use.7 The Religious Orders Study found no relationship between incident dementia, cognitive function, or neuropathologic changes and NSAIDs use in a population of Catholic Religious over a period of up to 12 years, although of interest, ibuprofen use was associated with higher levels of AD pathologic changes.8 Moreover, clinical trials of nonselective or COX2-selective NSAIDs in patients with AD dementia or with mild cognitive impairment have not demonstrated a neuroprotective effect, and one trial even suggested increased incidence of AD.9–12 A primary prevention trial with NSAIDs in AD was terminated early.13
One possible explanation for these apparently contradictory results is that patients with dementia, including those clinically classified as having AD dementia, are “contaminated” with comorbid disease processes that can contribute to dementia, such as vascular brain injury (VBI) from cerebral microinfarcts (CMIs) or Lewy body disease (LBD).14–17 This is especially true for cohorts of older participants, such as in ACT, where comorbidity is common and the population-attributable risk for dementia is 45% from AD and 33% from VBI.14 The effect of NSAIDs on these pathogenic processes that contribute to dementia in the elderly is unknown. NSAIDs might alter the pathogenesis of these diseases other than AD that commonly contribute to dementia. If so, any protective effects on one type of pathology might be masked in individuals with common comorbid conditions. We analyzed data from the ACT autopsy cohort to help understand effects of NSAIDs on multiple pathologic processes associated with dementia in the elderly.
METHODS
Patients.
ACT is an ongoing prospective, community-based study of brain aging and incident dementia described in detail elsewhere.18,19 Briefly, between 1994 and 2003, ACT enrolled 3,392 cognitively intact community-dwelling participants aged ≥65 years from a population of approximately 23,000 members of Group Health Cooperative (GHC), a large integrated health care delivery system in King County, WA. The ACT cohort was assembled in 2 phases. Between 1994 and 1996, a random sample was drawn to form an initial cohort of 2,581 individuals. Using similar methods, an expansion cohort of 811 was enrolled between 2001 and 2003, yielding a total study size of 3,392. Information on enrollees' demographics, medical history, and cognitive and functional status was collected at baseline and subsequent biennial follow-up visits, with any dementia diagnoses assigned using criteria from the DSM-IV.20
Standard protocol approvals, registrations, and patient consents.
Participants gave informed consent for all procedures as approved by the Institutional Review Boards of GHC and the University of Washington.
Neuropathologic evaluation.
At baseline, participants were asked for consent to brain autopsy. For those who were undecided, additional requests were made at biennial follow-up visits. In accordance with state law, next of kin were also required to file informed consent for autopsy after death. Among the 319 ACT participants who died and underwent study autopsy, 7 neuropathologic endpoints were evaluated as previously described.14 These included neuritic plaque (NP) score according to the Consortium to Establish a Registry for Alzheimer's Disease (CERAD); neurofibrillary tangle (NFTs) as measured by Braak stage21; number of CMIs; extent of neocortical LBD; number of macroscopic cystic infarcts; extent of amyloid angiopathy (AA); and severity of atherosclerotic disease. For all analyses, we dichotomized each neuropathology measure as high vs low/none. High measures (which are associated with increased risk of dementia) were defined as follows: intermediate or frequent NP score; Braak stage V or VI for NFTs; >2 CMIs; any presence of neocortical LBD; any cystic infarcts observed grossly; any presence of AA; and moderate or severe atherosclerotic disease. Archival neuropathology was reviewed in the period between 2005 and 2007 by Drs. Montine and Sonnen in order to form the current neuropathology database and limit interobserver variability.
NSAID exposure.
NSAID prescription data were available on ACT participants from the GHC pharmacy database. This database included all prescriptions dispensed from March 1977 to the present, including drug name, strength, and amount dispensed. To ensure a thorough accounting of longstanding NSAID exposure, only ACT participants with ≥10 years of enrollment in GHC before joining the original or expansion ACT cohorts were included (n = 3,026); this included 293 of the 319 ACT autopsies available at the time of this analysis. The nonselective COX inhibitor NSAIDs used by ACT participants were (in order of use from high to low): ibuprofen, naproxen, indomethacin, sulindac, meclofenamate, tolmetin, piroxicam, diclofenac, nabumetone, flurbiprofen, fenoprofen, etodolac, ketorolac, mefenamic acid, oxaprozin, and ketoprofen. Together, ibuprofen, naproxen, indomethacin, and sulindac accounted for approximately 80% of all prescriptions. For comparisons of dosages among NSAID types, we used standard drug references to assign a standard daily dose (SDD) for each medication type,22 similar to the approach used in the Rotterdam study.23 We then computed the number of SDDs dispensed for each prescription fill by multiplying the number of tablets dispensed by the tablet strength and dividing by the SDD of the NSAID type. For each ACT subject, we summed the cumulative number of SDDs ever dispensed up to time of death, or for those who had not died, up to time of last visit. We categorized levels of cumulative NSAID use as <60 (which included nonusers and would serve as the reference group for all analyses), 60–499, 500–999, 1,000–1,999, and ≥2,000 SDD. The choice of cutpoints for classifying exposure levels was motivated in part by cutoffs used in the prior ACT NSAIDs and clinical dementia study,7 as well as by the need to facilitate comparisons of high levels of cumulative use (e.g., 1,000–1,999, ≥2,000 SDD) to light/nonuse (<60 SDD).
Statistical analyses.
Starting with all eligible ACT study participants in the greater cohort, we classified individuals by their last known study and mortality status (living and enrolled in ACT, living but withdrawn from ACT, deceased but nonconsenting to autopsy, deceased and autopsied). Then we generated descriptive tables of our sample of interest: ACT participants who died and underwent autopsy. We provided demographic and neuropathologic outcome information for these individuals, overall and stratified by levels of cumulative NSAID use. From these individuals, we estimated the association between each of the neuropathologic endpoints and cumulative NSAID use using log-linear models, adjusted for age at death, gender, education, race, and baseline measures of diabetes mellitus, hypertension, and regular exercise. Adjustment variables were selected based on prior literature and plausibility of associations with neuropathologic endpoints and NSAID use. For each endpoint, the relative risk (RR) of a high neuropathology measure is presented for each level of cumulative NSAID use relative to a reference group of <60 SDD. Each neuropathologic outcome represents a different process of interest; as such, no adjustment for multiple comparisons is needed.24 Since autopsy studies are vulnerable to selection bias related to withdrawal, death, and autopsy consent,25 we adjusted all analyses using inverse-probability weighting to broaden the generalizability of our study from the autopsied cases to the entire ACT cohort.26 Weights were obtained by fitting a logistic selection model to all ACT participants, with the outcome of interest being whether or not a subject died and consented to autopsy. Covariates chosen for the selection model included baseline comorbidities such as diabetes, hypertension, heart or cerebrovascular disease, congestive heart failure, and exercise, as well as other characteristics previously reported as being related to consent, including age, race, gender, education, marital status, depression, and dementia status.25 To ensure accurate classification of dementia status at time of death, patients not previously diagnosed with dementia were excluded from analyses if their last clinical evaluation occurred more than 24 months prior to death. This resulted in the exclusion of 30 autopsies, with 6 additional autopsies being excluded due to other missing covariates for the selection model. Sensitivity analyses for the covariates included in the selection model were performed to ensure robustness of results. Further, to ensure adequate accounting of uncertainty due to the introduction of the selection weights, inference for all selection bias models was performed using the bootstrap; bias-corrected and accelerated confidence intervals were constructed for all RRs.14
RESULTS
Table 1 provides baseline characteristics and comorbidity information for the 293 autopsied ACT participants who met the ≥10 years of GHC enrollment criteria. Characteristics are presented stratified by levels of cumulative NSAID exposure. Autopsied participants who were heavy NSAID users (1,000+ SDD) had higher rates of dementia, were more frequently female, and possessed a greater prevalence of hypertension, heart disease, and low self-rated health at ACT study baseline than participants who were light/nonusers (<60 SDD). These relationships across NSAID exposures were also reflected in the broader ACT cohort (table e-1 on the Neurology® Web site at www.neurology.org).
Table 1 Characteristics of autopsied ACT participants, overall and stratified by total cumulative SDD of NSAID use
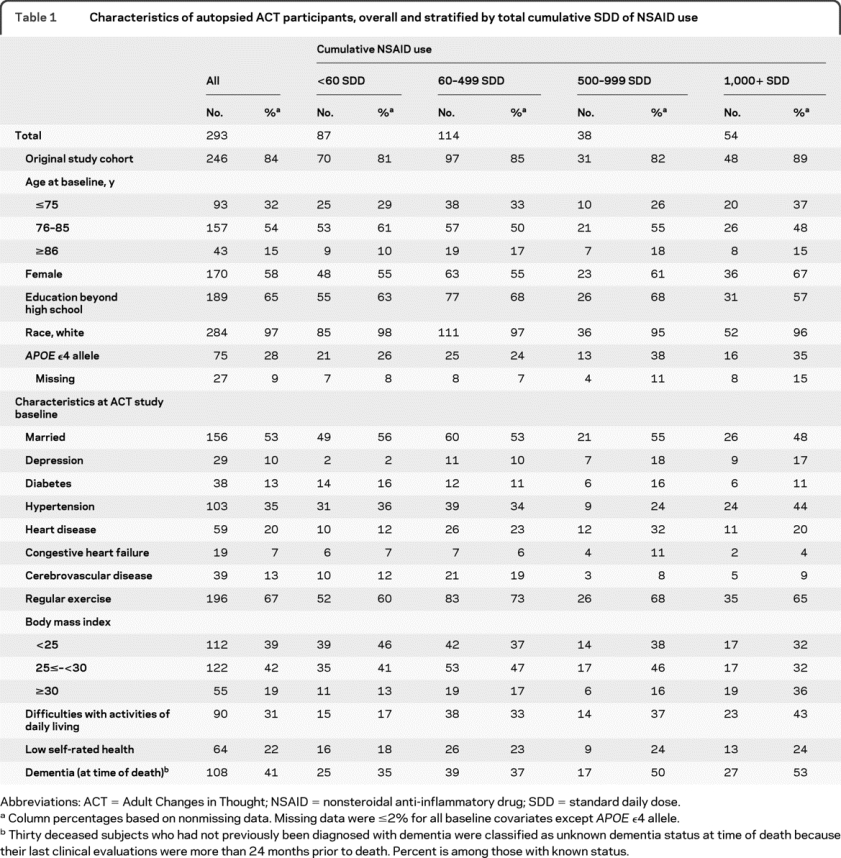
Table 2 presents the dichotomized neuropathologic measures for the included ACT autopsies stratified by cumulative NSAID use. Heavy NSAID users had greater prevalence of high NP scores and advanced NFT stage than light/nonusers. Heavy users also tended to have more cerebral microinfarcts and cystic infarcts, and more severe atherosclerotic disease than light/nonusers. Amyloid angiopathy did not tend to differ across NSAID exposure groups.
Table 2 Neuropathologic measures among eligible ACT autopsies,a overall and stratified by total cumulative SDD of NSAID use
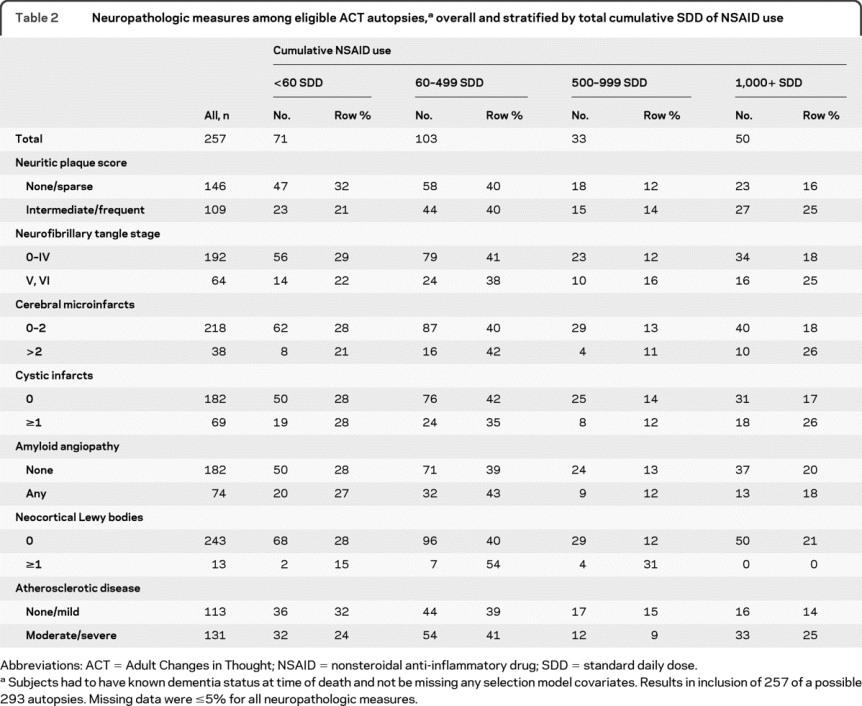
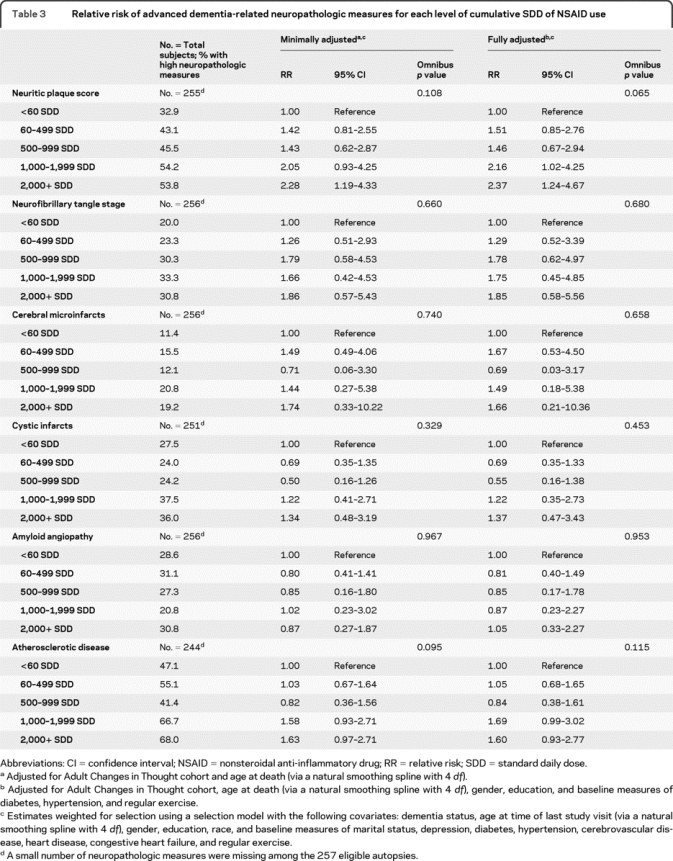
Table 3 presents the RR estimates for the association between the neuropathologic measures and cumulative NSAID use, based on the 257 eligible autopsy cases and adjusted for potential confounding and selection using the entire ACT sample as described above. We did not include the LBD neuropathologic endpoint in these analyses due to the rarity of this outcome in our sample. Of the remaining 6 neuropathologic outcomes, our analyses suggested that only the risk of a high NP score increased with increasing NSAID use (omnibus test: p = 0.065) and that this was significant for the 1,000 to 1,999 SDD (adjusted RR 2.16, 95% CI 1.02–4.25, compared to light/nonuse [<60 SDD]) and ≥2,000 SDD (adjusted RR 2.37, 95% CI 1.24–4.67) groups. Risk of moderate or severe atherosclerosis also tended to be greater (borderline significant) for the 1,000 to 1,999 SDD (adjusted RR 1.69, 95% CI 0.99–3.02, compared to light/nonuse [<60 SDD]) and ≥2,000 SDD (adjusted RR 1.60, 95% CI 0.93–2.77) groups. No other neuropathologic endpoint was significantly associated with NSAID use.
Table 3 Relative risk of advanced dementia-related neuropathologic measures for each level of cumulative SDD of NSAID use
DISCUSSION
Our results indicate that heavy use of NSAIDs by ACT participants was associated with increased NP score, a hallmark feature of AD that contains amyloid β peptides, abnormal neurites, and reactive glia. While the identified association has concordance with results of a recently published study that associated heavy NSAID use among the general population of ACT participants with increased incidence of all-cause dementia and probable AD,7 it is challenging to reconcile our findings with other observational studies that suggest protracted NSAID use reduces the risk of AD. One might speculate that a possible reason for differences in the apparent association with NSAIDs in this study vs earlier epidemiologic studies is that the ACT cohort is older on average. However, it is not possible to test this hypothesis with our data because an ACT study inclusion criterion was that subjects were at least 65 years of age and dementia-free at time of study entry, and most of our subjects were older than 65 at time of entry. Thus, we have no younger cohort that can be used for comparison. Another difference in our study is that because of the extensive pharmacy database in ACT, we measured chronic exposure more fully than previous studies. Also, the specific NSAIDs reported differ between studies. For example, in ACT and the Religious Orders Study, ibuprofen was the most commonly used NSAID, but in the Rotterdam study diclofenac was the most common.8,30 Although no pharmacologic effect of NSAIDs has been demonstrated in vivo other than suppression of COX activity, NSAIDs have idiosyncratic actions in model systems,27 raising the possibility that particular NSAIDs, acting through unspecified means, may enhance the risk for NP accumulation. If true, then the specific ensemble of NSAIDs used may profoundly influence study outcome.
NSAIDs have been implicated in promoting atherogenesis through their impact on the relative concentrations of thromboxane A2 and prostacyclin.28 Indeed we observed increased ratio of urinary metabolites of thromboxane compared to prostacyclin and adverse cardiovascular outcomes in subjects randomized to naproxen or celecoxib in a primary prevention trial for AD.29 Both nonselective and selective NSAIDs have been associated with increased risk of stroke in epidemiologic studies30 while repeated metaanalyses of clinical trials have shown no such association.31,32 Our results suggest that nonselective NSAID use might also be associated with increased atherosclerosis. Nonselective NSAID use may not be associated with altered burden of other common dementia-related neuropathology in the elderly. Despite robust experimental evidence for the neuroprotective effect of NSAIDs on Lewy-related pathology, epidemiologic studies have shown an inconsistent protective effect of chronic NSAID use.33 In a previous study in the ACT cohort, there was no association between risk for PD and NSAID use.34
Our study has unique strengths. We use detailed exposure data derived from a computerized pharmacy database begun in 1977. To our knowledge, this represents the most complete long-term NSAID exposure data applied to an autopsy neuropathology assessment. Also, the pharmacy record avoids common issues of recall bias that are inherent in self-report data. As outcomes, autopsy evaluations do not suffer from the inherent limitations of clinical dementia diagnoses, can confidently identify comorbid neuropathology, and may offer additional insight into the neuropathologic processes underlying dementia. Further, while autopsy studies are known to be prone to selection bias, we were in the unique position of being able to adjust our estimates for selection effects using weights estimated from a selection model using information from the entire ACT cohort.26 In this way we expand the interpretability of our results to a more meaningful population.
We also recognize that our study has several limitations. Our sample size, although relatively large for an autopsy study, precludes more detailed stratification of NSAID type (e.g., ibuprofen, naproxen sodium) at this time. Additionally, as we did not have access to detailed information about over-the-counter (OTC) use of NSAIDs, there likely exists misclassification of NSAID exposure due to any OTC use not captured by the GHC pharmacy database. Self-reported NSAID use (both prescription and OTC) was collected from ACT participants at baseline and biennial study follow-up visits; however, this self-reported information lacked detail on NSAID strength, amount, or duration of use, and instead was limited to a simple yes/no question regarding use in the prior 2 years. We note, however, that only 29 of the 293 autopsied subjects reported any OTC NSAID use at their study visits, and of these 29 subjects, our pharmacy-based NSAID classification already identified 19 of them as having high levels of use (1,000–1999, ≥2,000 SDD) and only classified 1 of them as having no/light use (<60 SDD). Further, in the previous ACT NSAIDs analyses investigating clinical dementia status, the addition of self-report NSAID exposure did not significantly alter results.7 Standardized neuropathologic criteria for the staging of disease also have several limitations including interobserver variability and poor linearity. We have attempted to address interobserver variability by limiting the number of neuropathologist observers. We were insufficiently powered to find associations with less common dementia-associated neuropathologic outcomes such as Lewy body disease or to do separate analyses for individual NSAIDs. Further, given our sample size, effect estimates had large variability (as expressed by the wide CIs); therefore, even though point estimates for neuropathologic endpoints such as advanced NFT stage may have suggested some elevated risk among heavy NSAID use, we lacked power to make more certain conclusions. Finally, while we attempted to address the issue of selection bias within the framework of a selection model, because this model includes dementia status (which is assessed every 2 years by ACT study design), we had to exclude individuals without dementia who had not been assessed within a 2-year window of death.
Another limitation of our study, as with any observational study, is the potential for biased estimates due to residual or unmeasured confounding. We noted in our descriptive tables that subjects with heavier NSAID exposure also had generally worse health status than subjects with lower levels of exposure. These heavy use groups had more heart disease, more hypertension, and overall worse health than light/nonusers; however, it is not clear that more extensive comorbidity among these heavy users would also be associated with increased burden of dementia-related neuropathologic changes. We attempted to adjust for these differences by including comorbid conditions in both our main risk model and our selection model, but there still exists the possibility that our observed elevated risk of an increased NP score is related to the overall poorer health status among heavy NSAID users.
Our study is in accord with the previous finding from ACT that heavy NSAID use is associated with increased risk of dementia7 by demonstrating increased risk of NP accumulation with heavy NSAID use. Furthermore, our findings suggest minimal impact of NSAIDs on other pathologic findings associated with dementia with the exception of possibly atherosclerotic disease, reducing the likelihood that such effects may explain disparate findings in prior studies. Given the frequency with which NSAIDs are used among the elderly, it will be critical for other investigators to validate in other populations the observations made here.
AUTHOR CONTRIBUTIONS
Statistical analysis was conducted by R.L. Walker under the supervision of Dr. Haneuse.
DISCLOSURE
Dr. Sonnen serves as an Associate Editor of the Journal of Alzheimer's Disease; receives royalties from the publication of Molecular Pathology (Elsevier, 2009) and Essentials of Molecular Pathology (Elsevier, 2009); serves as a consultant for the Allen Brain Institute; and receives research support from the NIH/NIA (AG02380 [coinvestigator], AG05136 [coinvestigator], AG19349 [coinvestigator], and AG06781 [coinvestigator]). Dr. Larson served as Chair of the editorial board for the Journal of General Internal Medicine; receives publishing royalties from UpToDate, Inc.; and receives research support from the NIH (AG 06781 [PI]). Mr. Walker receives research support from the NIH (AG05136 [biostatistician], AG23801 [biostatistician], AG000258 [biostatistician], AG06781 [biostatistician], NCI CA63740 [biostatistician], NCI U01CA86076 [biostatistician], NCI U01CA86082 [biostatistician], NCI U01CA63736 [biostatistician], NCI U01CA70013 [biostatistician], NCI U01CA69976 [biostatistician], NCI U01CA63731 [biostatistician], and NCI U01CA70040 [biostatistician]). Dr. Haneuse serves as an Associate Editor for the Journal of Alzheimer's Disease and the International Society for Bayesian Analysis; and receives research support from the NIH (AG05136 [biostatistician], AG23801 [biostatistician], AG000258 [biostatistician], and AG06781 [biostatistician]), and the US Department of Veterans Affairs. Dr. Crane receives research support from the NIH (AG06781 [coinvestigator], AG024180 [coinvestigator], AG 029672-01 [PI], HG004610 [coinvestigator]; AG030618 [coinvestigator], AG010220 [coinvestigator], AR057954 [PI]. Dr. Gray serves on the editorial boards of the Journal of the American Geriatrics Society, Research in Gerontological Nursing, the American Journal of Geriatric Pharmacotherapy, and the Annals of Pharmacotherapy; and has received research support from the NIH (AG06781 [coinvestigator]). Dr. Breitner serves on scientific advisory boards for the N. Bud Grossman Center for Research in Memory and Care, the Florida ADRC (NIA and State of FL), and for the Chicago Health and Aging Project; serves on the editorial boards of Alzheimer's Disease and Associated Disorders, PLOS-One, Academics, Alzheimer's and Dementia, and the American Journal of Alzheimer's Disease; receives royalties from the publication of Fast Facts—Dementia, 2nd ed. (Health Press, 2009); and receives research support from the NIH (AG15477 [PI]) and from the Minnesota Medical Foundation. Dr. Montine serves on the editorial boards of the American Journal of Pathology, Brain Pathology, and the Journal of Alzheimer's Disease; and receives research support from the NIH (NS062684 [PI], AG32984 [co-PI], ES16754 [PI], ES007032 [PI], AG031892 [PI], AG023801 [PI], AG05136 [core leader], AG05136 [project leader], GM015431 [project leader], AG10880 [coinvestigator]) and from the Nancy and Buster Alvord Endowment.
Supplementary Material
Address correspondence and reprint requests to Dr. Joshua Sonnen, Department of Pathology, University of Washington, Seattle, WA 98104 jsonnen@u.washington.edu
Supplemental data at www.neurology.org
e-Pub ahead of print on September 1, 2010, at www.neurology.org.
Study funding: Supported by the NIH/NIA (AG05136, AG23801, AG000258, and U01AG06781), the Nancy and Buster Alvord Endowment, and the US Department of Veterans Affairs.
Disclosure: Author disclosures are provided at the end of the article.
Received January 27, 2010. Accepted in final form June 8, 2010.
REFERENCES
- 1.Szekely CA, Thorne JE, Zandi PP, et al. Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer's disease: a systematic review. Neuroepidemiology 2004;23:159–169. [DOI] [PubMed] [Google Scholar]
- 2.Yip AG, Green RC, Huyck M, Cupples LA, Farrer LA. Nonsteroidal anti-inflammatory drug use and Alzheimer's disease risk: the MIRAGE Study. BMC Geriatr 2005;5:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Szekely CA, Breitner JC, Fitzpatrick AL, et al. NSAID use and dementia risk in the Cardiovascular Health Study: role of APOE and NSAID type. Neurology 2008;70:17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lim GP, Yang F, Chu T, et al. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci 2000;20:5709–5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lim GP, Yang F, Chu T, et al. Ibuprofen effects on Alzheimer pathology and open field activity in APPsw transgenic mice. Neurobiol Aging 2001;22:983–991. [DOI] [PubMed] [Google Scholar]
- 6.McKee AC, Carreras I, Hossain L, et al. Ibuprofen reduces Abeta, hyperphosphorylated tau and memory deficits in Alzheimer mice. Brain Res 2008;1207:225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Breitner JC, Haneuse SJ, Walker R, et al. Risk of dementia and AD with prior exposure to NSAIDs in an elderly community-based cohort. Neurology 2009;72:1899–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arvanitakis Z, Grodstein F, Bienias JL, et al. Relation of NSAIDs to incident AD, change in cognitive function, and AD pathology. Neurology 2008;70:2219–2225. [DOI] [PubMed] [Google Scholar]
- 9.de Jong D, Jansen R, Hoefnagels W, et al. No effect of one-year treatment with indomethacin on Alzheimer's disease progression: a randomized controlled trial. PLoS One 2008;3:e1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reines SA, Block GA, Morris JC, et al. Rofecoxib: no effect on Alzheimer's disease in a 1-year, randomized, blinded, controlled study. Neurology 2004;62:66–71. [DOI] [PubMed] [Google Scholar]
- 11.Aisen PS, Schafer KA, Grundman M, et al. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA 2003;289:2819–2826. [DOI] [PubMed] [Google Scholar]
- 12.Thal LJ, Ferris SH, Kirby L, et al. A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology 2005;30:1204–1215. [DOI] [PubMed] [Google Scholar]
- 13.Group AR, Lyketsos CG, Breitner JC, et al. Naproxen and celecoxib do not prevent AD in early results from a randomized controlled trial. Neurology 2007;68:1800–1808. [DOI] [PubMed] [Google Scholar]
- 14.Sonnen JA, Larson EB, Crane PK, et al. Pathological correlates of dementia in a longitudinal, population-based sample of aging. Ann Neurol 2007;62:406–413. [DOI] [PubMed] [Google Scholar]
- 15.White L, Petrovitch H, Hardman J, et al. Cerebrovascular pathology and dementia in autopsied Honolulu-Asia Aging Study participants. Ann NY Acad Sci 2002;977:9–23. [DOI] [PubMed] [Google Scholar]
- 16.Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 2006;66:1837–1844. [DOI] [PubMed] [Google Scholar]
- 17.Savva GM, Wharton SB, Ince PG, Forster G, Matthews FE, Brayne C. Age, neuropathology, and dementia. N Engl J Med 2009;360:2302–2309. [DOI] [PubMed] [Google Scholar]
- 18.Kukull WA, Higdon R, Bowen JD, et al. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch Neurol 2002;59:1737–1746. [DOI] [PubMed] [Google Scholar]
- 19.Larson EB, Wang L, Bowen JD, et al. Exercise is associated with reduced risk for incident dementia among persons 65 years of age and older. Ann Intern Med 2006;144:73–81. [DOI] [PubMed] [Google Scholar]
- 20.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-IV. Washington, DC: American Psychiatric Association; 1994. [Google Scholar]
- 21.Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 22.Drug Facts and Comparisons. St. Louis, MO: Facts & Comparisons; 2008. [Google Scholar]
- 23.in t'Veld BA, Ruitenberg A, Hofman A, et al. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease. N Engl J Med 2001;345:1515–1521. [DOI] [PubMed] [Google Scholar]
- 24.Rothman KJ. No adjustments are needed for multiple comparisons. Epidemiology 1990;1:43–46. [PubMed] [Google Scholar]
- 25.Zaccai J, Ince P, Brayne C. Population-based neuropathological studies of dementia: design, methods and areas of investigation–a systematic review. BMC Neurol 2006;6:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haneuse S, Schildcrout J, Crane P, Sonnen J, Breitner J, Larson E. Adjustment for selection bias in observational studies with application to the analysis of autopsy data. Neuroepidemiology 2009;32:229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shie FS, Nivison M, Hsu PC, Montine TJ. Modulation of microglial innate immunity in Alzheimer's disease by activation of peroxisome proliferator-activated receptor gamma. Curr Med Chem 2009;16:643–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pratico D, Dogne JM. Vascular biology of eicosanoids and atherogenesis. Expert Rev Cardiovasc Ther 2009;7:1079–1089. [DOI] [PubMed] [Google Scholar]
- 29.Montine TJ, Sonnen JA, Milne G, Baker LD, Breitner JC. Elevated ratio of urinary metabolites of thromboxane and prostacyclin is associated with adverse cardiovascular events in ADAPT. PLoS One;5:e9340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abraham NS, El-Serag HB, Hartman C, Richardson P, Deswal A. Cyclooxygenase-2 selectivity of non-steroidal anti-inflammatory drugs and the risk of myocardial infarction and cerebrovascular accident. Aliment Pharmacol Ther 2007;25:913–924. [DOI] [PubMed] [Google Scholar]
- 31.White WB, West CR, Borer JS, et al. Risk of cardiovascular events in patients receiving celecoxib: a meta-analysis of randomized clinical trials. Am J Cardiol 2007;99:91–98. [DOI] [PubMed] [Google Scholar]
- 32.Chen LC, Ashcroft DM. Do selective COX-2 inhibitors increase the risk of cerebrovascular events? A meta-analysis of randomized controlled trials. J Clin Pharm Ther 2006;31:565–576. [DOI] [PubMed] [Google Scholar]
- 33.Esposito E, Di Matteo V, Benigno A, Pierucci M, Crescimanno G, Di Giovanni G. Non-steroidal anti-inflammatory drugs in Parkinson's disease. Exp Neurol 2007;205:295–312. [DOI] [PubMed] [Google Scholar]
- 34.Ton TG, Heckbert SR, Longstreth WT Jr, et al. Nonsteroidal anti-inflammatory drugs and risk of Parkinson's disease. Mov Disord 2006;21:964–969. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.