

Abstract
Tools to selectively and reversibly control gene expression are useful to study and model cellular functions. When optimized, these cellular switches can turn a protein's function “on” and “off” based on cues designated by the researcher. These cues include small molecules, drugs, hormones, and even temperature variations. Here we review three distinct areas in gene expression that are commonly targeted when designing cellular switches. Transcriptional switches target gene expression at the level of mRNA polymerization, with examples including the tetracycline gene induction system as well as nuclear receptors. Translational switches target the process of turning the mRNA signal into protein, with examples including riboswitches and RNA interference. Post-translational switches control how proteins interact with one another to attenuate or relay signals. Examples of post-translational modification include dimerization and intein splicing. In general, the delay times between switch and effect decreases from transcription to translation to post-translation; furthermore, the fastest switches may offer the most elegant opportunities to influence and study cell behavior. We discuss the pros and cons of these strategies, which directly influence their usefulness to study and implement drug targeting at the tissue and cellular level.
Keywords: Cell switch, Molecular switch, Tet-On, Tet-Off, Nuclear receptor, Riboswitch, RNAi, siRNA, Intein, Rapamycin
There is a sustained need to target specific cellular pathways in a timely and efficient manner, for which new tools must be developed. The ability to selectively control cellular function is advantageous, be it for modeling a disease state or for evaluating pathway-specific therapeutics. To achieve this, various cellular switches have been developed that turn isolated molecular pathways “on” or “off” depending on the selected signal (Figure 1). In our view, an ideal cell switch is: i) reversible, possessing the ability to return to its original state; ii) accurate, targeting only the action of interest; iii) rapid, works quickly enough to minimize the confounding actions of compensatory pathways; and iv) adaptable, the general technology should be robust enough to address multiple cellular targets.
Figure 1.

Cellular switches act at different points on protein expression. The central dogma of molecular biology is evident: DNA is transcribed into mRNA, and mRNA is translated into protein. These three categories of biopolymers interact with each other to do work within the cell. Representative cellular switches are indicated that act on these processes.
Cellular switches are widely used to study cell and molecular biology; however, different approaches meet the above criteria with various strengths and deficiencies. From the perspective of basic science, modulation of an isolated protein can help to determine its role in a chain of events in which it participates. From a translational perspective, cellular switches can be used to mimic pathological behavior, such as those involved in cancer, or to screen therapeutics for participation in a targeted pathway. For example, pathways associated with aggressive cancers may be switched from the “off” position to a pathologic state in the “on” position. These switches enable control over in vitro and in vivo models of cancer and aid in the identification of agents that interact with specific molecular pathways. While multiple switching approaches are used in research, an ideal switch remains a goal on the horizon. Here we discuss the state-of-the art in cellular switching and identify areas for improvement.
I. Transcriptional Switches
Cells naturally respond to their environment by altering gene transcription levels making transcription an excellent place to design a cellular switch. Transcriptional control signals increase or decrease the production of mRNA, which is related to the level of protein expression. Subsequently, this transcriptional response can be magnified many-fold by downstream effectors. Unfortunately, response to transcriptional regulation cannot be described as quick. It can take upwards of 24 h from transcriptional initiation for a mammalian gene to be fully functional [1]. In contrast, when deactivating a target gene product, transcriptional approaches are unable to influence the degradation rate of functional gene products. Despite the temporal delays inherent in transcriptional switches, they can be robust, reversible, and accurate.
1. Tetracycline (Tet) controlled systems
The Tet transcriptional regulators are among the most commonly used cell switches that can be controlled externally. Briefly, the Tet systems are drug-mediated switches that can be fused to genes of interest to control expression [2]. Two distinct flavors of Tet switches are exploited to repress or activate genes, respectively referred to as Tet-Off and Tet-On systems.
The Tet-Off approach utilizes the Tet transactivator (tTA) dimeric DNA binding protein as a regulator of gene expression (Figure 2A, B). This dimeric protein, tTA, is created by fusing the DNA binding domain of Tet-repressor (TetR) with the promoter sequence of viron protein 16 (VP16) produced by the Herpes Simplex virus [2]. tTA is placed under the control of a constitutive promoter so it is always transcribed. The gene of interest is put downstream of the human cytomegalovirus promoter (p-CMV), compatible with the potent transactivator VP16 as well as the Tet operator sequence (tetO) of E. coli transposon Tn10. In the absence of the Tet repressor, tTA binds tetO, activates p-CMV, and transcribes the target gene. When Tet is added to cells, it binds tTA, causing a conformational change in the DNA binding domain, dissociating it from tetO, and stopping gene transcription. Therefore, Tet addition turns the CMV promoter controlled gene off, hence the name Tet-Off. Removal of Tet from the cells then turns the gene back to high expression levels. Depending on the amount of Tet added to the media, up to 1 μg/mL, the gene of interest can be regulated up to 5 orders of magnitude.
Figure 2.
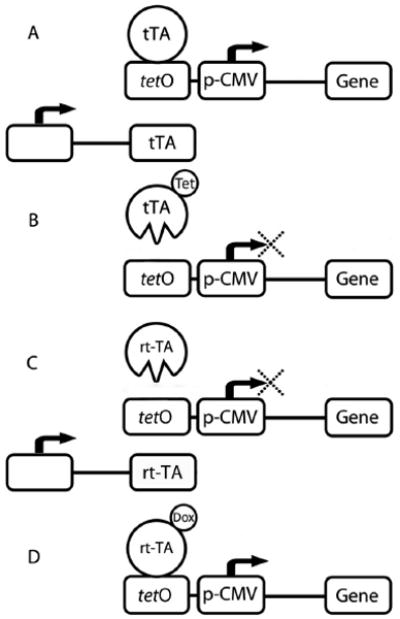
Transcriptional inactivation and activation by the Tet-Off and Tet-On switches. A) In the absence of Tet, tTA complex binds Tet operon, allowing for transcription of the target gene downstream. B) Tet addition causes a conformational change in tTA, inhibiting the complex from binding the Tet operon. Transcription of the target gene is inhibited. C) In the presence of Dox, rtTA is able to bind, and transcription of the target gene proceeds. D) In the absence of Dox, rtTA is unable to bind the Tet operon, inhibiting the transcription of the target gene.
The Tet-On system utilizes similar proteins as the Tet-Off approach, but yields the opposite effect (Figure 2C, D). A reverse Tet-controlled transcriptional activator (rtTA) is transcribed by a constitutive promoter and the gene of interest, linked to p-CMV, is again placed under the control of tetO response element. In comparison with tTA, rtTA has four amino acid substitutions that are responsible for its opposite phenotype, three of which are located at the DNA binding domain [3]. These substitutions were generated using random mutagenesis and changed Glu71 to Lys71, Asp95 to Asn95, Leu101 to Ser101, and Gly102 to Asp102. In place of Tet, the structurally similar antibiotic doxycycline (Dox) is used to induce gene transcription. In the absence of Dox, rtTA does not bind tetO and the target gene remains silent. When Dox is added, it binds rtTA causing a conformational change in the DNA binding domain giving it high affinity for tetO, which turns on the p-CMV promoter and transcribes the target gene, effectively turning the switch on.
A number of improvements have been made since the tTA/rtTA systems were first introduced by Bujard and Gossen in 1992 [2]. Freundlieb et al. have addressed the problem of basal gene expression of the rtTA by developing a Tet controlled transcriptional silencer (tTS). Leaky expression of the gene in the repressed state can occur due to nearby enhancers activating alternative gene promoters [4]. The engineered silencer tTS binds the promoter region of the rtTA gene in the absence of Dox and blocks residual gene expression in the off state. Dox addition prevents tTS binding and allows the gene to be turned on. In the absence of Dox, transfection of cells with tTS decreases basal gene expression by 10 to 200-fold depending on the cell type. This allows for tighter control of gene transcription and increases the regulatory range of rtTA to 3 orders of magnitude [5].
Recently, Hillen and Suess discovered a 50 nucleotide long RNA aptamer that can act in place of Tet [6]. Aptamers are oligonucleotides with three dimensional shapes that can bind specifically to target molecules, in this case tTA. By binding tTA, this RNA aptamer represses transcription. The aptamer is stable in vivo, possesses high induction efficiency, and can be used in combination with Tet to modulate gene activation [6]. Therefore gene expression can be controlled by the concentration of the aptamer, the stability of the aptamer, as well as the amount of Tet added to the system. More recently, the Hillen group has developed novel Tet systems that control multiple genes using novel effectors that bind to new tetO sites. They described a single chain rtTA (sc-rtTA), which is less effected by other Tet-based transregulators. This new sc-rtTA is more sensitive to Dox and has less background activity in its absence[7]. The downside is that expression enhancement of a gene of interest using sc-rtTA is reduced compared to rtTA, likely due to having one activation domain compared to two on a rtTA dimer.
2. Nuclear receptors
Another way to control gene expression at the DNA level is by exploiting naturally occurring nuclear receptors found in the cytosol. Nuclear receptors have the dual role of acting as cellular receptors as well as transcription factors. Steroid and thyroid hormones bind their cognate nuclear receptors, thereby causing a conformational change in the receptor DNA binding domain. This conformational change provides a higher binding affinity for DNA, which induces redistribution to the nucleus, DNA binding, and targeted transcription. Naturally occurring nuclear receptors include the estrogen receptor and the peroxisome proliferator-activated receptor (PPAR) [8, 9].
To utilize nuclear receptors as biological switches, the specificity of the activation domain must be altered to respond to a molecule of interest while avoiding activation by natural analogues. The outstanding challenge of producing orthogonal receptor-ligand pairs is the labor-intensive process[10]. Molecular modeling is used to determine residues responsible for the loss of activity of the receptor, followed by the mutation of these residues, and the selection of mutants with high affinity for the desired ligand [11]. The Koh group describes TRB(H435A), a mutant of the thyroid hormone receptor that responds to the synthetic hormone QH2 61-fold higher over the natural ligand triiodothyronine(T3) [12]. Additionally, the TRB(H435A) mutant is non-responsive to physiological levels of T3 yet QH2 has low nanomolar activity at 6.4 nM. QH2 has been used to drive the expression of thyroid hormone response elements downstream of the thyroid hormone promoter. Alternatively, Schwimmer et al. describe a retinoid X receptor (RXR) mutant that has 300-fold higher affinity for the synthetic ligand LG335 than for retinoic acid [13]. Activation of this RXR mutant selectively drives the transcription of retinoic acid response elements.
To avoid basal transcription induced by natural hormones, insect-derived molting hormones, ecdysteroids, have been successfully used as gene switches in mammals [14, 15]. The ecdysteroid receptor (EcR) gene naturally binds ultraspiracle protein (USP) to form a heterodimer that binds DNA. To increase transduction efficiency, the EcR is genetically modified with a VP16 activation domain. In addition, the natural USP heterobinding protein can be replaced with the mammalian retinoic X receptor (RXR). The VP16/EcR fusion protein in conjunction with RXR binding, increases induction 212-fold over the native USP domain [14]. This switch can be induced by muristerone hormone that causes EcR-RXR to dimerize, translocate to the nucleus, and bind to an ecdysone-response element upstream of the gene of interest. Therefore, muristerone addition turns the gene of interest on. The gene remains off in the absence of muristerone. Recently, the potency of this gene-switch has been increased by methylating key groups on the ecdysteroid binding site [15].
3. Pros and cons of transcriptional switches
Many cell-switch approaches that target transcription rely on the interaction of a ligand and a receptor, commonly resulting in dimerization that induces nuclear localization and binding to a DNA-response element upstream of the gene of interest. The TetR systems are advantageous over nuclear receptors in that they require the expression of only one control protein, TetR, to modulate transcription. In contrast, nuclear receptors, as targeted by ecdysteroid, require the constitutive expression of two proteins, VP16/EcR and RXR. Another advantage, TetR approaches can either up-regulate or down-regulate in the presence of drug, while nuclear receptors are currently limited to up regulation.
However, TetR has limitations when used in animal models. TetR has been found to induce inefficient expression in certain tissues, and the drugs themselves can induce toxicity [16]. The use of nuclear receptors, ecdysteroids in particular, offer advantages over other transcription induction systems for in vivo studies. Steroids penetrate tissue and cellular barriers; therefore, controlling cellular expression with a steroid ligand avoids some cellular penetration problems observed with alternative approaches. Also, mouse studies have shown that ecdysteroid pharmacokinetics are favorable over TetR since they have faster distribution and clearance [14]. Lastly, ecdysteroid-controlled genes have low basal expression and high inducibility. While the EcR switch has yet to be thoroughly developed for animal models, preliminary studies suggest this approach may be superior for in vivo cellular switching [16].
II. Translational Switches
Protein translation is another node along the central dogma of modern biology that can be targeted for cellular switching. Two common approaches include riboswitches and RNA interference. Riboswitches are enzymatically active mRNA that regulate their own genetic code. Currently, the most ubiquitous cellular switches are based on RNA interference, which use small RNA molecules to target and destroy mRNA, reducing protein translation. Switches installed at the level of protein translation have temporal advantages over transcriptional switching since they act further down the gene expression pathway. Translational switches have the potential to relay on and off signals more quickly because they directly target mRNA and avoid the cellular transcriptional machinery. In addition, the translational machinery can increase or decrease protein expression, even under constant transcription; therefore, direct targeting of translation has broad potential for controlling the cellular levels of active proteins.
1. Riboswitches
RNA-mediated control of translation naturally occurs in prokaryotes as a response to metabolite fluctuations within the cell [17]. This observation has led to the engineering of RNA in eukaryotes that translate their protein products in response to the addition of target ligands. These riboswitches are found in the endogenous noncoding 5′ untranslated region (5′UTR) of mRNAs in bacteria. Riboswitches have two components, an aptamer region that binds a specific ligand, such as amino acids, coenzymes, vitamins, and purine bases, as well as an expression cassette that can translate a target gene sequence (Figure 3). These expression platforms function most commonly through conformational changes of the mRNA structure, changes that can result in early termination or inhibition of translation by blocking of ribosomal docking [18]. RNA aptamer domains are typically 70-170 nucleotides long. An example of a naturally occurring riboswitch is the thiamine pyrophosphate riboswitch. Thiamine pyrophosphate, acting as the ligand, binds the aptamer domain, which causes a conformational change in the downstream mRNA that blocks initiation of translation up to 16-fold for genes involved in thiamine synthesis. In the presence of thiamine pyrophosphate, this riboswitch turns protein translation off. Conversely, a glycine dependent riboswitch has been described that up regulates mRNA translation into protein. For these riboswitches, the glycine-binding aptamer is found upstream on the mRNA of several genes involved in glycine metabolism. When glycine concentrations increase within the cell, glycine binds the upstream riboswitch aptamer and causes translation of proteins that process glycine, presumably so that excess glycine can be used as an energy source [19]. In prokaryotes, a dissociation constant of 20-30 μM has been reported [20].
Figure 3.
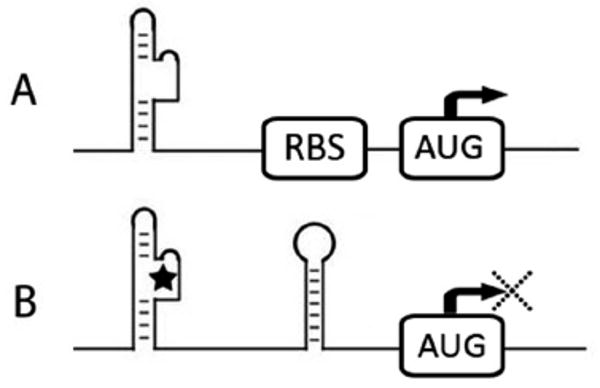
Riboswitch inactivation of ribosomal docking and initiation of translation. A) A RNA aptamer in a three dimensional conformation open for ribosomal docking in 5′UTR. B) A RNA aptamer binds to ligand, which causes a conformational change, formation of a hairpin turn, and sequestration of the ribosomal docking site upstream of the AUG start codon. This inhibits the initiation of protein translation.
These naturally occurring riboswitches in prokaryotes have been re-engineered to selectively control protein translation in eukaryotic cells. Guar et al. [21] have developed an artificial riboswitch that responds to theophylline, a drug with good cell uptake properties. A theophylline-binding aptamer is designed into the 3′ splice site region of a model pre-mRNA. In the presence of theophylline, splicing of the pre-mRNA is inhibited. Guar et al. showed that the location of the AG sequence, a 3′ splice site found at the end of introns, plays a role in theophylline response. Also, the theophylline aptamer is specific for its target drug, which was demonstrated by a lack of response to small molecules with similar properties. Finally, the group showed that the addition of theophylline to cells lacking the aptamer elicited no response, showing that the inhibition of mRNA splicing and the subsequent inhibition of gene expression was due to the artificial theophylline riboswitch.
The development of new riboswitches appears to be increasingly robust. Selective Evolution of Ligands by Exponential Enrichment (SELEX) is one approach used to identify optimal mRNA sequences for riboswitch targeting [22]. For SELEX, a large oligonucleotide library is exposed to the desired ligand, and non-binding oligos are washed away using affinity chromatography. The remaining oligos are amplified using PCR and recursively selected for low equilibrium binding constants and slower disassociation rates [23]. Suess et al. have recently described a SELEX approach using an in vitro screen followed by an in vivo screen in order to select aptamers that control translation in yeast [24]. A library of SELEX derived sequences specific for the drug neomycin were cloned upstream of a GFP reporter gene. Riboswitches containing neomycin aptamers were developed that turn off target gene translation. First, the sequences were screened for GFP expression in the absence of neomycin. Then, the selected constructs were screened for decreases in fluorescence in response to neomycin. In total 50,000 sequences were screened, and 30 sequences were identified with neomycin-dependent decrease in GFP fluorescence. One construct decreased gene expression 7.5-fold. This robust approach enables the screening of large numbers of potential aptamers as translational regulators and has significant control a wide variety of aptamers for various applications.
2. RNA interference
Since its development in the late 90s [25], RNA interference technology has arguably become one of the most popular methods of expression knockdown. To reversibly arrest protein translation, double stranded RNA (dsRNA) homologous to the target mRNA is administered either directly using small interference RNA (siRNA) or via DNA plasmids that expresses a short hairpin RNA (shRNA) [26]. shRNA is later cleaved into siRNA.
RNA interference is typically administered in one of two ways: either the siRNA is chemically synthesized and delivered to cells, or shRNA is made by genes that are incorporated into the cell [27]. The transcription of RNA from a DNA plasmid can be induced externally by administration of drug/ligands as already discussed for the Tet system. Endogenous dsRNA is cleaved by Dicer into 21 nucleotide base sequences with 2 nucleotide overhangs at the 3′ ends. These small fragments are termed siRNAs. siRNAs can also be added exogenously, bypassing Dicer cleavage into smaller sequences. siR-NAs are comprised of 2 strands, the sense and the antisense strand relative to the target mRNA (Figure 4). The double stranded siRNA complexes with the RNA-induced silencing complex (RISC), and activates it. The activated RISC complex includes Argonaut, a protein with endonuclease activity that serves as the catalytic component of the RISC complex. After the sense strand dissociates, the antisense strand binds its complementary target mRNA using Watson-Crick base pairing [28]. Once bound, RISC cleaves the target mRNA into fragments, inhibiting its translation into protein. RNA interference is slowly reversible upon clearance of siRNA. Without the introduction of additional siRNA, normal translation resumes.
Figure 4.
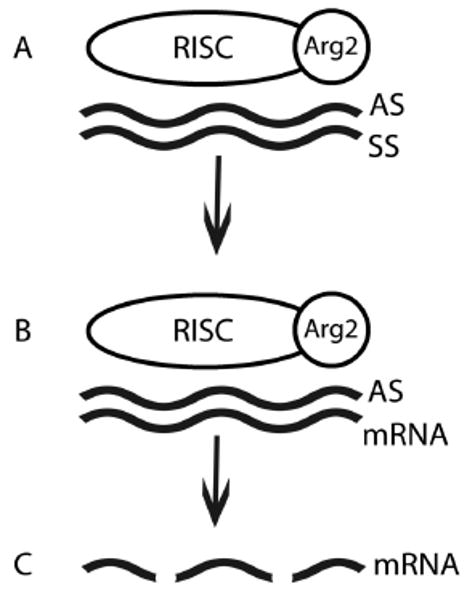
Translational control of gene expression via the RISC complex on siRNA. A) A double stranded RNA complex is added. The RISC complex binds to the anti-sense strand (AS) via the Argonaut protein. B) Complementary mRNA binds the anti-sense strand. C) Complementary mRNA is cleaved by RISC, which down regulates the translation of target mRNA.
One drawback for the use of RNA interference technology as a cellular switch is its off target effects. Not only does the time delay from administration to phenotype expression afford the cell the opportunity to compensate for the loss of protein function, but siRNA can potentially effect the output of hundreds of genes [29]. One of these effects is the decrease of endogenous microRNA effects due to si/shRNA's competition for incorporation into the RISC complex, which can cause cellular toxicity [30-32]. Such toxicity has been described by Grimm et al. in mice when shRNA is expressed long term in the liver [33]. Numerous modifications to the siRNA approach have been proposed to limit toxicity while still retaining high efficiency. For example, the non-guiding sense strand of the siRNA, the strand that does not bind RISC, should contain disfavored overhangs while the guiding anti-sense strand, the strand of RNA complementary to mRNA, should contain favored overhangs to ensure incorporation into RISC [34]. Lowering the concentration of siRNA administered is one way to mitigate off target effects; however, the level of knockdown is dose dependent [35]. To compensate for reduced doses, longer siRNA duplexes can be used (27 nucleotides as opposed to 21) that have higher target affinities. These longer siRNAs can be effective at lower doses, acting with 100-fold higher potency than corresponding 21 nucleotide siRNAs. The longer 27 nucleotide siRNAs start to show knockdown at 50 pM concentrations and full knockdown at 5 nM. Shorter 21 nucleotide siRNAs do not begin to show knockdown until 5 nM and never reached full knockdown. Since the off target effects are correlated to the amount of siRNA administered [36], the lower amount of the 27 nucleotide siRNA may decrease these side effects. By appropriate optimization, these 27-mer siRNAs may avoid activation of interferon or protein kinase R, substrates of the cellular immune response that induce toxicity due to siRNA [27].
A significant obstacle to in vitro siRNA silencing is due to the large molecular size and poor membrane permeability of oligonucleotides. Various delivery systems are required to introduce siRNA, which include lentiviral vectors and polyelectrolyte complexes [37]. Naturally, the uptake and cellular transformation efficiency limits the speed, effectiveness and reversibility of siRNA based switches.
3. Pros and cons of translation-mediated switches
Using translation to control cellular function has advantages as well as pitfalls. Riboswitches are unique because they do not require the constitutive expression of foreign proteins to control expression. When using proteins to control genetic expression, such as tTA in the Tet system, subtle increases or decreases in concentration may amplify changes in expression of the target protein. Since riboswitches do not utilize cell proteins, they offer good control over translation. Despite this, a major limitation of riboswitches is that they must be delivered by an expression platform base. Additionally, only 6 aptamers have been identified that can be engineered to function as riboswitches and only 2 of them, theophylline and Tet, have been used as binding domains in creating more complex riboswitches [24].
RNA interference has the advantage over other cellular switches in its ability to target genes of known sequence. Setting up a functional RNAi system requires little more than generating the desired oligonucleotide and administering it to cells. This circumvents the need to do large-scale selection screens such as those required to generate riboswitches. The speed as well as dose-dependent control of RNAi activity may be improved using the previously described Tet responsive approaches [38]. Despite this, off-target effects remain problematic with RNAi technology, which must be addressed when implementing and analyzing data from this approach.
III. Post-Translational Switching
Cellular switching at the post-translational level can be achieved by manipulating pre-proteins in order to control their activity. One advantage of such a switch over a transcriptional or translational modulator is its ability to operate on the scale of minutes as opposed to hours. This speed reduces the time for the cell to compensate for the loss or addition of function with confounding pathways. Also, it can avoid the potential toxicity that occurs due to prolonged absence of essential functions, such as RISC. And furthermore, cellular switch at the protein level may be more advantageous for study of cell cycle processes as they are temporally contingent. Two post-translational switches will be discussed: chemical inducers of dimerization (CIDs) and protein (intein) splicing.
1. Chemical inducers of dimerization
The term “chemical inducers of dimerization” (CID) was first coined by the Crabtree group to describe a bifunctional ligand that bridges two hetero- or homologous proteins [39]. Bifunctional ligands are inspired by the naturally occurring FKBP12-FK506-calcineurin system. When immunosuppressant drug, FK506, is added, it binds FK506-binding protein (FKBP12) with a nanomolar binding affinity of 0.4 nM [40]. This complex binds the serine/threonine phosphatase calcineurin and inactivates it by sequestering it from entering the nucleus. While FKBP12-FK506 binds and inhibits calcineurin, the FKBP12-rapamycin system binds and inactivates FRAP [41]. Rapamycin, an FK506 analogue, binds both FKBP12 and FKBP-rapamycin-associated protein (FRAP), a component of mTOR [42]. Rapamycin simultaneously binds both these proteins within two hydrophobic binding pockets. In the absence of dimerizing agents FK506 or rapamycin, the binding affinity of FKBP12 for either calcineurin or FRAP is absent.
Since its characterization, analogues to FK506 and rapamycin have been developed that display ligand inducible aggregation and disaggregation. In both instances, the protein of interest is genetically fused to a variant of the FK506 binding domain. In ligand inducible aggregation, addition of FK506 induces aggregation between proteins tagged with its binding domain. Such dimerization has been shown to be sufficient for a variety of cellular functions [40, 43-45]. For example, Moskowitz et al. showed that the assembly of clathrin endocytosis machinery was arrested by adding the cell-permeable crosslinker FK1012-A to cells with FK506-binding protein tagged clathrin light chain [46]. FK1012-A is a molecular conjugate of two FK506 moieties, and addition of this small molecule results in the homo-dimerization of FKBP12 domain (Figure 5). In this case, two clathrin light chains fused with FKBP12 moieties dimerize and lose function. Addition of FK1012-A to live cells reduced membrane budding events by 3.3-fold and transferrin internalization by 2.2-fold. This process was reversible, and clathrin-mediated endocytosis resumed within 2 h after the removal of drug [46]. Alternatively, ligand induced disaggregation was demonstrated by Rollin et al. By mutation of a single amino acid in the ligand-binding domain, the naturally monomeric FKBP dimerizes, which constitutively deactivates its fusion protein. Adding a ligand of FKBP, such as rapamycin, dissociates dimeric complexes into monomer form, which restores protein activity [47]. Another example has been demonstrated by Crabtree et al., who engineered the amyloid beta peptide to contain FKBP [48]. Upon addition of rapamycin, even in nanomolar ranges, amyloid beta, a protein whose aggregation is associated with Alzheimer's disease, was blocked from self aggregation.
Figure 5.
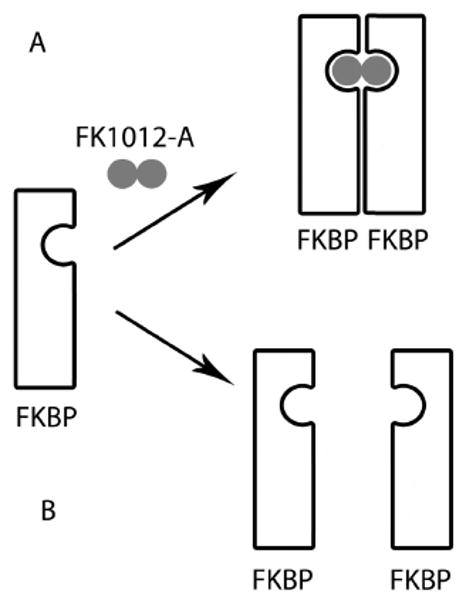
Post-translational protein deactivation using chemical inducers of dimerization. A rapamycin-analogue (FK1012-A) approach is depicted. A) In the presence of FK1012-A, FK506-binding protein (FKBP) forms dimeric complexes, which inhibits the activity of some proteins. B) In the absence of FK1012-A, FKBP becomes monomeric, which restores protein activity.
As discussed above, the rapamycin-mediated switch requires optimization of a fusion protein between a target protein and an active FKBP domain. To avoid the need to develop fusion constructs, an alternative is to design bi-functional ligands with two sites that bind directly to the unmodified target protein. For example, Kudus et al. showed that a derivative of geldanamycin functions as a bifunctional linker. This derivative was used to dimerize the estrogen receptor with the molecular chaperone Hsp90 inducing degradation of the receptor [49]. As an alternative to degradation, chemical inducers of dimerization can be engineered to fuse two proteins that do not interact under normal circumstances. By co-localizing functional protein domains, synthetic dimers can create new cellular functions. This strategy circumvents the possible perturbations that can occur with genetically manipulated proteins; however, the optimization of bifunctional ligands is akin to the development of a drug-like small molecule, which presents alternative challenges.
2. Intein splicing
The intein splicing strategy, also referred to as protein splicing, utilizes the post-translational gene product to function as a switch [50]. An intein is a region of amino acids within an inactivated protein that spontaneously excises itself while joining its two flanking exteins, thereby activating the protein (Figure 6). This natural function has been harnessed to respond to both temperature and small molecule inducers. Temperature sensitive mutants with conditionally active inteins have been identified [51]. At permissive temperatures, an intein splices itself out, yielding the functional protein. At non-permissive temperatures, the intein sterically blocks the functional association of flanking exteins and the protein remains inactive. To adapt this technology to other model systems and avoid time intensive mutant screens, Tan et al. devised a strategy that generates conditional temperature mutants [52]. A library of temperature sensitive inteins was constructed. These inteins were then inserted into a protein of interest, making fusion proteins responsive to the temperature-sensitivity of the intein. This approach generated mutants sensitive to temperatures ranging from 18 to 30 °C. This narrow window is critical to the development of useful switches because they must operate near physiological temperatures.
Figure 6.
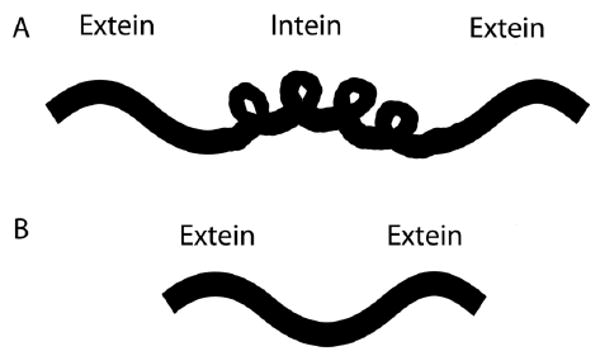
Post-translational protein activation by intein splicing. A) An intein is located between 2 exteins on a protein. B) Intein excision activates the protein. Protein remains inactive until intein excision is prompted.
To carry out cellular switching experiments at isothermal conditions, it is beneficial to use small molecules to control intein splicing. To achieve this, inteins have been developed that are ligand responsive. Yuen et al. developed an intein mutant that is responsive to hydroxytamoxifen (4HT), where 4HT initiates protein splicing and activation [53]. 4HT responsive inteins were genetically inserted into native mammalian proteins, from which active proteins could be developed from a variety of genes, including GFP and transcriptional mediators of hedgehog signaling such as Gli3 and Gli1 [53]. Addition of 1 μM of 4HT resulted in a 10-fold increase in fluorescence in cells expressing the GFP-intein. Muir et al. have shown intein splicing can be rapamycin inducible. FKBP and FRB binding domains are fused to two separate inteins, which are in turn fused to 2 extein proteins. In the presence of rapamycin, the FKBP and FRB domains dimerize and bring the inteins into close enough proximity to induce splicing, which activated the target protein vacuolar ATPase subunit A. In the absence of rapamycin, the protein remains inactive [54, 55]. As with ligand inducible disaggregation, when the homodimerizing F36M mutant of FKBP12 is attached to an intein, protein splicing occurs normally until the addition of rapamycin, which blocks splicing and activity. The FKBP12 protein is usually monomeric, but the F36M mutation causes it to homodimerize. In the absence of rapamycin, protein splicing and therefore protein function is constitutively on. Adding rapamycin stops F36M homodimerization, inhibiting the proximity of the two inteins, and stops splicing, thus inhibiting the production of functional protein [56].
3. Pros and cons of post-translational switches
The most pronounced advantage of post-translational control for a cell switch system is its ability to be temporally controlled. While transcriptional and translational control of protein production can take upwards to 24 h, protein dimerization and splicing can be successfully accomplished within minutes [46]. Speedy switching is essential, as it enables the modulation of essential genes without permitting excessive time for the accumulation of secondary and tertiary cellular responses that may easily confound the primary effects of the target or even lead to cell death. The rapamycin-FKBP strategy is rapid and controlled by a reversible interaction with a small molecule; however, this approach still requires genetic manipulation of the target protein. Similarly, the intein approach requires genetic engineering and can be drug mediated; however, a major drawback of the intein strategy is that it is not reversible.
A variety of tools exist that can reversibly switch cellular processes on and off; furthermore, protein activity can be manipulated at transcription, translation, or post-translation. By targeting the transcriptional machinery, its expression may be augmented many fold using both the TetR system and nuclear receptors. Both approaches have been evaluated using in vitro models; however, it is unclear which approach is superior in vivo. By targeting the translational machinery, a tighter control of protein expression can be achieved using RNA interference and riboswitches. RNA interference is widely used due to its ease of implementation, but remains leaky and potentially immunogenic. In contrast, riboswitches are challenging to implement since new models require labor-intensive mutational screens. By targeting post-translational modification, it is possible to induce more rapid effects using techniques based on chemically induced dimerization or intein excision. Directly controlling protein-protein interaction using dimerization and intein splicing may well be the fastest way to control intracellular processes; however, many post-translational approaches continue to require genetic manipulation of the target in order to optimize a model system. Ultimately, the simplicity of implementation and the necessity for speed as well as reversibility should determine which strategy to pursue.
References
- 1.Buskirk AR, Liu DR. Creating small-molecule-dependent switches to modulate biological functions. Chem Biol. 2005;12:151–61. doi: 10.1016/j.chembiol.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 2.Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA. 1992;89:5547–51. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gossen M, Freundlieb S, Bender G, Muller G, Hillen W, Bujard H. Transcriptional activation by tetracyclines in mammalian cells. Science. 1995;268:1766–1769. doi: 10.1126/science.7792603. [DOI] [PubMed] [Google Scholar]
- 4.Freundlieb S, Baron U, Bonin AL, Gossen M, Bujard H. Use of tetracycline-controlled gene expression systems to study mammalian cell cycle. Methods Enzymol. 1997;283:159–173. doi: 10.1016/s0076-6879(97)83014-5. [DOI] [PubMed] [Google Scholar]
- 5.Freundlieb S, Schirra-Muller C, Bujard H. A tetracycline controlled activation/repression system with increased potential for gene transfer into mammalian cells. J Gene Med. 1999;1:4–12. doi: 10.1002/(SICI)1521-2254(199901/02)1:1<4::AID-JGM4>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 6.Hunsicker A, Steber M, Mayer G, Meitert J, Klotzsche M, Blind M, Hillen W, Berens C, Suess B. An RNA aptamer that induces transcription. Chem Biol. 2009;16:173–180. doi: 10.1016/j.chembiol.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 7.Zhou X, Symons J, Hoppes R, Krueger C, Berens C, Hillen W, Berkhout B, Das AT. Improved single-chain transactivators of the Tet-On gene expression system. BMC Biotechnol. 2007;7:6. doi: 10.1186/1472-6750-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hurtado A, Holmes KA, Geistlinger TR, Hutcheson IR, Nicholson RI, Brown M, Jiang J, Howat WJ, Ali S, Carroll JS. Regulation of ERBB2 by oestrogen receptor-PAX2 determines response to tamoxifen. Nature. 2008;456:663–666. doi: 10.1038/nature07483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature. 2008;454:470–477. doi: 10.1038/nature07202. [DOI] [PubMed] [Google Scholar]
- 10.Chockalingam K, Chen Z, Katzenellenbogen JA, Zhao H. Directed evolution of specific receptor-ligand pairs for use in the creation of gene switches. Proc Natl Acad Sci USA. 2005;102:5691–5696. doi: 10.1073/pnas.0409206102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kinzel O, Fattori D, Muraglia E, Gallinari P, Nardi MC, Paolini C, Roscilli G, Toniatti C, Gonzalez Paz O, Laufer R, Lahm A, Tramontano A, Cortese R, De Francesco R, Ciliberto G, Koch U. A structure-guided approach to an orthogonal estrogen-receptor-based gene switch activated by ligands suitable for in vivo studies. J Med Chem. 2006;49:5404–5407. doi: 10.1021/jm060516e. [DOI] [PubMed] [Google Scholar]
- 12.Hassan AQ, Koh JT. A functionally orthogonal ligand-receptor pair created by targeting the allosteric mechanism of the thyroid hormone receptor. J Am Chem Soc. 2006;128:8868–8874. doi: 10.1021/ja060760v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwimmer LJ, Rohatgi P, Azizi B, Seley KL, Doyle DF. Creation and discovery of ligand-receptor pairs for transcriptional control with small molecules. Proc Natl Acad Sci USA. 2004;101:14707–14712. doi: 10.1073/pnas.0400884101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.No D, Yao TP, Evans RM. Ecdysone-inducible gene expression in mammalian cells and transgenic mice. Proc Natl Acad Sci USA. 1996;93:3346–3351. doi: 10.1073/pnas.93.8.3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lapenna S, Dinan L, Friz J, Hopfinger AJ, Liu J, Hormann RE. Semi-synthetic ecdysteroids as gene-switch actuators: synthesis, structure-activity relationships, and prospective ADME properties. Chem Med Chem. 2009;4:55–68. doi: 10.1002/cmdc.200800280. [DOI] [PubMed] [Google Scholar]
- 16.Albanese C, Hulit J, Sakamaki T, Pestell RG. Recent advances in inducible expression in transgenic mice. Semin Cell Dev Biol. 2002;13:129–141. doi: 10.1016/s1084-9521(02)00021-6. [DOI] [PubMed] [Google Scholar]
- 17.Nudler E, Mironov AS. The riboswitch control of bacterial metabolism. Trends Biochem Sci. 2004;29:11–17. doi: 10.1016/j.tibs.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 18.Link KH, Breaker RR. Engineering ligand-responsive gene-control elements: lessons learned from natural riboswitches. Gene Ther. 2009 doi: 10.1038/gt.2009.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mandal M, Lee M, Barrick JE, Weinberg Z, Emilsson GM, Ruzzo WL, Breaker RR. A glycine-dependent riboswitch that uses cooperative binding to control gene expression. Science. 2004;306:275–279. doi: 10.1126/science.1100829. [DOI] [PubMed] [Google Scholar]
- 20.Kwon M, Strobel SA. Chemical basis of glycine riboswitch cooperativity. RNA. 2008;14:25–34. doi: 10.1261/rna.771608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim DS, Gusti V, Pillai SG, Gaur RK. An artificial riboswitch for controlling pre-mRNA splicing. RNA. 2005;11:1667–1677. doi: 10.1261/rna.2162205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mayer G, Famulok M. In vitro selection of conformational probes for riboswitches. Methods Mol Biol. 2009;540:291–300. doi: 10.1007/978-1-59745-558-9_21. [DOI] [PubMed] [Google Scholar]
- 23.Mairal T, Ozalp VC, Lozano Sanchez P, Mir M, Katakis I, O'Sullivan CK. Aptamers: molecular tools for analytical applications. Anal Bioanal Chem. 2008;390:989–1007. doi: 10.1007/s00216-007-1346-4. [DOI] [PubMed] [Google Scholar]
- 24.Suess B, Weigand JE. Engineered riboswitches: overview, problems and trends. RNA Biol. 2008;5:24–29. doi: 10.4161/rna.5.1.5955. [DOI] [PubMed] [Google Scholar]
- 25.Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 26.Pratt AJ, Macrae IJ. The RNA-induced silencing complex: a versatile gene-silencing machine. J Biol Chem. 2009;284:17897–17901. doi: 10.1074/jbc.R900012200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Castanotto D, Rossi JJ. The promises and pitfalls of RNA-interference-based therapeutics. Nature. 2009;457:426–433. doi: 10.1038/nature07758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rana TM. Illuminating the silence: understanding the structure and function of small RNAs. Nat Rev Mol Cell Biol. 2007;8:23–36. doi: 10.1038/nrm2085. [DOI] [PubMed] [Google Scholar]
- 29.Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khan AA, Betel D, Miller ML, Sander C, Leslie CS, Marks DS. Transfection of small RNAs globally perturbs gene regulation by endogenous microRNAs. Nat Biotechnol. 2009;27:549–555. doi: 10.1038/nbt.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castanotto D, Sakurai K, Lingeman R, Li H, Shively L, Aagaard L, Soifer H, Gatignol A, Riggs A, Rossi JJ. Combinatorial delivery of small interfering RNAs reduces RNAi efficacy by selective incorporation into RISC. Nucleic Acids Res. 2007;35:5154–5164. doi: 10.1093/nar/gkm543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoo JW, Kim S, Lee DK. Competition potency of siRNA is specified by the 5′-half sequence of the guide strand. Biochem Biophys Res Commun. 2008;367:78–83. doi: 10.1016/j.bbrc.2007.12.099. [DOI] [PubMed] [Google Scholar]
- 33.Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, Marion P, Salazar F, Kay MA. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 34.Bramsen JB, Laursen MB, Nielsen AF, Hansen TB, Bus C, Langkjaer N, Babu BR, Hojland T, Abramov M, Van Aerschot A, Odadzic D, Smicius R, Haas J, Andree C, Barman J, Wenska M, Srivastava P, Zhou C, Honcharenko D, Hess S, Muller E, Bobkov GV, Mikhailov SN, Fava E, Meyer TF, Chattopadhyaya J, Zerial M, Engels JW, Herdewijn P, Wengel J, Kjems J. A large-scale chemical modification screen identifies design rules to generate siRNAs with high activity, high stability and low toxicity. Nucleic Acids Res. 2009;37:2867–2881. doi: 10.1093/nar/gkp106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim DH, Behlke MA, Rose SD, Chang MS, Choi S, Rossi JJ. Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat Biotechnol. 2005;23:222–226. doi: 10.1038/nbt1051. [DOI] [PubMed] [Google Scholar]
- 36.Persengiev SP, Zhu X, Green MR. Non specific, concentration-dependent stimulation and repression of mammalian gene expression by small interfering RNAs (siRNAs) RNA. 2004;10:12–18. doi: 10.1261/rna5160904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi SW, Lee SH, Mok H, Park TG. Multifunctional siRNA delivery system: polyelectrolyte complex micelles of six-arm PEG conjugate of siRNA and cell penetrating peptide with crosslinked fusogenic peptide. Biotechnol Prog. 2009 doi: 10.1002/btpr.310. [DOI] [PubMed] [Google Scholar]
- 38.Kappel S, Matthess Y, Kaufmann M, Strebhardt K. Silencing of mammalian genes by tetracycline-inducible shRNA expression. Nat Protoc. 2007;2:3257–3269. doi: 10.1038/nprot.2007.458. [DOI] [PubMed] [Google Scholar]
- 39.Spencer DM, Graef I, Austin DJ, Schreiber SL, Crabtree GR. A general strategy for producing conditional alleles of Src-like tyrosine kinases. Proc Natl Acad Sci USA. 1995;92:9805–9809. doi: 10.1073/pnas.92.21.9805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spencer DM, Wandless TJ, Schreiber SL, Crabtree GR. Controlling signal transduction with synthetic ligands. Science. 1993;262:1019–1024. doi: 10.1126/science.7694365. [DOI] [PubMed] [Google Scholar]
- 41.Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369:756–758. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- 42.Choi J, Chen J, Schreiber SL, Clardy J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science. 1996;273:239–242. doi: 10.1126/science.273.5272.239. [DOI] [PubMed] [Google Scholar]
- 43.Wehrman T, Kleaveland B, Her JH, Balint RF, Blau HM. Protein-protein interactions monitored in mammalian cells via complementation of beta -lactamase enzyme fragments. Proc Natl Acad Sci USA. 2002;99:3469–3474. doi: 10.1073/pnas.062043699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suh BC, Inoue T, Meyer T, Hille B. Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science. 2006;314:1454–1457. doi: 10.1126/science.1131163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rivera VM, Wang X, Wardwell S, Courage NL, Volchuk A, Keenan T, Holt DA, Gilman M, Orci L, Cerasoli F, Jr, Rothman JE, Clackson T. Regulation of protein secretion through controlled aggregation in the endoplasmic reticulum. Science. 2000;287:826–830. doi: 10.1126/science.287.5454.826. [DOI] [PubMed] [Google Scholar]
- 46.Moskowitz HS, Heuser J, McGraw TE, Ryan TA. Targeted chemical disruption of clathrin function in living cells. Mol Biol Cell. 2003;14:4437–4447. doi: 10.1091/mbc.E03-04-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rollins CT, Rivera VM, Woolfson DN, Keenan T, Hatada M, Adams SE, Andrade LJ, Yaeger D, van Schravendijk MR, Holt DA, Gilman M, Clackson T. A ligand-reversible dimerization system for controlling protein-protein interactions. Proc Natl Acad Sci USA. 2000;97:7096–7101. doi: 10.1073/pnas.100101997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gestwicki JE, Crabtree GR, Graef IA. Harnessing chaperones to generate small-molecule inhibitors of amyloid beta aggregation. Science. 2004;306:865–869. doi: 10.1126/science.1101262. [DOI] [PubMed] [Google Scholar]
- 49.Kuduk SD, Zheng FF, Sepp-Lorenzino L, Rosen N, Danishefsky SJ. Synthesis and evaluation of geldanamycin-estradiol hybrids. Bioorg Med Chem Lett. 1999;9:1233–1238. doi: 10.1016/s0960-894x(99)00185-7. [DOI] [PubMed] [Google Scholar]
- 50.Muir TW. Chemical biology: cutting out the middle man. Nature. 2006;442:517–518. doi: 10.1038/442517a. [DOI] [PubMed] [Google Scholar]
- 51.Zeidler MP, Tan C, Bellaiche Y, Cherry S, Hader S, Gayko U, Perrimon N. Temperature-sensitive control of protein activity by conditionally splicing inteins. Nat Biotechnol. 2004;22:871–876. doi: 10.1038/nbt979. [DOI] [PubMed] [Google Scholar]
- 52.Tan G, Chen M, Foote C, Tan C. Temperature-sensitive mutations made easy: generating conditional mutations by using temperature-sensitive inteins that function within different temperature ranges. Genetics. 2009;183:13–22. doi: 10.1534/genetics.109.104794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yuen CM, Rodda SJ, Vokes SA, McMahon AP, Liu DR. Control of transcription factor activity and osteoblast differentiation in mammalian cells using an evolved small-molecule-dependent intein. J Am Chem Soc. 2006;128:8939–8946. doi: 10.1021/ja062980e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mootz HD, Muir TW. Protein splicing triggered by a small molecule. J Am Chem Soc. 2002;124:9044–9045. doi: 10.1021/ja026769o. [DOI] [PubMed] [Google Scholar]
- 55.Schwartz EC, Saez L, Young MW, Muir TW. Post-translational enzyme activation in an animal via optimized conditional protein splicing. Nat Chem Biol. 2007;3:50–54. doi: 10.1038/nchembio832. [DOI] [PubMed] [Google Scholar]
- 56.Brenzel S, Mootz HD. Design of an intein that can be inhibited with a small molecule ligand. J Am Chem Soc. 2005;127:4176–4177. doi: 10.1021/ja043501j. [DOI] [PubMed] [Google Scholar]