

Abstract
Objective:
To determine whether TDP-43 type is associated with distinct patterns of brain atrophy on MRI in subjects with pathologically confirmed frontotemporal lobar degeneration (FTLD).
Methods:
In this case-control study, we identified all subjects with a pathologic diagnosis of FTLD with TDP-43 immunoreactive inclusions (FTLD-TDP) and at least one volumetric head MRI scan (n = 42). In each case we applied published criteria for subclassification of FTLD-TDP into FTLD-TDP types 1-3. Voxel-based morphometry was used to compare subjects with each of the different FTLD-TDP types to age- and gender-matched normal controls (n = 30). We also assessed different pathologic and genetic variants within, and across, the different types.
Results:
Twenty-two subjects were classified as FTLD-TDP type 1, 9 as type 2, and 11 as type 3. We identified different patterns of atrophy across the types with type 1 showing frontotemporal and parietal atrophy, type 2 predominantly anterior temporal lobe atrophy, and type 3 predominantly posterior frontal atrophy. Within the FTLD-TDP type 1 group, those with a progranulin mutation had significantly more lateral temporal lobe atrophy than those without. All type 2 subjects were diagnosed with semantic dementia. Subjects with a pathologic diagnosis of FTLD with motor neuron degeneration had a similar pattern of atrophy, regardless of whether they were type 1 or type 3.
Conclusions:
Although there are different patterns of atrophy across the different FTLD-TDP types, it appears that genetic and pathologic factors may also affect the patterns of atrophy.
GLOSSARY
- AD
= Alzheimer disease;
- ADRC
= Alzheimer's Disease Research Center;
- bvFTD
= behavioral variant frontotemporal dementia;
- CBS
= corticobasal syndrome;
- CDR-SB
= Clinical Dementia Rating scale sum of boxes;
- FTLD
= frontotemporal lobar degeneration;
- FTLD-MND
= frontotemporal lobar degeneration with motor neuron degeneration;
- FTLD-TDP
= frontotemporal lobar degeneration with TDP-43 immunoreactive inclusions;
- MMSE
= Mini-Mental State Examination;
- NCI
= neuronal cytoplasmic inclusion;
- PNFA
= progressive nonfluent aphasia;
- SMD
= semantic dementia;
- STMS
= Short Test of Mental Status;
- VBM
= voxel-based morphometry.
The frontotemporal lobar degenerations (FTLD) are a heterogeneous group of pathologies1 that can be subdivided essentially into 3 groups based on the immunohistochemical profile of the disease entity.2 Those with tau protein deposition are grouped together as FTLD-tau, those with TAR DNA-binding protein of 43 kDa deposition as FTLD-TDP, and those with fused in sarcoma deposition as FTLD-FUS.
Recent evidence demonstrates that FTLD-TDP can be subdivided, based on the morphologic appearances, and distribution of TDP-43 immunoreactive inclusions.3,4 Using the Mackenzie et al.4 scheme, which correlates with clinical diagnosis, FTLD-TDP type 1 is characterized by neuronal cytoplasmic inclusions (NCI) and short dystrophic neurites in superficial cortex, type 2 by long thin dystrophic neurites in superficial and deep cortex, and type 3 by predominance of NCIs and a paucity of dystrophic neurites. A rare fourth type is also recognized to be associated with mutations in the valosin-containing protein gene.5,6
We have previously demonstrated that patterns of atrophy in FTLD differ according to pathology, with the presence of motor neuron degeneration,7 and with genetics, with the presence of mutations in progranulin (GRN).8 However, it is unclear whether morphologic differences across FTLD-TDP types relate to specific atrophy patterns, and whether these differences affect the relationship between atrophy, pathology, and genetics. The ability to predict FTLD-TDP type during life could be critically important, particularly if type maps onto different genetic variants. We therefore aimed to determine whether patterns of atrophy are associated with FTLD-TDP type.
METHODS
Subjects.
We identified all subjects from the neuropathology files of the Mayo Clinic, Rochester, MN, which had a pathologic diagnosis of FTLD-TDP type 1, 2, or 3 and a volumetric MRI performed between 1993 and 2005. Of 47 subjects identified, 5 were excluded due to poor quality MRI, leaving 42 subjects in the study. The first usable MRI was used in all cases; this was also the first available MRI in all cases except 2, where the second available MRI was used. All patients were seen by a behavioral neurologist within the Department of Neurology, Mayo Clinic.
The medical records of all cases were reviewed by one behavioral neurologist (K.A.J.) blinded to pathologic diagnosis, for the abstraction of data, including clinical diagnosis, demographics, Short Test of Mental Status (STMS),9 Mini-Mental State Examination (MMSE),10 and the Clinical Dementia Rating scale sum of boxes (CDR-SB).11 Behavioral variant frontotemporal dementia (bvFTD), semantic dementia (SMD), and progressive nonfluent aphasia (PNFA) were diagnosed based on consensus criteria.12 Established criteria were used to diagnose corticobasal syndrome (CBS)13 and Alzheimer disease (AD).14 Subjects with features of frontotemporal dementia and motor neuron disease were diagnosed with FTD-MND.
Thirty healthy controls that have not yet come to postmortem with volumetric MRI were age- and gender-matched to the study cohort. Controls were recruited into the Alzheimer's Disease Research Center (ADRC), performed within normal limits on standardized neurologic and neuropsychological testing, including STMS, MMSE, and CDR-SB, and were selected from the ADRC database based purely on age and gender.
Standard protocol approvals and patient consents.
Informed consent was obtained from all subjects or proxies for participation in the studies, which were approved by the Mayo Institutional Review Board.
Pathologic methods.
Neuropathologic examinations were performed according to the recommendations of the Consortium to Establish a Registry for Alzheimer's Disease.15 All cases underwent routine staining methods with hematoxylin-eosin, Bielschowsky silver, and immunohistochemistry to phosphorylated neurofilament, phospho-tau, α-synuclein, β-amyloid, and TDP-43 as previously described.16 All cases were diagnosed as FTLD-TDP. Sections of frontal and medial temporal lobe including hippocampal dentate granule cells were restained using a DAKO autostainer with TDP-43 (rabbit polyclonal; 1:3,000, ProteinTech Group, Chicago, IL) and reviewed by a single neuropathologist (D.W.D.) for subclassification. A pathologic diagnosis of frontotemporal lobar degeneration with motor neuron degeneration (FTLD-MND) was made based on the presence of motor neuron degeneration affecting brainstem, cranial nerve XII, or anterior horn cells, in addition to other features as previously described in detail.17
Criteria for FTLD-TDP subclassification.
The Mackenzie classification scheme4 was chosen in this study to classify all 42 cases since it has been found to correlate with clinical diagnosis4,18 and be useful when subcortical brain regions were analyzed.18 This scheme4 fits well with the scheme proposed by Sampathu et al.3 as follows: Mackenzie type 1 = Sampathu type 3, Mackenzie type 2 = Sampathu type 1, and Mackenzie type 3 = Sampathu type 2. Cases was classified as type 1 if there were moderate to numerous TDP-43 immunoreactive NCI, as well as thin and short dystrophic neurites predominantly in layer II of neocortex and variable density of pleomorphic NCI in the dentate gyrus of the hippocampus; as type 2 if there were a predominance of large and thick dystrophic neurites not restricted to any layer of the neocortex with absent, or at most sparse, NCI and no intranuclear inclusions in the neocortex; and type 3 if there were NCI in neocortex and dentate granule cells of hippocampus with absent to sparse dystrophic neurites.
GRN sequencing.
All subjects with frozen brain tissue, and hence stored DNA, or blood samples provided antemortem for research purposes, underwent genetic screening for mutations in all 13 exons of GRN gene as previously described.19
Image acquisition.
All subjects had a coronal T1-weighted 3-dimensional volumetric spoiled gradient echo sequence with 124 contiguous partitions and 1.6-mm slice thickness (22 × 16.5 cm field of view, 25° flip angle). All images underwent preprocessing correction for gradient nonlinearity20 and intensity nonuniformity21 as previously described.22
Image analysis.
Voxel-level patterns of atrophy were assessed using voxel-based morphometry (VBM)23 and SPM5 (http://www.fil.ion.ucl.ac.uk/spm). Images were normalized and segmented into gray matter, white matter, and CSF using customized tissue probability maps created from all subjects in the study and the unified segmentation24 routine followed by the HMRF cleanup step. Gray matter images were modulated and smoothed with a Gaussian kernel of 8 mm full width at half maximum. Patterns of gray matter loss were assessed in each FTLD-TDP type compared to controls, and compared to each other. In addition, 2 subanalyses were performed: 1) to compare type 1 subjects who screened positive or negative for mutations in GRN, and 2) to compare subjects with and without pathologic FTLD-MND within types 1 and 3. Age and gender were included in the models as covariates, and time from disease onset to scan was included as a covariate in all analyses that compared disease groups. Analyses comparing disease groups to controls were assessed corrected for multiple comparisons using familywise error at p < 0.05, whereas all direct comparisons between disease groups were assessed uncorrected at p < 0.001.
An atlas-based parcellation technique was also employed using SPM5 and the automated anatomic labeling atlas in order to generate left and right gray matter volumes for lateral frontal and parietal lobes and lateral and medial temporal lobes. Regions were selected based on the VBM results. An asymmetry score was calculated for each region for each subject as follows: (left volume − right volume) × 2/left volume plus right volume. In addition, regional volumes were divided by total gray matter volume to correct for differences in global atrophy between subjects and to allow the assessment of relative regional atrophy in each subject.
Statistical analysis.
Statistical analyses were performed utilizing the JMP computer software (JMP Software, version 8.0; SAS Institute Inc, Cary, NC) with α set at 0.05. Binary data were compared across FTLD-TDP types with χ2 test while continuous data were compared using the Kruskal-Wallis test. Post hoc testing using Mann-Whitney U was performed for variables that showed significant differences across types.
RESULTS
Subject demographics.
Twenty-two of the FTLD-TDP subjects had type 1, 9 had type 2, and 11 had type 3 pathology (table 1). No differences were observed across all 4 subject groups (FTLD-TDP types and controls) in education (p = 0.29) or age at scan (p = 0.17), although differences were observed in STMS, MMSE, and CDR-SB (p < 0.0001). The only demographic variables that showed significant differences across the 3 FTLD-TDP types were disease duration and age at death, with shorter disease durations and younger ages observed in type 3.
Table 1 Subject demographics
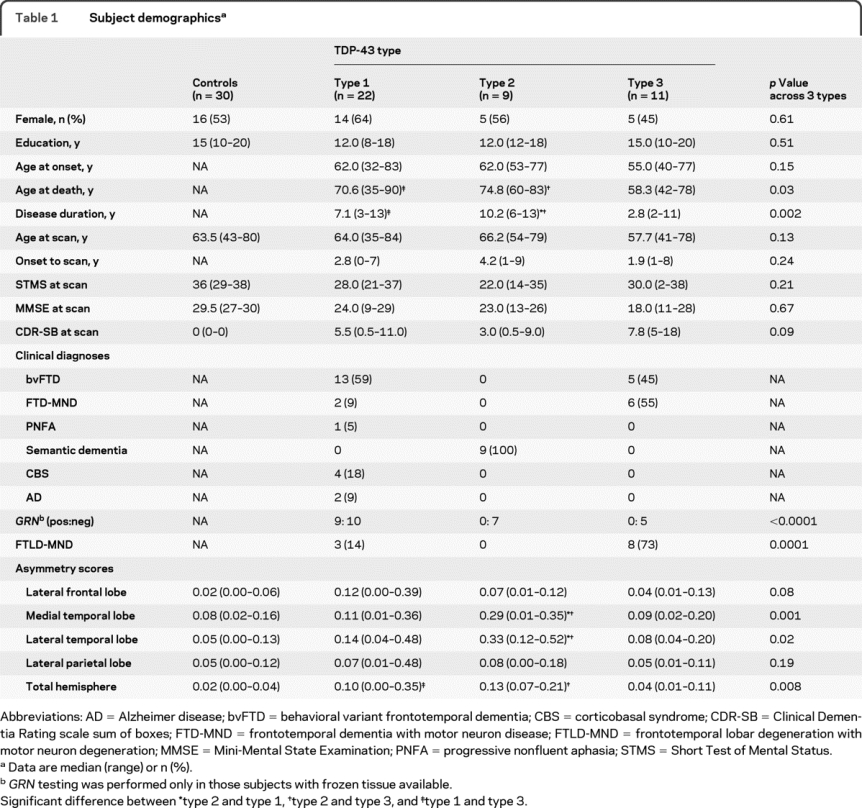
Type 1 subjects showed a range of clinical diagnoses, with the most common being bvFTD. All subjects with a clinical diagnosis of CBS or AD were type 1. All type 2 subjects had a clinical diagnosis of SMD. Type 3 subjects had clinical diagnoses of either bvFTD or FTD-MND. Mutations in GRN were only observed in type 1 subjects. All GRN subjects had a family history of dementia (3 subjects had pathologically confirmed FTLD in a relative). No demographic differences were observed between type 1 subjects with and without mutations in GRN. A pathologic diagnosis of FTLD-MND was observed in both types 1 and 3, although was more common in type 3. The 3 FTLD-MND type 1 subjects were significantly younger than type 1 subjects without MND (table e-1 on the Neurology® Web site at www.neurology.org), and had a higher proportion of subjects with family history than the FTLD-MND type 3 subjects (100% in type 1 vs 12.5% in type 3), although they all tested negative for GRN. Two type 1 FTLD-MND cases had clinical FTD-MND. The FTLD-MND type 3 subjects had shorter disease duration than type 3 subjects without MND (p = 0.01), with a similar trend observed within type 1 (p = 0.17).
Comparisons of FTLD-TDP types to controls.
Gray matter loss was observed throughout frontal, temporal, and parietal lobes in the entire cohort of FTLD-TDP subjects (figure e-1). Patterns of gray matter loss observed in each FTLD-TDP type compared to controls are shown in figure 1. Type 1 showed widespread patterns of loss, involving medial and lateral frontal, temporal, and parietal cortices and caudate nucleus. In contrast, types 2 and 3 were associated with more focal patterns of loss. Type 2 showed losses almost exclusively in anterior temporal lobes, involving medial, inferior, and middle temporal lobes, with greater loss observed in the left hemisphere. Loss was also observed in left anterior insula. Type 3 showed loss in frontal lobes, predominantly involving lateral and medial posterior frontal lobe, and bilateral insula, caudate nuclei, and hippocampi.

Figure 1 Patterns of gray matter loss in frontotemporal lobar degeneration with TDP-43 immunoreactive inclusions (FTLD-TDP) types 1, 2, and 3 compared to controls
Results are shown on 3-dimensional renders of the brain after correction for multiple comparisons using the familywise error at p < 0.05.
Comparisons between FTLD-TDP types.
Using VBM uncorrected at p < 0.001, type 1 showed greater loss in frontal and parietal lobes than type 2 and greater loss in temporal and parietal lobes than type 3. Type 2 showed greater involvement of anterior temporal lobes than types 1 and 3. Finally, type 3 showed greater loss in frontal lobes than type 2. Effect maps highlighting these differences are shown in figure 2. The gray matter-corrected atlas-based parcellation regional volumes demonstrate which regions show atrophy over and above the level of global loss, and showed similar differences across groups (figure 3). Significant differences were also observed across types in medial and lateral temporal lobe asymmetry scores, with greater asymmetry observed in type 2, and in total hemisphere asymmetry, with greater asymmetry observed in type 1 and type 2 (table 1).

Figure 2 Unthresholded t statistic effect maps highlighting differences between frontotemporal lobar degeneration with TDP-43 immunoreactive inclusions (FTLD-TDP) types
Type 1 subjects showed greater loss in the parietal lobes than both type 2 and type 3 subjects, with additional loss in the frontal lobes compared to type 2. Type 2 showed greater loss in the temporal lobes than both type 1 and type 3. Type 3 showed greater loss in the frontal lobes than type 2.

Figure 3 Box plots showing regional gray matter volume data for the 3 frontotemporal lobar degeneration with TDP-43 immunoreactive inclusions (FTLD-TDP) types
The boxes indicate the median and interquartile range (IQR) of the distributions while the horizontal lines extending from the boxes stop at the most extreme data points within 1.5 IQRs. All individual points are shown and points have been shifted randomly in the horizontal direction to avoid overlap. Subjects who screened positive for a mutation in GRN are indicated. Regional gray matter (GM) volumes were divided by total GM volume to correct for differences in global atrophy between subjects and to allow the assessment of relative regional atrophy in each subject, and converted to z scores showing deviation from controls. p Values represent differences across the 3 FTLD-TDP types. On post hoc testing, significant differences were observed between type 1 and 2 for all regions, between type 1 and 3 for the medial temporal and lateral parietal lobes, and between type 2 and 3 for lateral frontal, lateral temporal, and medial temporal lobes.
Analyses assessing GRN and FTLD-MND.
The FTLD-TDP type 1 subjects who screened positive or negative for mutations in GRN both showed gray matter loss in frontal, temporal, and parietal lobes compared to controls (figure 4A). A VBM direct comparison showed greater involvement of temporal and parietal lobes in GRN-positive subjects compared to GRN-negative subjects at an uncorrected threshold of p < 0.001 (figure e-2). Atlas-based parcellation showed greater lateral temporal atrophy in GRN-positive subjects than GRN-negative subjects (p = 0.007), with no significant difference observed in frontal and parietal regions (figure 2). No significant differences in asymmetry were observed between GRN-positive and -negative subjects.

Figure 4 Patterns of gray matter loss in frontotemporal lobar degeneration with TDP-43 immunoreactive inclusions (FTLD-TDP) type 1 and type 3 subjects depending on GRN mutation status and the presence of frontotemporal lobar degeneration with motor neuron degeneration (FTLD-MND)
(A) Patterns of loss in type 1 subjects who are positive and negative for GRN mutations. (B) Patterns of loss in type 1 and type 3 subjects with and without FTLD-MND. Results are shown on 3-dimensional renders of the brain after correction for multiple comparisons using the familywise error at p < 0.05.
Patterns of loss across types 1 and 3 divided based on the presence of FTLD-MND are shown in figure 4B. Type 1 and type 3 subjects with FTLD-MND showed almost identical patterns of gray matter loss involving posterior frontal lobes bilaterally compared to controls. The type 1 subjects without MND showed similar patterns of loss to the entire type 1 group (figure 1), and the type 3 subjects without MND showed no regions of loss after correction for multiple comparisons, and no discernable patterns were observed when uncorrected at p < 0.001, when compared to controls. Very few significant differences were observed across these groups on direct comparison in VBM or using regional volumes, although there were some nonsignificant trends for regional differences between subjects with and without FTLD-MND within type 1 and type 3, and for more severe loss in FTLD-MND subjects with type 1 compared to type 3 (figure e-3).
DISCUSSION
We identified different patterns of atrophy across the FTLD-TDP types, suggesting that there may be a distinct pattern or signature for each type. However, we found that the patterns of atrophy were affected by the presence of mutations in GRN and the presence of FTLD-MND. These findings therefore suggest that FTLD-TDP type alone does not determine patterns of atrophy.
FTLD-TDP type 1 was associated with a bilateral pattern of frontotemporal and parietal atrophy. Clinical phenotypes in this group were varied and included bvFTD, FTD-MND, PNFA, CBS, and AD. Other studies have similarly found varied clinical phenotypes in FTLD-TDP type 1.4,18,25,26 In contrast, type 2 showed a very focal pattern of asymmetric temporal lobe atrophy and was only associated with the clinical diagnosis of SMD. Many previous studies have linked this clinical phenotype to this specific pattern of atrophy,27–30 and some have linked this clinical phenotype to this pathologic type.4,18,26 Therefore, unlike type 1, there appears to be a tight correlation between pathology, anatomy, and clinical phenotype in type 2. FTLD-TDP type 3 was associated with a focal pattern of frontal atrophy, but unlike type 2, the clinical phenotype varied between bvFTD and FTD-MND. Indeed, over 50% of subjects had clinical evidence of motor neuron disease and the majority also had pathologic evidence of motor neuron degeneration. We have previously shown that this pattern of atrophy is associated with FTLD-MND.7 This group was also younger than the other FTLD-TDP types, and had shorter disease duration, features that are typical of subjects with FTLD-MND.31,32 Regional volumes provided good separation between the different FTLD-TDP types, suggesting that patterns of atrophy could be useful in differentiation. The type 1 subjects showed greater involvement of the parietal lobes than the other types and type 2 showed greater involvement of the temporal lobes. Type 3 was associated with focal frontal atrophy.
However, although one may speculate that these represent signature patterns of the FTLD-TDP pathologic types, we also found that genetic and pathologic features play a role in determining the patterns of atrophy. Almost half of the subjects in type 1 screened positive for mutations in GRN. We observed patterns of frontal, temporal, and parietal lobe volume loss in the type 1 subjects with and without mutations in GRN, although those with mutations in GRN showed significantly greater loss in the lateral temporal lobe than those without mutations. These results appear to be at odds with previous studies that have associated GRN mutations with parietal atrophy.8,33,34 We have previously shown that pathologically confirmed cases of FTLD with GRN mutations have greater frontal and parietal loss than those without GRN mutations.8 Subsequent TDP-43 typing has however revealed that the GRN-negative group in that study consisted of subjects with various different FTLD-TDP types, which would have influenced the group differences. The current study demonstrates that parietal lobe volume loss is actually a feature of FTLD-TDP type 1 pathology and that additional lateral temporal lobe atrophy distinguishes those with GRN mutations. Asymmetric patterns of atrophy have also been observed in subjects with mutations in GRN.33 We found no difference in the degree of asymmetry across the type 1 cases with and without mutations in GRN. This concurs with the fact that varied clinical phenotypes were also observed in both groups, and suggests that asymmetry is also a feature of type 1 pathology.
The pathologic diagnosis was also not consistent across the type 1 group, with 3 subjects showing pathologic FTLD-MND. This finding is somewhat unusual since FTLD-MND has been linked primarily to type 3.4,18,26 We also observed 3 subjects with type 3 who did not have pathologic or clinical MND. Although the number of subjects was small, we found that that the patterns of regional atrophy were the same in the 2 groups of FTLD-MND subjects, regardless of type, and that these patterns differed somewhat from the type 1 and type 3 subjects without FTLD-MND. Once again, this demonstrates that patterns of atrophy may not solely be driven by FTLD-TDP type, but that the pathology of FTLD-MND may have a signature, as we have previously suggested.7 The FTLD-MND subjects were also young and showed short disease duration, regardless of type, as is typical for FTLD-MND,31,32 and showed shorter disease duration and younger age than the type 1 and 3 subjects without FTLD-MND. We did however observe that a significantly greater proportion of subjects with FTLD-MND type 1 had a family history compared with FTLD-MND type 3. Given that the FTLD-MND type 1 subjects screened negative for GRN mutations, it is possible that another, as yet undiscovered, gene mutation could be responsible for FTLD. However, it is wise to be cautious in interpreting these findings since the number of type 1 subjects with FTLD-MND was very small. Further work is needed to better understand how FTLD-MND type 1 differs from FTLD-MND type 3.
The results of this study therefore suggest that patterns of atrophy may be useful to help predict FTLD-TDP type, and hence could be important clinically to improve patient prognosis. The presence of GRN mutations should however be considered, and although subjects with FTLD-MND are usually type 3, the presence of features that suggest FTLD-MND, such as clinical MND, short disease duration, and focal patterns of frontal atrophy, could also suggest type 1. The presence of family history may help increase the odds of type 1 pathology in these cases. The findings also increase our understanding of the relationship between protein deposition, pathologic features, and atrophy. Importantly, they demonstrate that FTLD-TDP is not an anatomically homogenous entity and hence the TDP-43 protein itself is not associated with a specific pattern of atrophy. The morphologic differences across the FTLD-TDP types are instead more tightly associated with the resultant pattern of atrophy.
AUTHOR CONTRIBUTIONS
Statistical analysis was conducted by Dr. Keith A. Josephs.
ACKNOWLEDGMENT
The authors thank Monica Casey-Castanedes for performing the immunohistochemistry and Stephen D. Weigand for constructing the box plots.
DISCLOSURE
Dr. Whitwell receives research support from the Dana Foundation and the NIH (R01- DC010367 [coinvestigator] and R01-AG037491 [coinvestigator]). Dr. Jack serves as a consultant for Elan Corporation and GE Healthcare; receives research support from Pfizer Inc., the NIH (NIA R01-AG11378 [PI] and U01 AG024904-01 [coinvestigator]), and the Alexander Family Alzheimer's Disease Research Professorship of the Mayo Foundation; and holds stock in GE Healthcare and Johnson & Johnson. Dr. Parisi serves on scientific advisory boards for the US Government Defense Health Board and the Subcommittee for Laboratory Services and Pathology; serves as a Section Editor for Neurology®; receives royalties from the publication of Principles & Practice of Neuropathology, 2nd ed. (Oxford University Press, 2003); and receives research support from the NIH (NS32352-13 [coinvestigator]). M.L. Senjem has received research support from Pfizer Inc. Dr. Knopman serves as Deputy Editor of Neurology®; has served on data safety monitoring boards for Sanofi-Aventis, GlaxoSmithKline, and Eli Lilly and Company; is an investigator in clinical trials sponsored by Elan Corporation, Baxter International Inc., and Forest Laboratories, Inc.; and receives research support from the NIH (R01-AG023195 [PI], R01-AG11378 [coinvestigator], P50 AG16574 [coinvestigator], U01 AG 06786 [coinvestigator], and R01 HL70825 [coinvestigator]). Dr. Boeve has served as a consultant to GE Healthcare; receives royalties from the publication of Behavioral Neurology of Dementia (Cambridge Medicine, 2009); and receives research support from Cephalon, Inc., the NIH (P50 AG16574 [coinvestigator], UO1 AG06786 [coinvestigator], and RO1 AG15866 [coinvestigator]), the Alzheimer's Association, and the Center for Inherited Disease Research (U24 AG026395 [coinvestigator]). Dr. Rademakers holds patents re: Methods and materials for detecting and treating dementia; and receives research support from the NIH (P50-AG16574 [PI on Project 2], RO1 NS065782-01 [PI], and R56 AG26251-03A1 [coinvestigator]), the Pacific Alzheimer Research Foundation (Canada), the Association for Frontotemporal Dementia, and the Amyotrophic Lateral Sclerosis Association. M. Baker holds patents re: Methods and materials for detecting and treating dementia. Dr. Petersen serves on scientific advisory boards for Elan Corporation, Wyeth, and GE Healthcare; receives royalties from publishing Mild Cognitive Impairment (Oxford University Press, 2003); and receives research support from the NIH/NIA (P50-AG16574 [PI], U01-AG06786 [PI], R01-AG11378 [coinvestigator], and U01-24904 [coinvestigator]). Dr. Dickson serves on the editorial boards of the American Journal of Pathology, Journal of Neuropathology and Experimental Neurology, Brain Pathology, Neurobiology of Aging, Journal of Neurology Neurosurgery and Psychiatry, Annals of Neurology, and Neuropathology; and receives research support from the NIH (P50-AG25711 [CL], P50-AG16574 [CL], P50-NS40256 [PI], P01-AG17216 [PI], P01-AG03949 [coinvestigator], and R01-AG15866 [coinvestigator]). Dr. Josephs receives research support from the NIH (NIDCD R01- DC010367 [PI], NIA R01-AG037491 [PI], and NINDS 2P50 NS040256-10 [coinvestigator]) and the Dana Foundation.
Supplementary Material
Address correspondence and reprint requests to Dr. Keith A. Josephs, Department of Neurology, Mayo Clinic, Rochester, MN 55905 josephs.keith@mayo.edu
Supplemental data at www.neurology.org
Study funding: Supported by The Dana Foundation, NIH grants R01-AG037491, R01-DC010367, P50-AG16574, U01-AG06786, R01-AG11378, and R01 NS065782, as well as the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer's Disease Research Program of the Mayo Foundation, the Alexander Family Alzheimer's Disease Research Professorship of the Mayo Foundation, and the NIH Construction Grant (NIH C06 RR018898).
Disclosure: Author disclosures are provided at the end of the article.
Received June 1, 2010. Accepted in final form August 24, 2010.
REFERENCES
- 1.Josephs KA. Frontotemporal dementia and related disorders: deciphering the enigma. Ann Neurol 2008;64:4–14. [DOI] [PubMed] [Google Scholar]
- 2.Mackenzie IR, Neumann M, Bigio EH, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol 2010;119:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sampathu DM, Neumann M, Kwong LK, et al. Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol 2006;169:1343–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mackenzie IR, Baborie A, Pickering-Brown S, et al. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol 2006;112:539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neumann M, Mackenzie IR, Cairns NJ, et al. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol 2007;66:152–157. [DOI] [PubMed] [Google Scholar]
- 6.Watts GD, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 2004;36:377–381. [DOI] [PubMed] [Google Scholar]
- 7.Whitwell JL, Jack CR Jr, Senjem ML, Josephs KA. Patterns of atrophy in pathologically confirmed FTLD with and without motor neuron degeneration. Neurology 2006;66:102–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whitwell JL, Jack CR Jr, Baker M, et al. Voxel-based morphometry in frontotemporal lobar degeneration with ubiquitin-positive inclusions with and without progranulin mutations. Arch Neurol 2007;64:371–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kokmen E, Naessens JM, Offord KP. A short test of mental status: description and preliminary results. Mayo Clin Proc 1987;62:281–288. [DOI] [PubMed] [Google Scholar]
- 10.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 11.Hughes CP, Berg L, Danziger WL, et al. A new clinical scale for the staging of dementia. Br J Psychiatry 1982;140:566–572. [DOI] [PubMed] [Google Scholar]
- 12.Neary D, Snowden JS, Gustafson L, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 1998;51:1546–1554. [DOI] [PubMed] [Google Scholar]
- 13.Boeve BF, Lang AE, Litvan I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol 2003;54(suppl 5):S15–S19. [DOI] [PubMed] [Google Scholar]
- 14.McKhann G, Drachman D, Folstein M, et al. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 15.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD): part II: standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991;41:479–486. [DOI] [PubMed] [Google Scholar]
- 16.Josephs KA, Petersen RC, Knopman DS, et al. Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology 2006;66:41–48. [DOI] [PubMed] [Google Scholar]
- 17.Josephs KA, Parisi JE, Knopman DS, et al. Clinically undetected motor neuron disease in pathologically proven frontotemporal lobar degeneration with motor neuron disease. Arch Neurol 2006;63:506–512. [DOI] [PubMed] [Google Scholar]
- 18.Josephs KA, Stroh A, Dugger B, Dickson DW. Evaluation of subcortical pathology and clinical correlations in FTLD-U subtypes. Acta Neuropathol 2009;118:349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baker M, Mackenzie IR, Pickering-Brown SM, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006;442:916–919. [DOI] [PubMed] [Google Scholar]
- 20.Jovicich J, Czanner S, Greve D, et al. Reliability in multi-site structural MRI studies: effects of gradient non-linearity correction on phantom and human data. NeuroImage 2006;30:436–443. [DOI] [PubMed] [Google Scholar]
- 21.Sled JG, Zijdenbos AP, Evans AC. A nonparametric method for automatic correction of intensity nonuniformity in MRI data. IEEE Trans Med Imaging 1998;17:87–97. [DOI] [PubMed] [Google Scholar]
- 22.Jack CR, Jr., Bernstein MA, Fox NC, et al. The Alzheimer's Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging 2008;27:685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ashburner J, Friston KJ. Voxel-based morphometry: the methods. Neuroimage 2000;11:805–821. [DOI] [PubMed] [Google Scholar]
- 24.Ashburner J, Friston KJ. Unified segmentation. Neuroimage 2005;26:839–851. [DOI] [PubMed] [Google Scholar]
- 25.Grossman M, Wood EM, Moore P, et al. TDP-43 pathologic lesions and clinical phenotype in frontotemporal lobar degeneration with ubiquitin-positive inclusions. Arch Neurol 2007;64:1449–1454. [DOI] [PubMed] [Google Scholar]
- 26.Snowden J, Neary D, Mann D. Frontotemporal lobar degeneration: clinical and pathological relationships. Acta Neuropathol 2007;114:31–38. [DOI] [PubMed] [Google Scholar]
- 27.Chan D, Fox NC, Scahill RI, et al. Patterns of temporal lobe atrophy in semantic dementia and Alzheimer's disease. Ann Neurol 2001;49:433–442. [PubMed] [Google Scholar]
- 28.Josephs KA, Whitwell JL, Knopman DS, et al. Two distinct subtypes of right temporal variant frontotemporal dementia. Neurology 2009;73:1443–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Josephs KA, Whitwell JL, Vemuri P, et al. The anatomic correlate of prosopagnosia in semantic dementia. Neurology 2008;71:1628–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mummery CJ, Patterson K, Price CJ, et al. A voxel-based morphometry study of semantic dementia: relationship between temporal lobe atrophy and semantic memory. Ann Neurol 2000;47:36–45. [PubMed] [Google Scholar]
- 31.Hu WT, Seelaar H, Josephs KA, et al. Survival profiles of patients with frontotemporal dementia and motor neuron disease. Arch Neurol 2009;66:1359–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Josephs KA, Knopman DS, Whitwell JL, et al. Survival in two variants of tau-negative frontotemporal lobar degeneration: FTLD-U vs FTLD-MND. Neurology 2005;65:645–647. [DOI] [PubMed] [Google Scholar]
- 33.Beck J, Rohrer JD, Campbell T, et al. A distinct clinical, neuropsychological and radiological phenotype is associated with progranulin gene mutations in a large UK series. Brain 2008;131:706–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whitwell JL, Jack CR Jr, Boeve BF, et al. Voxel-based morphometry patterns of atrophy in FTLD with mutations in MAPT or PGRN. Neurology 2009;72:813–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.