

Abstract
PBP 5 plays a critical role in maintaining normal cellular morphology in mutants of E. coli lacking multiple PBPs. The most closely related homologue, PBP 6, is 65% identical to PBP 5 but is unable to substitute for PBP 5 in returning these mutants to their wild type shape. The relevant differences between PBPs 5 and 6 are localized in a 20 amino acid stretch of domain I in these proteins, which includes the canonical KTG motif at the active site. We determined how these differences affected the enzymatic properties of PBPs 5 and 6 toward β-lactam binding and the binding and hydrolysis of two peptide substrates. We also investigated the enzymatic properties of recombinant fusion proteins in which active site segments were swapped between PBPs 5 and 6. The results suggest that the in vivo physiological role of PBP 5 is distinguished from PBP 6 by the higher degree of D, D-carboxypeptidase activity of the former.
Keywords: Penicillin-binding proteins, morphology maintenance, DD-carboxypeptidase, Escherichia coli
Introduction
Of the twelve known penicillin-binding proteins (PBPs) in Escherichia coli, four are D,D-carboxypeptidases (DD-CPases): PBPs 4, 5 and 6, and DacD (Holtje, 1998; Ghosh et al., 2008). These enzymes remove the terminal D-alanine from the pentapeptide side chains of muramic acid in peptidoglycan (PG), and the resulting tetrapeptides cannot act as donors during formation of peptide crosslinks in the cell wall (Ghuysen, 1991). Although the DD-CPases are usually the most abundant PBPs in the cell, they are not essential for bacterial survival (Denome et al., 1999) and the in vivo purposes of these seemingly nonessential and redundant enzymes are mostly unknown.
The exception to the above statement is the E. coli protein PBP 5, which helps maintain the normal morphology of this organism even in the absence of seven other PBPs (Nelson & Young, 2001). In the absence of PBP 5 by itself the cells exhibit small morphological aberrations, but as more PBPs are deleted the cells become greatly misshapen (Nelson & Young, 2000; Nelson & Young, 2001). PBP 5 consists of two major domains, I and II, oriented almost at right angles to one other (Davies et al., 2001; Nicholas et al., 2003). The DD-CPase active site is located in domain I and is responsible for maintaining normal cell shape (Ghosh & Young, 2003; Nelson et al., 2002). Domain II is composed mostly of β sheets and may lift the enzymatic domain away from the inner membrane and into the periplasm toward the peptidoglycan substrate (Ghosh & Young, 2003; Nelson et al., 2002). At its extreme carboxyl terminus, at the base of domain II, PBP 5 has a short 18 amino acid (Jackson & Pratt, 1987) amphipathic helix that tethers the protein to the outer face of the inner membrane (Ghosh & Young, 2003; Nelson et al., 2002). The closest homologue to PBP 5 from any organism is PBP 6, from E. coli itself. Interestingly, PBP 6, though ~65% identical to PBP 5, cannot restore normal shape to aberrant cells, as can PBP 5 (Ghosh & Young, 2003). Domain swap and mutagenesis experiments indicate that the relevant differences between the two enzymes localize to domain I, and, in fact, to a small stretch of 20 amino acids that surrounds the canonical KTG motif of the active site (Ghosh & Young, 2003; Nelson et al., 2002). For shorthand, we will refer to this 20 amino acid segment as the “morphology maintenance domain” (MMD) (Ghosh & Young, 2003) (Fig. 1). When PBP 6 is engineered so that its MMD is replaced by that from PBP 5, the mosaic protein (PBP 656) complements the shape defects of the E. coli mutants as well as wild type PBP 5 (Ghosh & Young, 2003).
Fig. 1. Location of the “morphology maintenance domain” (MMD) of PBP 5.
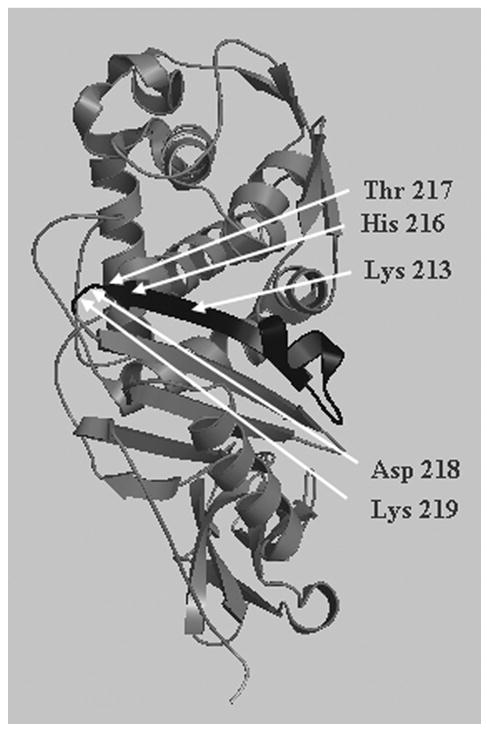
Top view: Arrows point to residues of sPBP 5 important for catalysis.
Conversely, replacing the MMD of PBP 5 with that from PBP 6 (creating PBP 565) abolishes the ability to complement. PBPs 5 and 6 differ by seven residues in this peptide fragment, but only two, Asp 218 and Lys 219, seem to be necessary to confer on PBP 656 the complementation attributes of PBP 5 (Ghosh & Young, 2003) (Fig. 1). However, mutation of these two amino acids does not eliminate the ability of PBP 5 to complement shape defects, suggesting that other structural features blunt the effects of these changes in the wild type protein.
The preceding results imply that differences in the abilities of PBPs 5 and 6 to complement cell shape defects can be explained by alterations in enzymatic activity due to minor changes in the amino acid residues near their active sites. However, the DD-CPases as a group share substantial homology in their primary structures and are believed to act on PG substrates via similar mechanisms in vitro (Baquero et al., 1996). What is not known is if the MMD residues affect the DD-CPase activities of these PBPs, and whether these changes explain the difference in the physiological functions of PBPs 5 and 6. To determine more exactly how the 20 amino acid MMDs of PBPs 5 and 6 contribute to the differences in the in vitro DD-CPase activities of these enzymes, we compared the enzymatic characteristics of four soluble PBPs (designated as “sPBPs”): sPBP 5, sPBP 6, and the mosaic proteins sPBP 656 and sPBP 565. The variations in enzymatic activities among these proteins help explain the basis of different biochemical and physiological properties of the DD-CPase PBPs.
Material and Methods
Bacterial strains, plasmids and chemicals
E. coli BL21* (Stratagene, West Cedar Creek, TX) was used to express recombinant proteins for purification in bulk. Plasmid pT7-cPBP5 was provided as gift by Robert A. Nicholas. Plasmids pAG6-4, pAG565-3 and pAG656-1 (9), were used to amplify the genes of soluble PBP 6, PBP 565 and PBP 656, respectively. Unless otherwise specified, restriction enzymes and DNA modifying enzymes were from New England Biolabs, (Ipswich, MA) and other chemicals and reagents were from Sigma-Aldrich, (St Louis, MO).
Construction of soluble PBP variants and fusion proteins
To create genes expressing soluble PBPs, the genes encoding the respective PBPs were amplified by using oligonucleotide primers (from MWG Biotech Inc., High Point, NC) in such a way that the resulting genes would express proteins devoid of their signal peptides and carboxyl-terminal amphipathic anchors. Primer pairs used for amplifications were: (A) P1 and P2 for sPBP 6, using pAG6-4 as the template; (B) P1 and P5 for sPBP 656, using pAG656-1 as the template; and (C) P3 and P4 for sPBP 565, using pAG565-3 as the template (Table 1). The conditions for amplification with Deep Vent DNA polymerase were: 94 °C for 5 min (initial denaturation), 94 °C for 1 min, 60 °C for 1 min and 72 °C for 1 min (for 30 cycles), followed by a final extension of 72 °C for 7 min. Each amplified product was cloned separately into the Nde I and Hind III sites of pT7-7K, creating pTA6-2S (expressing sPBP 6), pTA656-2S (expressing sPBP 656) and pTA565-3S (expressing sPBP 565). Plasmids were selected by including kanamycin (50 μg mL−1) in the medium and were sequenced to confirm that no mutations had arisen (sequencing was performed by MWG Biotech Inc., High Point, NC). Any sequence disparities in the constructs were removed by site directed mutagenesis and reconfirmed by sequencing.
Table 1.
Primer sequences for amplifying soluble PBPs 5 and 6, and their mosaics
| Primer | Primer sequence |
|---|---|
| P1 | 5′-CTC TCT CTC ATA TGG CGG AAC AAA CCG TTG AAG CGC CGA GC-3′ |
| P2 | 5′-CTC TCT CTA AGC TTT CAT CCG CCC TCT TCC ACA TTT TCC ATC AC -3′ |
| P3 | 5′-CTC TCT CTC ATA TGG ATG ACC TGA ATA TCA AAA CTA T -3′ |
| P4 | 5′-GCA TGC AAG CTT TCA ATC GAT CAC CGG ATC GCC GAA GTT ACC TTC CGG GAT TTC -3′ |
| P5 | 5′-GCA TGC AAG CTT TCA ATC CCA CAC CCG ACC AAA GAA TCC GCC-3′ |
Underlined sequences are complementary to the 5′ or 3′ end of the gene fragment in each construct. Boldfacing designates enzyme recognition sites: AAGCTT, Hind III; CATATG, Nde I.
Expression and purification of soluble PBPs
Plasmids encoding the sPBPs were transformed into E. coli BL21* and expressed under optimal conditions as determined beforehand (not shown). In short, for expression of sPBP5, overnight cultures were diluted 1:100 and grown at 30 °C in the presence of kanamycin (50 μg mL−1) to an A600 of 0.2, at which point isopropyl-β-D-thiogalactopyranoside (IPTG) was added to a final concentration of 0.5 mM and the cultures were incubated for an additional 12 h. For expression of all other sPBPs overnight cultures were grown at 26 °C to an A600 of 1.0 (stationary phase), at which time IPTG was added to a final concentration of 0.1 mM and the cultures were incubated for an additional 8 h. Cells were harvested at 5000 × g for 10 min (Beckman Avanti™ J25I, Fullerton, CA), the cell pellets were collected and resuspended in lysis buffer (400 μg mL−1 lysozyme, 50 mM Tris-HCl, 200 mM NaCl, 1 mM phenylmethylsulfonyl fluoride, pH 7.5) for 5 h at 4 °C with occasional stirring. Gross cell debris was removed by centrifugation at 8000 × g (Eppendorf 5810 R, Hamburg, Germany) for 10 min at 4 °C, and membrane vesicles were removed from the resulting supernatant by ultracentrifugation at 100,000 × g for 1 h at 4 °C (Sorvall Ultra Pro 80, Medcompare, San Francisco, CA). Soluble PBPs were purified from this final supernatant by ampicillin affinity chromatography, as described (Nicholas & Strominger, 1988), with slight modifications. sPBP supernatants were incubated with ampicillin-coupled activated CH-Sepharose 4B (Amersham Biosciences Piscataway, NJ) for 1 h at 30 °C. The resin was washed once with 50 mM Tris-HCl, pH 7.5, containing 1 M NaCl, and then washed once more with the same buffer lacking NaCl. The resin bound PBPs were eluted with 1 M NH2OH, 0.5 M Tris-HCl, pH 7.0 (Nicholas & Strominger, 1988). The purified PBP fractions (1.5 mL) were pooled and dialyzed against 20 mM Tris- HCl, 150 mM NaCl, pH 7.5, with three changes of buffer. Protein concentrations were determined by the Bradford assay kit (Sigma Chemical Co, St Louis, MO).
Fluorescent penicillin labeling
The activity of each purified sPBP was determined by labeling with 50 μM BOCILLIN FL(Invitrogen Inc., Carlsbad, CA) (Zhao et al., 1999). Reaction mixtures were incubated for 30 min at 35 °C, after which the proteins were denatured by adding 10 μL of denaturing solution to the reaction mixture and boiling for an additional 3 min. The proteins were separated and analyzed by electrophoresis through 12% SDS-PAGE gels. Labeled PBPs were visualized by washing the gel twice with deionized water and scanning immediately by using a Typhoon Trio variable imager (Amersham Biosciences, Piscataway, NJ) at an excitation wavelength of 488 nm and an emission wavelength of 526 nm.
Circular dichroism analyses
The far UV circular dichroism (Far UV CD) spectra of each soluble protein was determined by using a Jasco J-810 spectropolarimeter (Easton, MD), placing the samples in a quartz cell (path length = 0.2 cm) at 25 °C. Spectral data of sPBPs (2.5 μM) were collected with a 0.2 nm step resolution, 1 s time constant, 10 milli degrees sensitivity at a 2.0 nm spectral bandwidth, with a scanning speed of 50 nm min−1. Corrected spectra were obtained by subtracting the solvent spectrum. To reduce noise and random instrumental error, an average spectrum was compiled from four successive accumulations over a range of 200–240 nm. The recorded spectra in milli degrees of ellipticity (θ) were converted to mean residue ellipticity [θ] in degree cm2 deci mol−1 by using the following equation:
where c is the protein concentration in mg mL−1, l is the path length in cm, Mr is the protein molecular mass, and NA is the number of amino acids in the protein. Secondary structure was estimated by k2d software (Andrade et al., 1993).
Kinetic analysis of PBP interaction with β-lactam and peptide substrates
The kinetic parameters relating to the interaction of PBPs (E) with peptide (S) or β-lactam (S) were calculated by obeying the reaction,
where, K = (k−1 + k2)/k1, E • S is the non-covalent Michaelis complex, E–S is the covalent acyl enzyme complex, and P is the released product. The rate of acyl-enzyme complex was determined by the second order rate constant k2/K at sub-saturating concentrations of β-lactam antibiotics.
The acylation rate of sPBPs was assessed by incubating the enzymes (250 μg) for 30, 60, 90 or 120 s with BOCILLIN FLat different concentrations (25, 50 and 100 μM). Due to the poor binding of sPBP 565 with BOCILLIN FL, this protein was incubated with the substrate for longer times (1, 2, 4 and 6 h). The reaction was stopped by adding SDS sample buffer and denaturing the proteins by boiling for 5 min. Samples were analyzed by subjecting them to 12% SDS-PAGE and subsequently measuring the band intensities by densitometric scanning (UVP Gel documentation system, San Gabriel, CA) (Chambers et al., 1994). The second order rate constant (k2/K) was determined by calculating the pseudo-first-order rate constant, ka, using the following equation:
where PBPD denotes the density (experimentally derived) of the fluorophore associated with sPBP bound BOCILLIN FLobtained by incubating for time t at a given BOCILLIN FL concentration D, and PBPo denotes the density at which all binding sites of sPBPs were saturated with BOCILLIN FL. Values obtained for ka were plotted against the corresponding BOCILLIN FL concentration, and k2/K values were determined by calculating the slope.
The deacylation rate of purified sPBPs was determined by incubating proteins (50 μg) with BOCILLIN FL (50 μM) for 15 min at 37 °C. At t = 0, penicillin G was added to 3 mM, and the amount of fluorescent intensity remaining covalently bound to the protein was determined by removing the aliquots at various times (Guilmi et al., 2000). The labeled PBPs were quantified by densitometric scanning after separation by SDS-PAGE.
The deacylation reaction obeys the following equation:
where PBPt is the residual acyl-enzyme concentration at time t and PBPo is the initial concentration of acyl-enzyme complex. The deacylation constant (k3) was determined by quantifying the fluorescence of enzyme-bound BOCILLIN FL (the acyl-enzyme complex) that decreases over time, and was determined by using the above equation.
DD-carboxypeptidase activities of each sPBP were determined for the artificial substrate Nα, Nε-Diacetyl-Lys-D-Ala-D-Ala and for the peptidoglycan mimetic pentapeptide L-Ala-γ-D-Glu-L-Lys-D-Ala-D-Ala (USV custom peptide synthesis, Mumbai, India). Each sPBP (2 μg) was mixed with varying concentrations (0.25 to 12 mM) of the respective peptides, and the reaction volume was adjusted to 60 μL with 50 mM Tris HCl, pH 8.5. The mixture was incubated for 30 min at 37 °C, at which time 140 μL of freshly prepared enzyme-coenzyme mix was added (this solution was composed of a 20:10:5:1 ratio of the following: 50 mM Tris HCl, pH 8.5; 0.3 mg mL−1 of FAD; 10 μg mL−1 of horseradish peroxidase; and 5 mg mL−1 of D-amino acid oxidase). This final mixture was incubated for 5 min at 37 °C. Free D-alanine generated in this reaction was detected by the method of Frere et al (Frere et al., 1976), and compared to a standard D-alanine solution by using a Multiskan Spectrum-1500 Spectrophotometer (Thermo Scientific, Nyon-1, Switzerland) set at 460 nm.
Kinetic parameters for the DD-CPase assay were deduced from the linear regression of the double reciprocal plot (Lineweaver-Burk plot) (Lineweaver & Burk, 1934).
where, V is the reaction velocity (the reaction rate), Km is the Michaelis-Menten constant, Vmax is the maximum reaction velocity, and [S] is the substrate concentration. Further, to estimate the activity of the enzyme the turnover number (kcat) was measured by using the following equation:
Kinetic data were analyzed by using the Enzyme Kinetic Module of SIGMAPLOT v 9.0 (SYSTAT Software Inc., Chicago, IL) to determine Km and kcat.
Results
Previously, we constructed recombinant fusion proteins in which MMD segments were swapped between PBPs 5 and 6, creating the mosaic proteins PBP 656 and PBP 565 (9). PBP 656 complemented the shape defects of PBP mutants, but PBP 565 did not (Ghosh & Young, 2003). Here, we investigated the properties of the fusion proteins and their wild type counterparts to determine if enzymological differences among these PBPs might explain the disparities in their in vivo behaviors.
Creation and purification of soluble PBPs
To measure the biophysical and biochemical properties of PBPs 5 and 6 and their mosaic partners, it was necessary to create soluble versions of the enzymes. To do so, we constructed cloned genes that eliminated the signal peptide and C-terminal membrane anchor of each protein. Soluble PBP 5 can be prepared by deleting the C-terminal 10 amino acids that constitute the membrane-binding amphipathic helix (Pratt et al., 1986). Since the sequences of the C-terminal amphipathic anchors of PBPs 5 and 6 have substantial similarity (Nelson et al., 2002), we constructed soluble versions of these PBPs by removing 11 (PBP 565) or 15 amino acids (PBPs 6 and 656) from their carboxyl termini. In addition, we removed the 29 N-terminal amino acids that constitute the signal peptide for PBP 565 and 27 N-terminal amino acids for PBP 6 and PBP 656, so that the soluble proteins were not exported to the periplasm but remained cytoplasmic. The primer pairs used to create these soluble and truncated PBPs via PCR are listed in Table 1. The final proteins contained 359 amino acids (sPBP 6 and sPBP 656) or 364 aa (sPBP5 and sPBP 565). Genes encoding these soluble PBPs were cloned and the proteins were overproduced by inducing gene expression under optimum conditions of incubation time, temperature and IPTG concentration. No inclusion body was accumulated upon overexpression.
The sPBPs were purified by ampicillin-linked affinity chromatography, which generated a significant amount of product for sPBP 5 (0.4 mg mL−1), sPBP 6 (0.3 mg mL−1) and sPBP 656 (0.8 mg mL−1). The average total amounts of these purified proteins represented approximately 3–5 mg per liter of culture. However, the concentration of purified sPBP 565 was much less, so it was necessary to concentrate this protein 200-fold by ultra-filtration (Nicholas & Strominger, 1988) to give a final concentration of 0.4 mg mL−1. Molecular masses of the sPBPs, as determined by separation on 12% SDS-PAGE gels, were ~ 40 kDa (sPBP 5 and sPBP 565) and ~ 39 kDa (sPBP 6 and sPBP 656) (Fig. 2). The proteins were stable for months after storing at −80° C with 50 % glycerol and were functionally active because they all bound BOCILLIN FL(Zhao et al., 1999), a fluorescent version of penicillin V, even after long storage. The presence of labeled bands after SDS-PAGE indicated that BOCILLIN FL bound covalently to each sPBP (Fig. 2B), though less BOCILLIN FLbound to an equivalent amount of sPBP 565 than to the other three proteins (Fig. 2B, lane 4).
Fig. 2. SDS-PAGE analysis of purified sPBPs and their mosaics.
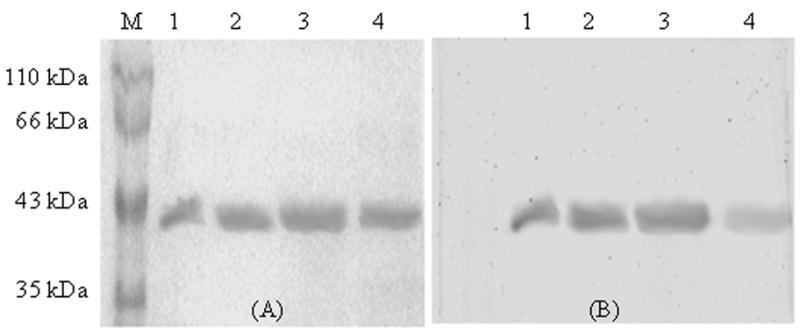
Purified sPBPs (3 μg) were stained with Coomassie blue (A), or with BOCILLIN FL (B). Lane M, Protein molecular weight markers; Lane 1, sPBP 5; Lane 2, sPBP 6; Lane 3, sPBP 656; and Lane 4, sPBP 565.
Gross secondary structures of the PBP variants are indistinguishable
It was possible that moving the MMD from one PBP to another could have altered the overall secondary structure of the recipient protein so much that one or more of its functions were disturbed. To determine if there were gross changes to the secondary structures of the mosaic PBPs, we analyzed the sPBPs by low-resolution circular dichroism (CD) spectroscopy to estimate the distribution of α-helical and β-sheet structures (Sreerama et al., 1999; Venyaminov & Yang, 1996). The predicted secondary structures indicated there were no substantial differences among any of the sPBPs (Table 2), suggesting that their overall folding patterns remained intact. The results eliminated this trivial explanation for the inability of PBP 6 and PBP 565 to complement shape defects in vivo.
Table 2.
Percentage distribution of secondary structures obtained from CD
| PBPs | % of α-Helix | % of β-sheet | % of Random coil |
|---|---|---|---|
| sPBP 5 | 23 | 27 | 50 |
| sPBP 6 | 22 | 27 | 51 |
| sPBP 656 | 24 | 25 | 51 |
| sPBP 565 | 24 | 24 | 52 |
The morphology maintenance domain (MMD) alters acylation and deacylation rate toward penicillin
β-lactam antibiotics bind covalently to a serine residue at the active site of PBPs, thereby inactivating the enzymes. Because β-lactams are substrate analogues of the D-alanyl-D-alanine terminus of the peptide side chain in peptidoglycan (Park & Strominger, 1957; Park, 1996), the rate of acylation by penicillin measures one facet of the enzymatic activity of the PBPs. To determine how efficiently sPBPs bound penicillin, we assessed the interaction of each sPBP with BOCILLIN FL. The acylation rate (k2/K) for sPBP 5 was approximately 40% of the rate observed for sPBP 6 (Table 3). The rate for mosaic protein sPBP 656 was ~70% of that for sPBP 6, which was more than 50% greater than that of sPBP 5. Thus, grafting the MMD of PBP 5 into PBP 6 decreased the penicillin acylation rate of sPBP 6, though the rate remained higher than that of wild type sPBP 5 (Table 3). This indicates that the MMD of PBP 5 is important but does not by itself determine the efficiency of acylation in the context of PBP 6. On the other hand, the acylation rate for sPBP 565 was drastically lower than that of PBP 5. Therefore, placing the MMD of PBP 6 into PBP 5 decreased the acylation rate of PBP 5 to 98% of its former value.
Table 3.
Kinetic parameters with BOCILLIN FL
| Protein | k2/K | k3 |
|---|---|---|
| M−1 s−1 | s−1 × 10 −5 | |
| sPBP 5 | 800 ± 50.3 | 18.9 ± 1.7 |
| sPBP 6 | 1900 ± 65.5 | 1.7 ± 0.1 |
| sPBP 656 | 1300 ± 45.6 | 6.8 ± 0.1 |
| sPBP 565 | 20 ± 5.2 | ND |
ND = Not detected
To understand how efficiently the sPBPs released bound penicillin from the acyl-enzyme complex (a measure of the catalytic efficiency), k3 values were determined for each of the constructs. The acylation rate for sPBP 6 was about 10 times less than that of sPBP 5 (Table 3). However, upon grafting the stretch of amino acids that corresponds to the MMD of PBP 5 into PBP 6 (i.e., sPBP 656), the deacylation efficiency of sPBP 6 increased four-fold. In contrast, the hydrolysis of BOCILLIN FL by sPBP 565 was too slow to measure in laboratory conditions, indicating that although the PBP 5 MMD was partially efficient in influencing deacylation of the BOCILLIN substrate, the corresponding stretch of amino acids from PBP 6 had no such effect. Taken together, the influence of the PBP 5 MMD on acylation and deacylation is noteworthy, and the rates of penicillin acylation or deacylation can serve as good predictors for the ability of PBPs 5 or 6 or of their mosaic counterparts to complement morphological defects of E. coli shape mutants.
The DD-carboxypeptidase activity of sPBP 5 is higher than that of sPBP 6
The acylation rates as measured by penicillin binding may not reflect the true enzymatic activities of the PBPs toward their natural D-ala-D-ala peptidoglycan substrates. The conventional substrate used to assay the DD-CPase activity of PBPs is Nα,Nε-diacetyl-Lys-D-Ala-D-Ala (AcLAA) (Supplementary Fig. 1A), and the activity of PBP 5 toward this substrate is significant (Nicholas et al., 2003). To determine if the in vivo differences of the PBPs coincided with differences in their native DD-CPase activities, we measure the kinetic properties of the soluble versions of PBPs 5 and 6 and their mosaic constructs toward AcLAA. The Km of sPBP 6 for AcLAA was 7 times lower than that of sPBP 5, indicating that PBP 6 formed the acyl-enzyme complex at a much faster rate than that of PBP 5 (Table 4).
Table 4.
Kinetic parameters with peptide substrates
| Protein | Nα, Nε-Diacetyl-Lys-D-Ala-D-Ala | L-Ala-γ-D-Glu-L-Lys-D-Ala-D-Ala | ||
|---|---|---|---|---|
| Km (mM) | kcat (s−1) | Km (mM) | kcat (s−1) | |
| sPBP 5 | 8.98 ± 1.79 | 2.70 ± 0.30 | 1.38 ± 0.43 | 1.41 ± 0.11 |
| sPBP 6 | 1.28 ± 0.26 | 0.56 ± 0.03 | ND | ND |
| sPBP 656 | 4.28 ± 1.34 | 0.71 ± 0.09 | 0.81 ± 0.21 | 0.20 ± 0.01 |
| sPBP 565 | ND | ND | ND | ND |
ND = Not detected
For sPBP 656 the Km was increased by a factor of ~3 compared to that of PBP 6, but sPBP 565 displayed no DD-CPase activity whatsoever (Table 4). These results were qualitatively equivalent to those observed for β-lactam binding among these proteins. sPBP 6 bound substrate significantly better than did sPBP 5; grafting the MMD of PBP 5 into PBP 6 reduced the affinity of sPBP 6 for its substrate, though the affinity of the mosaic protein was still higher than that of sPBP 5; and inserting the MMD of PBP 6 into sPBP 5 completely abrogated its DD-CPase activity, indicating that this active site segment of PBP 6 does not function in the PBP 5 background.
In contrast to what might be expected from the order of binding affinities, the DD-CPase activities did not correlate with higher binding of the AcLAA substrate. Instead, the turnover number (kcat) of sPBP 5 was ~5 times higher than that of sPBP 6; replacing the MMD of PBP 6 with that of PBP 5 increased the kcat of sPBP 656 by about 25%; but sPBP 565 remained inactive on this substrate (Table 4). Here, the degree of substrate binding was inversely correlated with the rate at which substrate was converted into product.
Only sPBP 5 acts as a DD-CPase on a pentapeptide substrate
Although AcLAA is routinely used for DD-CPase measurements, it is an artificial compound that does not exist in peptidoglycan. To analyze DD-CPase activity more appropriately, we assayed the activities of the PBPs toward a peptidoglycan mimetic pentapeptide substrate, LAla-γ-D-Glu-L-Lys-D-Ala-D-Ala (AGLAA) (Supplementary Fig. 1B). sPBP 5 exhibited significant DD-CPase activity, but sPBP 6 was inactive on this substrate (Table 4). Grafting the MMD of PBP 5 into PBP 6 produced DD-CPase activity in sPBP 656 (Table 4), indicating that this portion of the PBP 5 active site could impart to PBP 6 a measurable fraction of DD-CPase activity (about 14% that of sPBP 5). Once again, inserting the MMD of PBP 6 into PBP 5 completely eliminated the DD-CPase activity from the sPBP 565 mosaic protein (Table 4). Both the Km and kcat of sPBP 5 toward AGLAA were lower than when AcLAA was the substrate. This was in line with the behavior of sPBPs 5 and 6, in that a lower Km for the substrate was accompanied by a reduced rate of product formation.
Discussion
PBP 5 helps maintain the normal rod shape of E. coli and can restore the wild type shape to E. coli mutants deprived of three to seven PBPs (Nelson & Young, 2000; Nelson & Young, 2001; Nelson et al., 2002), and the mechanism by which it does so is probably related to its D,D-carboxypeptidase activity (Ghosh & Young, 2003; Nelson et al., 2002; Ghosh et al., 2008;). However, E. coli also expresses PBP 6, which exhibits DD-CPase activity and is the most closely related homologue of PBP 5 (Goffin & Ghuysen, 1998; Ghosh et al., 2008). However, despite these resemblances, PBP 6 cannot substitute for PBP 5 in maintaining or restoring normal cell shape to PBP mutants (Nelson & Young, 2001; Nelson et al., 2002; Ghosh & Young, 2003). At least some of the relevant differences in the in vivo functions of these two PBPs lie in a short stretch of residues in and near the active site (Ghosh & Young, 2003), but it is not known how these sequence differences affect the enzymatic activities of these enzymes. Here we investigated the kinetic properties of PBPs 5 and 6 and two mosaic proteins and find that the enzymes differ in their substrate preferences and in the rates at which they remove the terminal D-alanine from these substrates. The results suggest that these differences correlate with the in vivo phenotypes of shape maintenance.
The DD-carboxypeptidase activity of PBP 5 is responsible for maintaining cell shape
PBP 5 is clearly a better DD-CPase than PBP 6. For example, depending on the substrate, the DD-CPase activity of PBP 5 was previously shown to be 3 to 5 times greater than that of PBP 6 (Amanuma & Strominger, 1980). In our assays, the DD-CPase activity of sPBP 5 was five times greater than that of sPBP 6 when tested against the substrate Nα,Nε-diacetyl-Lys-D-Ala-D-Ala (AcLAA). An even greater difference was observed when the enzymes were tested against the peptidoglycan mimetic substrate L-Ala-γ-D-Glu-L-Lys-D-Ala-D-Ala (AGLAA), on which PBP 5 was active but to which PBP 6 may not bind covalently or else it may bind but not cleave. The failure of PBP 6 to act on this latter substrate is consistent with the observations of van der Linden and his co-workers (van der Linden et al., 1992). However, no DD-CPase activity on AcLAA and UDP-muramyl pentapeptide substrates was reported for either the membrane bound or the soluble form of PBP 6 (van der Linden et al., 1992). We speculate that sPBP 6 exhibited a low level of DD-CPase activity toward the artificial substrate, AcLAA, possibly because the active site cleft of sPBP 6 might accommodate smaller substrates like penicillin and AcLAA while being unable to bind a bulkier substrate such as AGLAA. The phenomenon of complete inertness of sPBP 6 toward pentapeptide substrate is interesting that simultaneously raises doubt whether it functions as DD-CPase in vivo at all.
Previously, we found that the differences between PBPs 5 and 6 in complementing shape defects in vivo could be narrowed down to a short stretch of 20 contiguous residues within the active site (the “morphology maintenance domain,” or MMD), where the two PBPs differ from one another by only 7 amino acids (Ghosh & Young, 2003). Shape complementation was associated with the MMD from PBP 5 and not with that from PBP 6 (Ghosh & Young, 2003). Here, we assayed soluble versions of mosaic proteins in which the MMDs from the two PBPs were switched with one another and found that DD-CPase activity was associated primarily with the MMD from PBP 5 but not with that from PBP 6. That is, sPBP 656 (PBP 6 containing the MMD of PBP 5) exhibited a DD-CPase activity toward both substrates that was more like PBP 5 than PBP 6, but sPBP 565 (PBP 5 containing the MMD of PBP 6) was not active on either of two substrates. In addition to its decreased DD-CPase activity, PBP 565 also bound and hydrolyzed the β-lactam BOCILLIN FLmuch less well than any of the other proteins. These behavioral changes of sPBP 565 must not be due to the improper folding of the molecule because CD spectral analyses predict that there is no gross alteration in the composition of overall secondary structure of sPBP 565 compared to sPBP 5, except that there is 3% less β-sheet structure in the former. Although, the reason for altered behavior of sPBP 565 is not clear, a gross change in the micro-architecture of the active site cannot be ruled out. Because β-sheet structures are not usual components of active sites, the lower percentage of β-sheet structure in sPBP 565 is not likely the key feature for its altered behavior. Nevertheless, the possible changes include altering the size and volume of the substrate binding pocket that may change the affinity or activity of the chimeric protein toward specific substrates. In any case, the ability of each PBP to act as a DD-CPase correlates exactly with its ability to complement cell shape changes in vivo, strongly suggesting that this activity is responsible for the shape maintenance phenotype.
Supplementary Material
(A) Nα,Nε-Diacetyl-Lys-D-Ala-D-Ala (AcLAA). (B) L-Ala-γ-D-Glu-L-Lys-D-Ala-D-Ala (AGLAA).
Acknowledgments
We thank Robert A. Nicholas for providing the plasmid pT7-cPBP5 and for suggestions regarding the construction of soluble PBPs. We also acknowledge Rakesh Sikder for initiating the computational work. This work was supported by a grant from the Department of Science and Technology, the Government of India, to ASG, and KDY was supported by a grant R01-GM061019 from the U. S. National Institutes of Health and by the Arkansas Biosciences Institute, the major research component of the Arkansas Tobacco Settlement Proceeds Act of 2000.
References
- Amanuma H, Strominger JL. Purification and properties of penicillin-binding proteins 5 and 6 from Escherichia coli membranes. J Biol Chem. 1980;255:1173–1180. [PubMed] [Google Scholar]
- Andrade MA, Chacon P, Merelo JJ, Moran F. Evaluation of secondary structure of proteins from UV circular dichroism spectra using an unsupervised learning neural network. Protein Eng. 1993;6:83–90. doi: 10.1093/protein/6.4.383. [DOI] [PubMed] [Google Scholar]
- Baquero MR, Bouzon M, Quintela JC, Ayala JA, Moreno F. dacD, an Escherichia coli gene encoding a novel penicillin-binding protein (PBP6b) with DD-carboxypeptidase activity. J Bacteriol. 1996;178:106–111. doi: 10.1128/jb.178.24.7106-7111.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers HF, Sachdeva MJ, Hackbarth CJ. Kinetics of penicillin binding to penicillin-binding proteins of Staphylococcus aureus. Biochem J. 1994;301 (Pt 1):139–144. doi: 10.1042/bj3010139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies C, White SW, Nicholas RA. Crystal structure of a deacylation-defective mutant of penicillin-binding protein 5 at 2.3-A resolution. J Biol Chem. 2001;276:616–623. doi: 10.1074/jbc.M004471200. [DOI] [PubMed] [Google Scholar]
- Denome SA, Elf PK, Henderson TA, Nelson DE, Young KD. Escherichia coli mutants lacking all possible combinations of eight penicillin binding proteins: viability, characteristics, and implications for peptidoglycan synthesis. J Bacteriol. 1999;181:3981–3993. doi: 10.1128/jb.181.13.3981-3993.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frere JM, Leyh-Bouille M, Ghuysen JM, Nieto M, Perkins HR. Exocellular DD-carboxypeptidases-transpeptidases from Streptomyces. Methods Enzymol. 1976;45:610–636. doi: 10.1016/s0076-6879(76)45054-1. [DOI] [PubMed] [Google Scholar]
- Ghosh AS, Young KD. Sequences near the active site in chimeric penicillin binding proteins 5 and 6 affect uniform morphology of Escherichia coli. J Bacteriol. 2003;185:2178–2186. doi: 10.1128/JB.185.7.2178-2186.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AS, Chowdhury C, Nelson DE. Physiological functions of D-alanine carboxypeptidases in Escherichia coli. Trends Microbiol. 2008;16:309–317. doi: 10.1016/j.tim.2008.04.006. [DOI] [PubMed] [Google Scholar]
- Ghuysen JM. Serine beta-lactamases and penicillin-binding proteins. Annu Rev Microbiol. 1991;45:37–67. doi: 10.1146/annurev.mi.45.100191.000345. [DOI] [PubMed] [Google Scholar]
- Goffin C, Ghuysen JM. Multimodular penicillin-binding proteins: an enigmatic family of orthologs and paralogs. Microbiol Mol Biol Rev. 1998;62:1079–1093. doi: 10.1128/mmbr.62.4.1079-1093.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilmi AMD, Mouz N, Petillot Y, Forest E, Dideberg O, Vernet T. Deacylation kinetics analysis of Streptococcus pneumoniae penicillin-binding protein 2x mutants resistant to β-lactam antibiotics using electrospray ionization-mass spectrometry. Anal Biochem. 2000;284:240–246. doi: 10.1006/abio.2000.4735. [DOI] [PubMed] [Google Scholar]
- Holtje JV. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol Mol Biol Rev. 1998;62:181–203. doi: 10.1128/mmbr.62.1.181-203.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson ME, Pratt JM. An 18 amino acid amphiphilic helix forms the membrane-anchoring domain of the Escherichia coli penicillin-binding protein 5. Mol Microbiol. 1987;1:23–28. doi: 10.1111/j.1365-2958.1987.tb00522.x. [DOI] [PubMed] [Google Scholar]
- Lineweaver H, Burk D. The Determination of Enzyme Dissociation Constants. J Am Chem Soc. 1934;56:658–666. [Google Scholar]
- Nelson DE, Young KD. Penicillin binding protein 5 affects cell diameter, contour, and morphology of Escherichia coli. J Bacteriol. 2000;182:1714–1721. doi: 10.1128/jb.182.6.1714-1721.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DE, Young KD. Contributions of PBP 5 and DD-carboxypeptidase penicillin binding proteins to maintenance of cell shape in Escherichia coli. J Bacteriol. 2001;183:3055–3064. doi: 10.1128/JB.183.10.3055-3064.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DE, Ghosh AS, Paulson AL, Young KD. Contribution of membrane-binding and enzymatic domains of penicillin binding protein 5 to maintenance of uniform cellular morphology of Escherichia coli. J Bacteriol. 2002;184:3630–3639. doi: 10.1128/JB.184.13.3630-3639.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas RA, Strominger JL. Site-directed mutants of a soluble form of penicillin-binding protein 5 from Escherichia coli and their catalytic properties. J Biol Chem. 1988;263:2034–2040. [PubMed] [Google Scholar]
- Nicholas RA, Krings S, Tomberg J, Nicola G, Davies C. Crystal structure of wild-type penicillin-binding protein 5 from Escherichia coli: implications for deacylation of the acyl-enzyme complex. J Biol Chem. 2003;278:52826–52833. doi: 10.1074/jbc.M310177200. [DOI] [PubMed] [Google Scholar]
- Park JT, Strominger JL. Mode of action of penicillin. Science. 1957;125:99–101. doi: 10.1126/science.125.3238.99. [DOI] [PubMed] [Google Scholar]
- Park JT. The murein sacculus. In: Neidhardt FC, Curtiss R III, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE, editors. Escherichia coli and Salmonella typhimurium: cellular and molecular biology. ASM Press; Washington, DC: 1996. pp. 48–57. [Google Scholar]
- Pratt JM, Jackson ME, Holland IB. The C terminus of penicillin-binding protein 5 is essential for localisation to the E. coli inner membrane. Embo J. 1986;5:2399–2405. doi: 10.1002/j.1460-2075.1986.tb04510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreerama N, Venyaminov SY, Woody RW. Estimation of the number of alpha-helical and beta-strand segments in proteins using circular dichroism spectroscopy. Protein Sci. 1999;8:370–380. doi: 10.1110/ps.8.2.370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Linden MP, de Haan L, Hoyer MA, Keck W. Possible role of Escherichia coli penicillin-binding protein 6 in stabilization of stationary-phase peptidoglycan. J Bacteriol. 1992;174:7572–7578. doi: 10.1128/jb.174.23.7572-7578.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venyaminov SY, Yang JT. Circular dichroism and the conformational analysis of biomolecules. Plenum; NY: 1996. [Google Scholar]
- Zhao G, Meier TI, Kahl SD, Gee KR, Blaszczak LC. BOCILLIN FL, a sensitive and commercially available reagent for detection of penicillin-binding proteins. Antimicrob Agents Chemother. 1999;43:1124–1128. doi: 10.1128/aac.43.5.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Nα,Nε-Diacetyl-Lys-D-Ala-D-Ala (AcLAA). (B) L-Ala-γ-D-Glu-L-Lys-D-Ala-D-Ala (AGLAA).