


Abstract
The protein common interface database (ProtCID) is a database that contains clusters of similar homodimeric and heterodimeric interfaces observed in multiple crystal forms (CFs). Such interfaces, especially of homologous but non-identical proteins, have been associated with biologically relevant interactions. In ProtCID, protein chains in the protein data bank (PDB) are grouped based on their PFAM domain architectures. For a single PFAM architecture, all the dimers present in each CF are constructed and compared with those in other CFs that contain the same domain architecture. Interfaces occurring in two or more CFs comprise an interface cluster in the database. The same process is used to compare heterodimers of chains with different domain architectures. By examining interfaces that are shared by many homologous proteins in different CFs, we find that the PDB and the Protein Interfaces, Surfaces, and Assemblies (PISA) are not always consistent in their annotations of biological assemblies in a homologous family. Our data therefore provide an independent check on publicly available annotations of the structures of biological interactions for PDB entries. Common interfaces may also be useful in studies of protein evolution. Coordinates for all interfaces in a cluster are downloadable for further analysis. ProtCiD is available at http://dunbrack2.fccc.edu/protcid.
INTRODUCTION
Many proteins function as homo- and heterooligomers, but in most cases the size and actual structures of these multimers are not known from direct solution experiments such as analytical ultracentrifugation and Nuclear Magnetic Resonance (NMR). Instead, they are based only on what is observed in X-ray crystal structures, sometimes even a single crystal structure. Both the Protein Data Bank (PDB) (1) and the European Bioinformatics Institute (EBI) (2) provide data on ‘biological assemblies’ that are derived from protein interactions in single crystals. The PDB’s author-defined biological units are based on what authors believe to be the biologically relevant structures, while the Protein Interfaces, Surfaces and Assemblies (PISA) (2) server from the EBI contains predicted oligomeric structures based on chemical thermodynamic calculations of complex stability. In addition, the EBI provides the Protein Quaternary Server (PQS), although this site is no longer updated (3). The PINS database at Oak Ridge National Laboratory also contains predicted biological assemblies for structures in the PDB, but only those released prior to mid-2007 (4).
Many online databases have used the PDB, PQS and PISA biological assemblies to examine the interfaces between proteins, including PIBASE (5), PSIMAP/PSIBASE (6), SNAPPI-DB (7), SCOPPI (8), PRINT (9) and iPfam (10). These sites present snapshots of the PDB at the time they were developed, and are not regularly updated. These databases analyze pairwise interactions between chains or between domains as defined by SCOP (11), CATH (12) or PFAM (13). Those databases based on SCOP and CATH are likely to be as behind on the PDB as SCOP and CATH are, on the order of 1–2 years. The current version of SCOP (1.75) covers only 38 221 entries or about 56% of the PDB. CATH (v. 3.3) currently contains 53 132 entries from the PDB or 79%. Those databases that use only PDB, PQS and PISA provide search tools but usually no additional information on which interfaces in X-ray crystallographic structures are likely to be biologically relevant.
Several other servers, such as PreBI (14), NOXClass (15), IBIS (16), PITA (17) and DiMoVo (18) also analyze interfaces in PDB entries, and try to predict which interfaces may be biologically relevant. Unlike PISA and PINS, they characterize individual interfaces and do not try to predict assemblies larger than dimers. An important feature of such servers is whether they are able to examine interfaces between monomers in different copies of the asymmetric unit and/or in different unit cells, since biologically relevant interactions may not be in the asymmetric unit itself (19). For instance, PreBI identifies biological interfaces by analyzing the electrostatic potential, hydrophobicity, the shape and the area of the interfaces, including those between asymmetric units and 26 neighboring unit cells (14). The NOXClass server uses support vector machines with features such as surface area, amino acid composition and conservation scores to infer biological relevance of interfaces, but only those within the asymmetric units of PDB entries (15). The Inferred Biomolecular Interaction Server (IBIS) predicts biologically relevant interactions for the chains in individual PDB entries by examining interfaces in the asymmetric units and the biological assemblies [as annotated in PISA or the PDB or the Conserved Domain Database (20)] of proteins closely related to the query (16). Even if the shared interface is present in the query crystal, it is not annotated as ‘observed’ unless it is present in the asymmetric unit of the query. PITA works on PDB entries and uploaded PDB files and uses symmetry operators to find potential biological interfaces within crystals (17). However, it failed to find interfaces in structures with known oligomeric assemblies that we tried. DiMoVo operates on uploaded PDB files containing dimers and uses solvation and Voronoi tessellation to predict whether the interface is biological or not (18). It does not build additional unit cells and interfaces have to be uploaded one by one by the user.
We have previously shown that if an interface is present in a number of crystal forms (CFs), especially when the proteins are homologous and not identical, then such interfaces are very likely to be biologically relevant structures (21). The data we analyzed previously were restricted to single-domain proteins separated into families as defined by SCOP and homologues of these identified with PSI-BLAST. In this article, we exploit this method for providing evidence in favor of the biological relevance of an interface across distant evolutionary relationships, and provide access to aligned structures of homo- and heterodimeric interfaces observed in multiple CFs. Our database is called Protein Common Interface Database (ProtCID).
We first use PFAM (13) to assign ‘chain architectures’ to each protein sequence in all PDB entries. The term architecture here is used in the same sense in which the PFAM website uses it to denote the ordered PFAM domain assignments along a protein sequence. Each PDB entry then has an ‘entry architecture’ that comprises the chain architectures in that entry. For each chain architecture observed in the PDB, we find all of the PDB entries and CFs with that chain architecture, and compare all of the homodimeric interfaces in representative entries of the different CFs. We cluster these interfaces based on similarity of pairwise amino acid contacts using average linkage hierarchical clustering. We perform the same kind of analysis on pairs of different chain architectures, comparing and clustering heterodimeric interfaces of single- and multi-domain protein chains. The results are stored in ProtCID.
A query to ProtCID is either a PDB entry code or a protein sequence or sequences. The server returns the chain architecture(s) for the query and asks the user to select one or two of these to search the database. Alternatively a user can browse a list of PFAM families, which will identify all PDB entries with a particular PFAM and their chain and entry architectures. If a single chain architecture is chosen from the query, then the server returns a list of clusters of homodimeric interfaces (and heterodimeric interfaces, if two chains have different sequences but both have the same chain architecture as the query). If the query is a pair of chain architectures, the server returns a list of clusters of heterodimeric interfaces. In both cases, for each cluster the server reports the number of CFs and PDB entries that contain the interface, the number of entries for which the PDB and PISA biological assemblies contain the interface, the average surface area and the minimum sequence identity between homologous chains in the different PDB entries in the cluster. An expandable table for each cluster provides the list of PDB entries, and for each entry the CF, whether the PDB and PISA biological unit contains the interface, the interface surface area, and the name and species of the protein(s) are provided.
ProtCID may be used to achieve a number of goals. First, it may be used to provide evidence in favor of the biological relevance of specific interfaces. Such interfaces are often conserved in different members of a family and may occur in different CFs. ProtCID does not designate specific interfaces as biologically relevant or not. Rather, it provides data on common interfaces (number of CFs, sequence information, PDB and PISA annotations, surface areas, etc.) that may be used to complement biophysical analysis and experimental data. In benchmarking, we found earlier that the more CFs that an interface is observed in, especially for non-identical proteins (<90%) and interfaces over 400 Å2, the more likely an interface was to be part of a biologically relevant assembly.
Second, a user may be interested in using a particular PDB entry as a template for comparative modeling or for analysis of existing experimental data. It commonly occurs that an interface shared by many PDB entries in a family is annotated in most but not all of the biological assemblies in the PDB and PISA. It may still be present in the crystals of entries that are missing the annotation. If this is the PDB entry of interest to a user, ProtCID may be used to identify the error and to provide coordinates for the interface from that entry. It may sometimes occur that a protein–protein interaction is not stable under the crystallization conditions used for a particular PDB entry, and a common interface observed in family members related to the query may be of interest.
Third, one of of ProtCID’s most useful features is that the user can download coordinate files for each cluster with a single click. We provide a program to produce a PyMOL script for each cluster to load the files, display them as rainbow-colored cartoon representations, and to align them to a common dimer in the cluster. The resulting visual display is highly informative of the variation in the common interfaces within the protein family, and can be used as a basis for further study of the evolution of biologically important protein–protein interactions.
METHODS
ProtCiD is compiled from entries in the PDB using the PDBML (XML) formatted files (22). From these files we obtain coordinates, crystallographic symmetry, and the biological assemblies defined by the authors of each structure (if available) in terms of symmetry operators and asym_ids. Since SCOP and CATH are 1–2 years behind on the PDB, we annotate each sequence in the PDB with its PFAM domains. We obtain PFAM domain assignments to sequences in the PDB from the PFAM website (13). Because the PFAM website does not update its assignments to the PDB frequently, we use PFAM’s RESTful web service to assign PFAMs to entries missing from PFAM itself.
We require sequence alignments of homologous chains from different PDB entries. We use the data from our PISCES database (23,24) as well as the structure alignment program FATCAT (23). PISCES contains PSI-BLAST (24) sequence alignments for all PDB chain pairs with E-values better than 0.001 [based on profiles constructed from NCBI’s non-redundant sequence database (25)] and structure alignments using combinatorial extension (CE) (26) for those PSI-BLAST detected homologues with sequence identity <50% or alignment coverage <80% of the shorter sequence. FATCAT allows flexibility between protein domains so that two homologous multi-domain proteins may be aligned, even if the domains have somewhat different orientations in the two structures. If both CE and FATCAT alignments are available, we choose the alignment with higher score calculated by summing the BLOSUM62 (27) substitution matrix scores over the aligned pairs.
Biological units from the PDB are generated from information given in the PDB XML files, while PISA assemblies are generated from the XML descriptions from the PISA website. The PDB’s XML files contain biological assemblies from the authors and in many cases from PISA and PQS. At our suggestion, the PDB now indicates the source of its biological assemblies––whether these are from the authors or from PISA or PQS. In some cases, the PISA website does not have a predicted assembly. Although the PDB’s XML file does contain a prediction from PISA (from running the software locally at the PDB). In those cases, we take the PISA assembly from the PDB file. About 4% of PDB structures do not contain an author-approved biological assembly, and in these cases we use the software-generated assembly given in the PDB XML file that is from PISA or PQS.
For each PDB entry, there are two levels of PFAM architectures: chain architecture and entry architecture. A ‘chain architecture’ denotes the PFAM domains in a protein chain with each PFAM in parentheses and multiple PFAMs concatenated by’_’ in the order of their starting locations. For instance, PDB entry 1E9H contains the sequence of cyclin A3, which has the chain architecture ‘(cyclin_N)_(cyclin_C)’. Chains in a single entry with the same protein sequence (and same entity_id in the PDB XML file) are only represented once. An ‘entry architecture’ is composed of the PFAM architectures of all unique sequences in a PDB entry, sorted in alphabetical order and separated by semi-colons. For instance, the PFAM entry architecture of PDB entry 1E9H is ‘(cyclin_N)_(cyclin_C);(Pkinase)’.
To analyze homo- and heterodimeric interfaces, we create groups of PDB entries that contain particular chain architectures or pairs of chain architectures. First, we define a group for each unique chain architecture found in one or more PDB entries. All PDB entries that contain a particular chain architecture are added to that group. Entries that contain more than one chain architecture thus will appear in multiple groups. These groups are used for analyzing homodimeric interfaces and some heterodimeric interfaces, those where both sequences are of the same chain architecture. For instance, there is a group’ (Cyclin_N)_(Cyclin_C)’ that contains 82 PDB entries. Some of these entries have other proteins as well (such as Pkinase proteins) but they all share the chain architecture ‘(Cyclin_N)_(Cyclin_C)’. Second, there is a group for each pair of chain architectures that occur together in at least one PDB entry. So there is a group ‘(Cyclin_N)_(Cyclin_C);(Pkinase)’ containing 73 PDB entries that have these two proteins
We divide each group into CFs, which are subsets of entries in the group with the same entry architectures, space group, the same asymmetric unit and similar crystal cell dimensions and angles (≤1%). Additionally we merged some CFs with the same entry architecture but different space groups or crystal cell dimensions or angles (>1%), if they contained highly similar interfaces, using a procedure described in our earlier work (21). We did not merge any CFs if the proteins had sequence identity less than 70%. We used the entry in each CF with the best X-ray resolution as the representative entry of the CF to compare interfaces between CFs.
We build crystals from the asymmetric unit and space group defined in PDB XML files as described earlier (21). Protein–protein interactions are identified and analyzed from the crystals, and defined on the level of chains. Two chains are considered to be interacting if and only if they have at least 10 pairs of Cβ atoms (Cα for Gly) with distance ≤ 12Å and at least one atomic contact ≤5Å.
A ‘common interface’ indicates that a similar chain–chain interaction pattern occurs in at least two CFs. The similarity of interface pairs was calculated by the Q function described by Xu et al. (21), which is equal to a weighted count of the common contacting residue pairs in two interfaces divided by the total number of unique pairs. A value of Q of 1 means all contacts in one interface exist in the other and at identical distances and vice versa. A value of Q of 0 means there are no common contacts. We have found that a value of Q of 0.2 or higher usually indicates a common interface and common orientations of the two monomers. We cluster interfaces of representative entries with surface area >200 Å2 using a hierarchical average linkage clustering algorithm (28). In this method, each interface is initialized to be a cluster. At each step, the two clusters with the highest average Q-score are merged, as long as 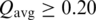 , where
, where 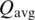 is the average Q-score between two clusters:
is the average Q-score between two clusters:
RESULTS
Summary statistics of the ProtCiD database
There are a total of 6067 PFAM (v24.0) families represented in protein sequences in the PDB. These exist in single and multi-domain proteins to form 7463 different chain architectures with known structure. The available data are summarized in Table 1. We construct overlapping ‘groups’ of PDB entries based on individual chain architectures or pairs of chain architectures to investigate interfaces in homo- and heterodimers, respectively. In this way, there are 7463 groups with a single architecture and 5768 pair architectures for a total of 13 231 PFAM architecture groups. There are a total of 3224 PFAM architecture groups with common interfaces (‘clusters’) in more than one CF, comprising 39 871 distinct PDB entries and 11 402 clusters. A total of 11% of these (1223 clusters) contain heterodimeric interfaces showing the interactions between different PFAM architectures. Figure 1 shows the overview of PFAM architecture groups and CFs in the database. The number of CFs in a group ranges from 1 to 591 [the antibody group (V-Set)_(C1-set)] and 50% of groups have two or three CFs (Figure 1 top). In this study, we do not analyze the groups that contain only V-set and C1-set. A total of 133 groups contain at least one common interface that exists in all of 10 or more distinct CFs (Figure 1 bottom).
Table 1.
Summary of data in ProtCID
| Single chain architectures | Pairs of chain architectures | Total | |
|---|---|---|---|
| # groups | 7463 | 5768 | 13 231 |
| # groups with M ≥ 2 | 2458 | 813 | 3271 |
| # groups with M ≥ 2, seqid<90 | 1461 | 317 | 1778 |
| # groups with M ≥ 5, seqid<90 | 615 | 116 | 731 |
| # groups with M ≥ 10, seqid<90 | 255 | 45 | 300 |
| # groups with M ≥ 20, seqid<90 | 88 | 4 | 92 |
| # entries | 62 499 | 5982 | 62 499 |
| # entries with M ≥ 2 | 39 076 | 4270 | 40 216 |
| # entries with M ≥ 2, seqid<90 | 29 282 | 2984 | 30 382 |
| # entries with M ≥ 5, seqid<90 | 19 935 | 2166 | 20 877 |
| # entries with M ≥ 10, seqid<90 | 13 301 | 1380 | 14 294 |
| # entries with M ≥ 20, seqid<90 | 7754 | 732 | 8408 |
| # clusters with M ≥ 2 | 10 761 | 1308 | 12 069 |
| # clusters with M ≥ 2,seqid<90 | 5696 | 511 | 6207 |
| # clusters with M ≥ 5,seqid<90 | 1403 | 161 | 1195 |
| # clusters with M ≥ 10,seqid<90 | 355 | 55 | 410 |
| # clusters with M ≥ 20,seqid<90 | 124 | 4 | 128 |
M is the number of CFs that contain a common interface.
Figure 1.

Data in ProtCID. Top, the number of groups given for each of the number of CFs in that group (only when #CF>1). Bottom, the number of groups that contain interfaces present in all N out of N CFs for that group.
Searching ProtCID
An example of a web search of ProtCID is presented in Figure 2. There are three types of queries: (i) a PDB ID; (ii) a PFAM ID or accession code; and (iii) one or two protein sequences. A PDB ID query, such as 1M54 as shown in Figure 2 (top), a crystal structure of cystathionine β-synthase (29), will return a list of PFAM chain architectures present in that PDB, in this case simply ‘(PALP)’ for pyridoxal-phosphate dependent enzymes (Figure 2, just below the first arrow). A user can select this PFAM chain architecture using the checkbox, and the server will return the interface clusters and the details about each cluster for the architecture (Figure 2, just below the second arrow). The (PALP) group consists of 128 PDB entries in 52 different CFs. The first cluster of interfaces occurs in 37 CFs, or 71% of 52 CFs for the chain architecture (PALP), and 109 PDB entries. This interface occurs in the asymmetric units of only 42 of these 109 entries (39%), demonstrating the need for investigating interfaces induced by symmetry relationships in a crystal. Clicking on the cluster number in the second column downloads a gzipped tar file with the coordinates of every dimer in the cluster. ProtCID provides a perl script for creating a structure superposition in Pymol (Delano,W.L., http://pymol.org) from the files in a folder containing the PDB files with the dimer interfaces. The script is available from the Help menu. The results of running the script are shown in Figure 3A. This dimer is well annotated with the interface observed in 103 and 105 of the PDB and PISA biological assemblies, respectively. Notably a search of the IBIS site at NCBI using 1M54 as a query provides the same dimer interface shown in Figure 3A but deems the entry a ‘singleton’ and does not show any of the interfaces in the 25 different but related proteins shown in Figure 3A. The reason for this is that IBIS has a relatively conservative cutoff for sequence identity with respect to the query of 50% for homodimers and 30% for heterodimers. IBIS does show this dimer for almost all of the other entries in this family. PISA can be used to find similar interfaces in homologues in the PDB but a search with 1M54 took several hours to complete, and PISA does not provide information on CFs.
Figure 2.

Screenshots of ProtCID. A search beginning with PDB entry 1M54 is shown at top. The search returns the PFAM architecture of 1M54, (PALP), or pyridoxal-dependent enzymes. Clicking the box for this PFAM architecture and clicking the button labeled ‘Retrieve Common Interface Clusters’ produces a table of clusters in the lower part of the figure. Clicking on ‘+’ next to a cluster produces a table of the PDB entries that contain the common interface in that cluster.
Figure 3.

Homodimers of PFAM architecture (PALP). (A) A cluster of interfaces present in 37 CFs and 109 PDB entries (only one structure per CF is shown). These structures include cysteine synthase, tryptophan synthase alpha subunit, threonine synthase, cystathionine beta synthase, 1-aminocyclopropane-1-carboxylate deaminase and O-acetylserine sulfhydrylase from 18 different species. (B) A cluster of interfaces present in 13 CFs and 17 PDB entries. These proteins include catabolic threonine dehydratase, serine racemase, threonine deaminase, l-serine dehydratase and two uncharacterized proteins from Schizosaccharomyces pombe. Chains are colored from blue to red from N to C terminus, respectively. Chains were aligned with Pymol to one representative structure.
A second large cluster for (PALP) is listed in the screenshot of ProtCID in Figure 2, which exists in 13 CFs and 17 PDB entries. It is present in the asymmetric units of only 7 of these 17 entries. These structures do not overlap with the set in Cluster 1 for (PALP) and form a different branch on a phylogenetic tree as shown in Figure 4. The PDB and PISA have 11 and 13 of these interfaces in their biological assemblies for these entries, respectively. Both miss this interface for the human and rat l-serine dehydratases in three different CFs. A superposition of these dimers is shown in Figure 3B. An IBIS search with PDB entry 3HMK in this cluster provides 5 of the 15 PDB entries (and 3 of 7 genes) shown by ProtCID.
Figure 4.

A phylogenetic tree of the proteins in the PDB with the PFAM architecture (PALP). Each color represents a different CF for Clusters 1 and 2 (inset). Only two threonine synthase structures, PDB entries 1VB3 (Escherichia coli) and 1KL7 (yeast), are missing either common dimer in their X-ray structures. Only one entry per CF is shown. The numbers after the PDB codes indicate the PDB’s entity_id number.
We previously analyzed single-domain proteins as defined by SCOP (11), but these results were limited to that portion of the PDB covered by SCOP and proteins related to SCOP-defined domains as calculated by PSI-BLAST (21). By using PFAM to identify multi-domain proteins, instead of SCOP, we are able to classify the domains in 98% of PDB entries and 98% of sequences in the PDB. In order to achieve this coverage, we used PFAM’s RestFUL web service (13), since PFAM itself is several months behind on the PDB. While in general SCOP is able to identify more remote relationships than PFAM, we are interested in common interfaces related by evolution. At longer evolutionary distances, some interfaces are not conserved (30) and so we do not necessarily require the identification of the most remote relationships.
Of the 83 539 (redundant) entity sequences in the PDB, 26% of them contain multiple PFAM domains. ProtCID can be searched (or browsed) by PFAM families and thus multi-domain proteins that share a particular domain can be readily identified. For instance, if the PFAM ‘PDZ’ is entered, a list of 293 PDB entries is returned along with their PFAM chain and entry architectures. There are nine different chain architectures in the PDB that contain a PDZ domain: (PDZ), 240 entries; (Trypsin)_(PDZ), 14 entries; (PDZ)_(PDZ), 11 entries; (PDZ_assoc)_(PDZ), 8 entries; (Trypsin)_(PDZ)_(PDZ), 6 entries; (MAGUK_N_PEST)_(PDZ), 6 entries; (PDZ)_(Peptidase_S41), 5 entries; (PDZ)_(fn3), 2 entries; and (PDZ)_(EBP50_C-term), 1 entry.
If we select one of the entries that has a chain architecture (Trypsin)_(PDZ), we find a cluster of homodimeric interfaces that occurs in all seven CFs and 14 entries for this architecture. A superposition of these dimers is shown in Figure 5. ProtCID shows that these proteins are annotated mostly as trimers in the PDB and PISA, and the interfaces in the figure are in fact the trimer interface. In the PDB, two of these entries are annotated as monomers. One of these, the structure of human HtrA2 serine protease, a mitochondrial protein involved in apoptosis (31), is described as a trimer in the published paper (32), while the biological assembly in the PDB (PDB entry 1LCY) was deposited as a monomer. This is an example where the author-deposited biological assemblies in the PDB do not necessarily coincide with what the authors themselves may view as the biologically relevant structure. The reasons for this are not clear but the phenomenon is not uncommon in our experience. ProtCID is valuable in checking whether a PDB entry of interest contains a biologically relevant interface present in many CFs of related proteins but for some reason is missing from the publicly available annotations. In this case, ProtCID provides the relevant interface. The structure of the htrA2 trimer is available from PISA or can be built in PyMol or other programs from the crystal symmetry operators. IBIS does not have this interaction for PDB entry 1LCY because of its conservative sequence identity cutoff.
Figure 5.

Homodimers of a multi-domain protein with PFAM architecture (trypsin)_(PDZ) from seven CFs and 14 PDB entries. The proteins include E. coli proteases degS and DO, Mycobacterium tuberculosis hypothetical protein Rv0983 and human HtrA2. The chains are colored blue to red from N to C terminus. The PDZ domains are in orange and red.
In some cases, PISA underannotates well-documented biologically relevant interfaces. In the case of another multi-domain protein, with PFAM chain architecture (ATP-gua_PtransN)_(ATP-gua_Ptrans), a single interface is present in 14 of 18 CFs and 18 PDB entries with minimum sequence identity of 49%. This interface is in the asymmetric units of 11 of these 18 entries. A superposition of these dimers is shown in Figure 6. Of these entries, the PDB’s biological units contain this dimer in 14 of 18 entries, while PISA has this dimer in only five cases. Three of the incorrect dimers in the PDB are in a single CF of human creatine kinase B. In their paper (33), the authors describe the common dimer shown by ProtCID, but the deposited biological assembly for all three (PDB entries 3B6R, 3DRB and 3DRE) is the same as the asymmetric unit for this CF, another example of apparently accidental misannotations in the PDB. IBIS annotates this interface as ‘biologically validated’ for four PDB entries (3JU5, 3JU6, 3L2D, 1U6R) and as ‘putative but not biologically validated’ in five more (2GL6, 3L2F, 1QH4 1QK1, 1VRP).
Figure 6.

Homodimers of PFAM chain architecture (ATP_gua_PtransN)_(ATP_gua_Ptrans) from 14 CFs and 18 PDB entries. The proteins include human, rabbit, chicken, bovine and electric ray creatine kinase, a worm glycocyamine kinase and a sea cucumber arginine kinase. Chains are colored blue to red from N to C terminus.
ProtCID is also able to show common interfaces in heterodimers across multiple CFs. A search starting with PDB entry 2GTP (34) reveals that this entry has two sequences, one with PFAM architecture, (RGS), the regulator of G-protein signaling, and one with PFAM (G-alpha), the G-protein α-subunit. Selecting both of these PFAMs produces a list of two common interfaces. The first of these is present in eight PDB entries and all six CFs with a minimum sequence identity of 36%. The second cluster is in only two CFs with 100% sequence identity and is not likely to be biologically relevant. We found previously that identical proteins may form the same interface in different CFs, although this is rare for homologous but not identical proteins. The dimer in all six CFs is shown in Figure 7. It has a mean surface area of 836 Å2. This dimer is annotated in the deposited biological units of all eight PDB entries in these CFs but only one of the PISA assemblies contains this interface. Six of the entries missing the heterodimer in PISA are oddly monomeric, and not heterooligomers at all. One is a heterodimer when it should be a heterotrimer (PDB entry 1FQJ).
Figure 7.

Heterodimers of PFAM domains (G-alpha), the G-protein alpha subunit, and (RGS), the regulator of G-protein signaling, from six CFs and eight PDB entries. RGS is in the upper left of the figure. The RGS proteins include human RGS1, RGS2, RGS8, RGS9, RGS10 and RGS16 as well as bovine RGS9 and mouse RGS16. The G-protein alpha subunits include human Gi and Gk, bovine Gt and mouse Go. Both proteins are colored from blue to red from N- to C-terminus.
IBIS shows this same dimer in all of the eight PDB entries that contain it. IBIS also has results for 2GTP that include many of the interactions for (G-alpha) proteins in ProtCID, including interactions with (RGS_like), (RhoGEF), (Guanylate_cyc) and (PDE6_gamma). IBIS presents some results for the query 2GTP that ProtCID does not due to the way PFAM divides the AAA superfamily of proteins. In PFAM, (G-alpha) and (Arf) are two families in the AAA clan. Since we do not yet compare chains with different PFAMs even when they are in the same clan, (Arf)-containing proteins are not included in the results for any G-alpha entries. IBIS does include some Arf proteins in the results for 2GTP, including interaction with Pleckstrin homology domains. IBIS also provides an intraprotein interaction between an Arf domain and an ArfGap domain in PDB entry 3LVQ, which ProtCID does not provide, since we do not currently analyze intraprotein domain–domain interfaces.
DISCUSSION
Identifying the structures of biologically relevant protein–protein interactions whether in homooligomers or between different proteins remains a challenging task. Some interactions are weak or transient and methods designed to identify stable protein–protein interfaces in crystals using physical chemical considerations may not identify these easily. In our earlier work, we showed that these methods also tend to identify some very large surface areas in crystals as biologically relevant even when the proteins are monomeric by all available experimental data (21).
We provide ProtCID as an alternative and complementary source of information on the biologically relevant interfaces between proteins in X-ray crystal structures. When there is some doubt about an interface, observing the same interface in homologous proteins may provide evolutionary and physical evidence in favor of that interface. As more structures are determined in large protein families, the comparison of CFs may play an important role in suggesting which interfaces are biologically relevant interactions. Identifying such common interfaces is difficult to perform manually for many structures, and therefore ProtCID provides a useful tool. We hope that the common interfaces identified by ProtCID can be used in further studies of the biophysical characteristics and evolutionary conservation of interaction surfaces for specific biological systems.
ProtCID has a number of limitations. First, in cases where there is only one CF for a protein or protein complex, ProtCID will not have any common interfaces to report. This occurs for 36% of the PDB, of which 32% are annotated as multimeric by PISA. Second, it sometimes occurs that similar interfaces are present in more than one CF but are not biologically relevant. This usually occurs when the entries are for identical sequences, for small interfaces less than 400 Å2, and usually for only two or three CFs. It may also occur for families with large numbers of structures, where through thorough sampling some interfaces may show high similarity by chance. This happens, for instance, among protein kinases. Third, any such server needs to define such relationships and strike a balance between clustering related proteins without introducing false relationships. We have used PFAM to define protein domains and architectures and therefore the evolutionary relationships among proteins. PFAM breaks up some large superfamilies into a number of families. We have not so far compared interfaces in different PFAMs within these superfamilies or ‘clans’ as PFAM calls them. Thus some distant evolutionary relationships may be missed in ProtCID. Finally, we compared proteins with the same architectures (either one or two at a time). So, for instance, we would not compare homodimeric interfaces between entries with architecture (Pkinase_Tyr)_(SH2) and those with (Pkinase_Tyr). The reason for this is that while PFAM may identify most proteins that belong to its defined domain families, it often aligns regions shorter than the full domain as observed in the three-dimensional structure. This makes it difficult to compare interfaces in different entries.
ProtCID will be updated monthly in order to keep up with the rapidly expanding size of the PDB. A comparison of interactions on the domain level, instead of the chain level, will be presented later.
FUNDING
National Institutes of Health (R01-GM73784 and R01-GM84453 to R.D.). Funding for open access charge: National Institutes of Health (R01-GM73784).
REFERENCES
- 1.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 3.Henrick K, Thornton JM. PQS: a protein quaternary structure file server. Trends Biochem. Sci. 1998;23:358–361. doi: 10.1016/s0968-0004(98)01253-5. [DOI] [PubMed] [Google Scholar]
- 4.Bordner AJ, Gorin AA. Comprehensive inventory of protein complexes in the protein data bank from consistent classification of interfaces. BMC Bioinformatics. 2008;9:234. doi: 10.1186/1471-2105-9-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davis FP, Sali A. PIBASE: a comprehensive database of structurally defined protein interfaces. Bioinformatics. 2005;21:1901–1907. doi: 10.1093/bioinformatics/bti277. [DOI] [PubMed] [Google Scholar]
- 6.Gong S, Yoon G, Jang I, Bolser D, Dafas P, Schroeder M, Choi H, Cho Y, Han K, Lee S, et al. PSIbase: a database of protein structural interactome map (PSIMAP) Bioinformatics. 2005;21:2541–2543. doi: 10.1093/bioinformatics/bti366. [DOI] [PubMed] [Google Scholar]
- 7.Jefferson ER, Walsh TP, Roberts TJ, Barton GJ. SNAPPI-DB: a database and API of structures, interfaces and alignments for protein–protein interactions. Nucleic Acids Res. 2007;35:D580–D589. doi: 10.1093/nar/gkl836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Winter C, Henschel A, Kim WK, Schroeder M. SCOPPI: a structural classification of protein-protein interfaces. Nucleic Acids Res. 2006;34:D310–D314. doi: 10.1093/nar/gkj099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tuncbag N, Gursoy A, Guney E, Nussinov R, Keskin O. Architectures and functional coverage of protein-protein interfaces. J. Mol. Biol. 2008;381:785–802. doi: 10.1016/j.jmb.2008.04.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Finn RD, Marshall M, Bateman A. iPfam: visualization of protein–protein interactions in PDB at domain and amino acid resolutions. Bioinformatics. 2005;21:410–412. doi: 10.1093/bioinformatics/bti011. [DOI] [PubMed] [Google Scholar]
- 11.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 12.Orengo CA, Michie AD, Jones S, Jones DT, Swindells MB, Thornton JM. CATH–a hierarchic classification of protein domain structures. Structure. 1997;5:1093–1108. doi: 10.1016/s0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- 13.Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, et al. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsuchiya Y, Kinoshita K, Ito N, Nakamura H. PreBI: prediction of biological interfaces of proteins in crystals. Nucleic Acids Res. 2006;34:W320–W324. doi: 10.1093/nar/gkl267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu H, Domingues FS, Sommer I, Lengauer T. NOXclass: prediction of protein–protein interaction types. BMC Bioinformatics. 2006;7:27. doi: 10.1186/1471-2105-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shoemaker BA, Zhang D, Thangudu RR, Tyagi M, Fong JH, Marchler-Bauer A, Bryant SH, Madej T, Panchenko AR. Inferred biomolecular interaction server––a web server to analyze and predict protein interacting partners and binding sites. Nucleic Acids Res. 2010;38:D518–D524. doi: 10.1093/nar/gkp842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ponstingl H, Kabir T, Thornton JM. Automatic inference of protein quaternary structure from crystals. J. Appl. Cryst. 2003;36:1116–1122. [Google Scholar]
- 18.Bernauer J, Bahadur RP, Rodier F, Janin J, Poupon A. DiMoVo: a Voronoi tessellation-based method for discriminating crystallographic and biological protein–protein interactions. Bioinformatics. 2008;24:652–658. doi: 10.1093/bioinformatics/btn022. [DOI] [PubMed] [Google Scholar]
- 19.Xu Q, Canutescu A, Obradovic Z, Dunbrack RL., Jr ProtBuD: a database of biological unit structures of protein families and superfamilies. Bioinformatics. 2006;22:2876–2882. doi: 10.1093/bioinformatics/btl490. [DOI] [PubMed] [Google Scholar]
- 20.Marchler-Bauer A, Anderson JB, Cherukuri PF, DeWeese-Scott C, Geer LY, Gwadz M, He S, Hurwitz DI, Jackson JD, Ke Z, et al. CDD: a Conserved domain database for protein classification. Nucleic Acids Res. 2005;33:D192–D196. doi: 10.1093/nar/gki069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu Q, Canutescu AA, Wang G, Shapovalov M, Obradovic Z, Dunbrack RL., Jr Statistical analysis of interface similarity in crystals of homologous proteins. J. Mol. Biol. 2008;381:487–507. doi: 10.1016/j.jmb.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Westbrook J, Ito N, Nakamura H, Henrick K, Berman HM. PDBML: the representation of archival macromolecular structure data in XML. Bioinformatics. 2005;21:988–992. doi: 10.1093/bioinformatics/bti082. [DOI] [PubMed] [Google Scholar]
- 23.Ye Y, Godzik A. Flexible structure alignment by chaining aligned fragment pairs allowing twists. Bioinformatics. 2003;19(Suppl. 2):II246–II255. doi: 10.1093/bioinformatics/btg1086. [DOI] [PubMed] [Google Scholar]
- 24.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of database programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Edgar R, Federhen S, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2008;36:D13–D21. doi: 10.1093/nar/gkm1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shindyalov IN, Bourne PE. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998;11:739–747. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- 27.Henikoff S, Henikoff JG. Performance evaluation of amino acid substitution matrices. Prot.: Struct., Funct. Genet. 1993;17:49–61. doi: 10.1002/prot.340170108. [DOI] [PubMed] [Google Scholar]
- 28.Legendre P, Legendre L. Numerical Ecology. 2nd edn. Amsterdam: Elsevier; 1998. [Google Scholar]
- 29.Taoka S, Lepore BW, Kabil O, Ojha S, Ringe D, Banerjee R. Human cystathionine beta-synthase is a heme sensor protein. Evidence that the redox sensor is heme and not the vicinal cysteines in the CXXC motif seen in the crystal structure of the truncated enzyme. Biochemistry. 2002;41:10454–10461. doi: 10.1021/bi026052d. [DOI] [PubMed] [Google Scholar]
- 30.Aloy P, Ceulemans H, Stark A, Russell RB. The relationship between sequence and interaction divergence in proteins. J. Mol. Biol. 2003;332:989–998. doi: 10.1016/j.jmb.2003.07.006. [DOI] [PubMed] [Google Scholar]
- 31.Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K, Takahashi R. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol. Cell. 2001;8:613–621. doi: 10.1016/s1097-2765(01)00341-0. [DOI] [PubMed] [Google Scholar]
- 32.Li W, Srinivasula SM, Chai J, Li P, Wu JW, Zhang Z, Alnemri ES, Shi Y. Structural insights into the pro-apoptotic function of mitochondrial serine protease HtrA2/Omi. Nat. Struct. Biol. 2002;9:436–441. doi: 10.1038/nsb795. [DOI] [PubMed] [Google Scholar]
- 33.Bong SM, Moon JH, Nam KH, Lee KS, Chi YM, Hwang KY. Structural studies of human brain-type creatine kinase complexed with the ADP-Mg2+-NO3-creatine transition-state analogue complex. FEBS Lett. 2008;582:3959–3965. doi: 10.1016/j.febslet.2008.10.039. [DOI] [PubMed] [Google Scholar]
- 34.Soundararajan M, Willard FS, Kimple AJ, Turnbull AP, Ball LJ, Schoch GA, Gileadi C, Fedorov OY, Dowler EF, Higman VA, et al. Structural diversity in the RGS domain and its interaction with heterotrimeric G protein alpha-subunits. Proc. Natl. Acad. Sci. USA. 2008;105:6457–6462. doi: 10.1073/pnas.0801508105. [DOI] [PMC free article] [PubMed] [Google Scholar]