


Abstract
Protein secretion is a key virulence mechanism of pathogenic and symbiotic bacteria, which makes the investigation of secreted proteins (‘effectors’) crucial for understanding the molecular bacterium–host interactions. Effective (http://effectors.org) is a database of predicted bacterial secreted proteins, implementing two complementary prediction strategies for protein secretion: the identification of eukaryotic-like protein domains and the recognition of signal peptides in amino acid sequences. The Effective web portal provides user-friendly tools for browsing and retrieving comprehensive precalculated predictions for whole bacterial genomes as well as for the interactive prediction of effectors in user-provided protein sequences.
INTRODUCTION
Bacterial protein secretion is a key virulence mechanism of pathogens and symbionts (1). Thereby effector proteins are transported from the bacterial cytosol into the extracellular medium as well as directly into the eukaryotic host cell (2). Eased uptake of the pathogen, manipulation of the immune response and preventing apoptosis of the infected host cell (3) are examples of the complex effects that are triggered by secreted bacterial proteins. For a better understanding of pathogenic organisms and the processes associated with virulence and infection on the molecular level, effectors are among the most important research targets. Their investigation promises novel routes for diagnostics and drug development, as they may offer pathogen-specific treatment of infections (4). Up to now, seven different bacterial secretion systems and the Sec pathway have been so far described as molecular ways of transport (1). Each of them is specific in terms of molecular structure and mechanism of translocation. Whereas, e.g. the Sec pathway is capable of transporting proteins into the extracellular medium, the syringe-like type III secretion system (TTSS) injects effectors directly into the host cell. The molecular recognition of effectors by the secretion machineries is not completely solved. For systems of types I, II, III, IV, V and the Sec pathway, it has been shown that signal peptides in effectors are important for their secretion (5).
To date, characterized effectors show high evolutionary divergence (6). Therefore, similarity-based approaches of transferring the functional annotation from well-characterized proteins to homologous sequences in other species have only limited power. Consequently, effectors have to be experimentally detected and functionally characterized for each pathogen. With only a small number of known effectors (7), there is a high demand for computer-based approaches for the prediction of candidate effector proteins. These bioinformatics methods can provide valuable support for experiments and target pre-selection (8). Starting points for bioinformatics analyses are the unique features of effector protein sequences. For two secretion systems, it is possible to predict effectors with high accuracy based on the associated signal peptide. For effectors secreted by the Sec pathway, this signal is accurately detected by SignalP (9). Also for the TTSS, a signal peptide in the N-terminal sequences of the effector proteins could be found (10–12). For secretion systems I, II, IV and V, there is to date no general method capable of identifying the respective signal sequence, nor is any method available that offers a large-scale signal-independent approach to effector identification.
In a number of single-organism studies, many effectors have been shown to contain diverse protein domain signatures that are typically found in eukaryotes (13–15). An example is the ankyrin-rich repeat domain that was found in proteins secreted via the type IV secretion system to disrupt the function of various eukaryotic factors in host cells (16). To date, the knowledge about these eukaryotic-like domains occurring in effector proteins is restricted to a small number of exemplary organisms and domains. The bioinformatic identification of eukaryotic-like domains can be performed by evaluating the taxonomic distributions of protein domains in a representative number of genomes of pathogens, non-pathogens and eukaryotes. All protein domains that occur in eukaryotes and pathogens, but not or only rarely in non-pathogens, could be detected this way. Despite its great potential, there has no large-scale bioinformatic resource yet been described making use of eukaryotic-like protein domains for the identification of novel effectors.
Here, we describe Effective, a novel and unique database of predicted secreted proteins in bacteria. Effective predicts putative effectors comprehensively by a combination of two complementary approaches: (i) independently from the mechanism of transport by identifying eukaryotic-like protein domains and (ii) by detecting the two known types of signal peptides regardless the presence of well-conserved protein domains. As SignalP (9) is the most widely used method predicting Sec pathway signals and EffectiveT3 represents the only type-III signal prediction method that was explicitly shown to be taxonomically universal (10), these two programs have been utilized.
METHODS AND IMPLEMENTATION
The Effective database and its web portal have been implemented using the JAVA programming language, a MySQL relational database and Java Server Pages.
The genome repository
Proteome data and features from a variety of different resources are integrated into the portal and structured in a genome repository: we derived the annotated proteins of all publicly available completely sequenced genomes listed in the RefSeq database (17), as well as all 11 912 domain signatures detected by Pfam (18) using the Simap database (19). For genomes included in the eggNOG Clusters of Orthologous Groups (20), we have additionally stored reduced proteomes containing only evolutionary conserved sequences being member of a COG or NOG. This approach eliminates ORFans representing possible gene over-predictions and thus improves the determination of domain enrichment scores (see below). All organisms covered by the genome repository are classified into 147 eukaryotes and, according to the Resource on Microbial Genomes (21), into 466 pathogenic, 121 symbiotic and 358 non-pathogenic bacteria. For all genomes, all genomic sequences including plasmids were considered. Furthermore, we identified those Gram-negative bacteria that harbor a probably functional TTSS. For this purpose, we used annotations from the KEGG database (22) to identify the proteomes that contain at least 2/3 of the proteins being part of the molecular TTSS apparatus. Regular updates of the genome repository are performed every 3 months.
Integration of signal-based effector prediction methods
The prediction of Sec pathway signal peptides was performed using the program SignalP (9), whereas EffectiveT3 has been used to predict type-III secreted proteins (10). Precalculations of the TTSS predictions have been performed for all 89 genomes in the repository harboring a probably functional TTSS.
Large-scale identification of eukaryotic-like protein domains
We implemented a systematic, large-scale approach to identify eukaryotic-like protein domains based on the comparison of domain frequencies in all genomes contained in the genome repository. The domain score enrichment score allows distinguishing between protein domains that are uniformly distributed over different classes of organisms and eukaryotic-like domains that are enriched in the proteomes of pathogenic bacteria. In order to eliminate the influence of bacterial contaminations in eukaryotic genomes, the calculation was restricted to protein domains that are detected in pathogenic genomes as well as in at least 10 eukaryotic genomes. To estimate the background model for each remaining domain, the average and standard deviation of its frequencies in all non-pathogenic genomes are calculated. For genomes included in the eggNOG Clusters of Orthologous Groups (20), frequencies according to the reduced proteomes containing only evolutionary conserved sequences have been determined additionally. For each pathogen genome, the domain enrichment score S of each domain has been calculated as the number of standard deviations σ in which the domain frequency in that particular pathogenic genome n differs from the average background frequency η in non-pathogen genomes: S = (n − η)/σ. Thereby it directly reflects the enrichment of a particular eukaryotic-like domain in proteins of a particular pathogenic genome. Although the distribution of domain occurrences across genomes has varying shapes, manual inspection of domain enrichment scores has shown that the domain enrichment scores typically show the characteristics of Z-scores and can be considered significant if higher than 3–5.
For user-provided data, sequences are scanned for Pfam domain signatures using HmmScan (18). Detected domains are evaluated based on the precalculated domain enrichment scores in the Effective database, considering the maximum score that is achieved by the particular domain in all pathogen genomes.
RESULTS
Effective is a database of predicted secreted proteins in bacterial genomes. We have implemented two complementary prediction strategies for protein secretion: (i) the recognition of signal peptides in amino acid sequences and (ii) the identification of eukaryotic-like protein domains. The current version of Effective contains 587 complete genomes of pathogenic and symbiotic bacteria, in which 421 774 effector proteins have been predicted in total by at least one method. As to be expected from the limited coverage of any of the prediction methods, many predictions are only supported by one or two methods (Figure 1); e.g. many proteins containing eukaryotic-like domains do not have a detectable secretion signal for the Sec and type III pathways. Contrarily, many proteins having a secretion signal do not contain any well-conserved protein domain, preventing the detection of eukaryotic-like function. This limitation is an intrinsic consequence of signature-based approaches. Consequently, the prediction of protein secretion by Effective does not require any consensus between the complementary prediction approaches but considers any single positive prediction to be of biological relevance. The overlap between predictions of Sec and type-III signal peptides is surprisingly high, as these two types of signal peptides should be incompatible to each other. However, due to the limited knowledge about the molecular principles of effector recognition by these secretion systems, we cannot yet discard one positive signal in the presence of the other. As the real number of secreted proteins is unknown for all genomes of pathogens and symbionts, the amount of false positives contributing to the surprisingly high number of predictions, only supported by SignalP (334 629) or EffectiveT3 (31 350), cannot be reliably determined. Considering the limited accuracy of both programs, subsequent filtering of the predictions (e.g. discarding functionally well-annotated, probably not-secreted proteins) is highly recommended. The ongoing improvement of bioinformatic tools for the identification of signal peptides will probably improve the situation within the next years.
Figure 1.
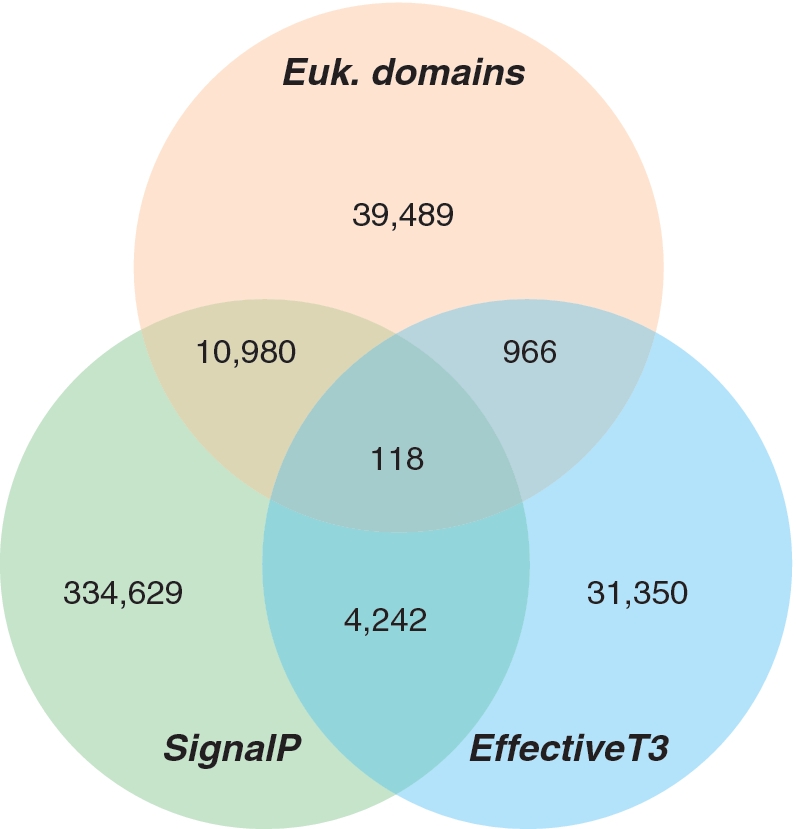
Numbers of predicted effectors in 587 genomes of pathogenic and symbiotic bacteria, indicated by supporting prediction method. EffectiveT3 has only been applied to 89 genomes of Gram-negative bacteria that encode a type III secretion system.
Predicted effectors in complete genomes of pathogenic and symbiotic bacteria
The applicability of the prediction approaches implemented in Effective is different: whereas Sec dependent secretion is a widespread feature of pathogens and symbionts and eukaryotic-like domains can be encoded in any of their genomes, type-III secreted proteins can only be expected in genomes of Gram-negative bacteria encoding a type-III secretion system that is likely to be functional. Effective therefore provides different tools accessing precalculated secreted proteins.
For all 89 genomes of Gram-negative bacteria that encode a probably functional type III secretion system, the results of EffectiveT3 predicting type-III effectors are included in the precalculated files available for download. These results contain the accessions and descriptions of all annotated proteins in the respective genomes, the EffectiveT3 scores and the secretion prediction for each protein according to the selective default threshold and standard prediction model.
By selecting the organism of interest, eukaryotic-like domains and signal peptides of the Sec pathway can be retrieved from the Effective database for each of the 587 genomes of pathogenic and symbiotic bacteria. In order to provide consistent scores across genomes, domain enrichment scores based on the total number of annotated proteins are always provided. If the genome is contained in the eggNOG database of Clusters of Orthologous Groups (20), additional domain enrichment scores according to the conserved proteome are given. As the further exploration and biologic interpretation of eukaryotic-like protein domains is facilitated by dedicated domain reports in Effective (see below), we provide these precalculated results online through interactive web pages that link any predicted domain to its status report page.
Comprehensive information about eukaryotic-like domains
The identification of eukaryotic-like domains, based on the score resulting from the taxonomic occurrence of each domain, is a unique feature of the Effective database. Specific report pages are therefore provided for each protein domain that has been detected in at least one pathogenic genome with a significant domain enrichment score of 4 or higher. For any domain, the numbers and lists of pathogenic, non-pathogenic and eukaryotic genomes encoding at least one protein having this domain are indicated. As the frequencies of the domain in these organisms determine the domain enrichment score, this information allows the user to understand why Effective has identified the particular domain as eukaryotic-like. For each listed pathogen genome, lists of proteins containing the particular eukaryotic-like domain, actually representing predicted effector proteins, can be obtained.
Interactive prediction of secreted proteins in user-provided sequences
The Effective database offers a user-friendly interface for the interactive prediction of secreted proteins in sequence data uploaded by the user. Compared to the precalculated data, this tool allows the user to control all prediction settings and to integrate the results from the different methods into one sorted table. Input data can contain even thousands of protein sequences; however, the user should be aware of the calculation time necessary to perform automatic domain annotation and signal peptide detection.
For the analysis of input sequence data, the user can choose from any combination of the three different prediction methods: (i) prediction of Sec pathway secreted proteins, (ii) prediction of type-III secreted proteins and (iii) identification of eukaryotic-like protein domains. The configurable parameters of these methods are explained in a detailed ‘Help’-section on the website. Proteins that have received positive predictions from at least one selected method are provided in tabular form for further visual inspection on the website and for download in Excel format.
Prediction tools for type-III secreted proteins and communication with the scientific community
The Effective database portal provides the EffectiveT3 program as a stand-alone application. By downloading and locally executing the program, or by starting it using Java WebStart, confidential sequence data can be analyzed without transferring it over the Internet to the Effective database. The list of known type-III secreted proteins that have been used for training the current EffectiveT3 classification models can be displayed or downloaded by the user. As further improvements of EffectiveT3 rely mostly on the availability of more comprehensive training data, users may submit the amino acid sequences newly characterized effectors via the Effective database to the developers of the EffectiveT3 software.
Update of data and prediction methods
Updates of the genome repository and all precalculated predictions in the Effective database are automatically conducted quarterly. If new prediction methods for secreted proteins become available, e.g. for the identification of signal sequences in type-IV secreted proteins, the portal architecture allows for their easy integration. All updates of the Effective database will be announced to the users via the ‘News’ section of the portal and the Effective mailing list.
CONCLUSIONS AND FUTURE WORK
The Effective database is the first bioinformatic resource combining two complementary approaches for the prediction of bacterial secreted proteins: (i) function-based prediction by identification of eukaryotic-like domains and (ii) prediction based on signal peptides leading to transport by protein secretion systems. None of the two strategies can, by principle, achieve complete coverage; therefore, their integration in a single resource is beneficial for the comprehensive annotation of putative effectors in genomes and proteomes. The user-friendly web portal of the Effective database offers a versatile toolbox for generating new effector candidates and for target selection toward experimental investigation of putative secreted proteins. As the development and improvement of computational methods for effector prediction is a vital area of research, new methods can be expected to become available within the next years. The Effective database provides a powerful framework for their easy integration and will therefore make relevant new methods accessible to the users of the database.
FUNDING
The ERA-NET PathoGenoMics (‘Pathomics’ to M.-A.J.); and the German Research Foundation (DFG RA 1719/1-1 to R.A.). Funding for open access charge: ERA-NET PathoGenoMics.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank Tanja Bieber and Dominik Lindner for their help in implementing and setting up the Effective web portal.
REFERENCES
- 1.Tseng TT, Tyler BM, Setubal JC. Protein secretion systems in bacterial–host associations, and their description in the Gene Ontology. BMC Microbiol. 2009;9(Suppl. 1):S2. doi: 10.1186/1471-2180-9-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rodrigues CD, Enninga J. The 'when and whereabouts' of injected pathogen effectors. Nat. Methods. 2010;7:267–269. doi: 10.1038/nmeth0410-267. [DOI] [PubMed] [Google Scholar]
- 3.Diacovich L, Gorvel JP. Bacterial manipulation of innate immunity to promote infection. Nat. Rev. 2010;8:117–128. doi: 10.1038/nrmicro2295. [DOI] [PubMed] [Google Scholar]
- 4.Rasko DA, Sperandio V. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discov. 2010;9:117–128. doi: 10.1038/nrd3013. [DOI] [PubMed] [Google Scholar]
- 5.Beeckman DS, Vanrompay DC. Bacterial secretion systems with an emphasis on the chlamydial Type III secretion system. Curr. Issues Mol. Biol. 2010;12:17–41. [PubMed] [Google Scholar]
- 6.Almeida NF, Yan S, Lindeberg M, Studholme DJ, Schneider DJ, Condon B, Liu H, Viana CJ, Warren A, Evans C, et al. A draft genome sequence of Pseudomonas syringae pv. tomato T1 reveals a type III effector repertoire significantly divergent from that of Pseudomonas syringae pv. tomato DC3000. Mol. Plant Microbe Interact. 2009;22:52–62. doi: 10.1094/MPMI-22-1-0052. [DOI] [PubMed] [Google Scholar]
- 7.Yang J, Chen L, Sun L, Yu J, Jin Q. VFDB 2008 release: an enhanced web-based resource for comparative pathogenomics. Nucleic Acids Res. 2008;36:D539–D542. doi: 10.1093/nar/gkm951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vizcaino C, Restrepo-Montoya D, Rodriguez D, Nino LF, Ocampo M, Vanegas M, Reguero MT, Martinez NL, Patarroyo ME, Patarroyo MA. Computational prediction and experimental assessment of secreted/surface proteins from Mycobacterium tuberculosis H37Rv. PLoS Computat. Biol. 2010;6:e1000824. doi: 10.1371/journal.pcbi.1000824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 2004;340:783–795. doi: 10.1016/j.jmb.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 10.Arnold R, Brandmaier S, Kleine F, Tischler P, Heinz E, Behrens S, Niinikoski A, Mewes HW, Horn M, Rattei T. Sequence-based prediction of type III secreted proteins. PLoS Pathog. 2009;5:e1000376. doi: 10.1371/journal.ppat.1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lower M, Schneider G. Prediction of type III secretion signals in genomes of Gram-negative bacteria. PloS one. 2009;4:e5917. doi: 10.1371/journal.pone.0005917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Samudrala R, Heffron F, McDermott JE. Accurate prediction of secreted substrates and identification of a conserved putative secretion signal for type III secretion systems. PLoS Pathog. 2009;5:e1000375. doi: 10.1371/journal.ppat.1000375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Voth DE, Heinzen RA. Coxiella type IV secretion and cellular microbiology. Curr. Opin. Microbiol. 2009;12:74–80. doi: 10.1016/j.mib.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cazalet C, Rusniok C, Bruggemann H, Zidane N, Magnier A, Ma L, Tichit M, Jarraud S, Bouchier C, Vandenesch F, et al. Evidence in the Legionella pneumophila genome for exploitation of host cell functions and high genome plasticity. Nat. Genet. 2004;36:1165–1173. doi: 10.1038/ng1447. [DOI] [PubMed] [Google Scholar]
- 15.Ponting CP, Aravind L, Schultz J, Bork P, Koonin EV. Eukaryotic signalling domain homologues in archaea and bacteria. Ancient ancestry and horizontal gene transfer. J. Mol. Biol. 1999;289:729–745. doi: 10.1006/jmbi.1999.2827. [DOI] [PubMed] [Google Scholar]
- 16.Pan X, Luhrmann A, Satoh A, Laskowski-Arce MA, Roy CR. Ankyrin repeat proteins comprise a diverse family of bacterial type IV effectors. Science. 2008;320:1651–1654. doi: 10.1126/science.1158160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, et al. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rattei T, Tischler P, Gotz S, Jehl MA, Hoser J, Arnold R, Conesa A, Mewes HW. SIMAP–a comprehensive database of pre-calculated protein sequence similarities, domains, annotations and clusters. Nucleic Acids Res. 2010;38:D223–D226. doi: 10.1093/nar/gkp949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muller J, Szklarczyk D, Julien P, Letunic I, Roth A, Kuhn M, Powell S, von Mering C, Doerks T, Jensen LJ, et al. eggNOG v2.0: extending the evolutionary genealogy of genes with enhanced non-supervised orthologous groups, species and functional annotations. Nucleic Acids Res. 2010;38:D190–D195. doi: 10.1093/nar/gkp951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Federhen S, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2010;38:D5–D16. doi: 10.1093/nar/gkp967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–D360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]