


Abstract
We describe IsoBase, a database identifying functionally related proteins, across five major eukaryotic model organisms: Saccharomyces cerevisiae, Drosophila melanogaster, Caenorhabditis elegans, Mus musculus and Homo Sapiens. Nearly all existing algorithms for orthology detection are based on sequence comparison. Although these have been successful in orthology prediction to some extent, we seek to go beyond these methods by the integration of sequence data and protein–protein interaction (PPI) networks to help in identifying true functionally related proteins. With that motivation, we introduce IsoBase, the first publicly available ortholog database that focuses on functionally related proteins. The groupings were computed using the IsoRankN algorithm that uses spectral methods to combine sequence and PPI data and produce clusters of functionally related proteins. These clusters compare favorably with those from existing approaches: proteins within an IsoBase cluster are more likely to share similar Gene Ontology (GO) annotation. A total of 48 120 proteins were clustered into 12 693 functionally related groups. The IsoBase database may be browsed for functionally related proteins across two or more species and may also be queried by accession numbers, species-specific identifiers, gene name or keyword. The database is freely available for download at http://isobase.csail.mit.edu/.
INTRODUCTION
The concept of gene homology, i.e. sets of genes across species that have been derived from a common ancestor, has been a powerful tool in comparative genomics research. In addition to its usefulness in understanding evolutionary relationships between genes, its practical application allows us to extrapolate experimentally derived insights from one species to another. In this article, we focus on discovering orthologs, which are homologous genes separated by speciation events (1). The concept of gene orthology encompasses two interpretations: phylogenetic and functional. The phylogenetic interpretation is that orthologs are genes/proteins in different species that have evolved from the same gene in a common ancestor. The functional interpretation is that orthologs are genes/proteins that perform functionally equivalent roles in different species. The two interpretations do not always yield exactly the same answer, but they usually yield similar answers (2). The functional interpretation of orthology has been extremely useful in annotation transfer tasks, for example, for identifying the human gene that performs the same role as a given fly gene. This practical use has also motivated a significant amount of work in the identification of orthologs.
The pioneering work of Tatusov et al. (3) introduced the Clusters of Orthologous Groups (COG) database, where clusters of orthologous genes were inferred using exhaustive sequence comparison of genes across multiple genomes. The basic approach described there continues to be used by much of the orthology detection community: perform pairwise sequence comparison between all the genes in the input set, and then cluster genes into groups where the intra-group sequence similarity is high while the between-group similarity is low. The differences between the various approaches lie in the details: how the sequences are compared (local versus global alignment); the heuristics for choosing the seed gene pairs for each cluster and how to combine/prune clusters (4–7). For example, InParanoid uses an ‘outgroup’ species to calibrate when the pairwise score is high enough for the genes to be co-clustered. As a pre-clustering step, OrthoMCL normalizes sequence comparison scores to adjust for differences in how far in the past speciation or gene duplication may have occurred.
In this article, we describe a different approach to the orthology detection problem. Our aim is to identify gene correspondences across species that maximize functional similarity. As our approach emphasizes functional similarity over phylogenetic relationships, we refer to our predictions as ‘isologs’, rather than ‘orthologs’. To compute isologs across species, we integrate sequence data with protein–protein interaction (PPI) data. It is now well established that PPI data capture significant functional information: proteins that interact with each other are likely to perform similar functions (8,9). Proteins that occupy the same topological position in their respective species-wide PPI networks are thus likely to perform the same function. In our approach, sequence comparisons still provide a strong signal, but they are supplemented with PPI similarity information. We believe that this provides a stronger approach to inferring functional similarity than the sequence-only methods currently used.
We introduce IsoBase, a web database of functionally related proteins based on the IsoRankN algorithm (10), currently covering the major eukaryotic model organisms: Saccharomyces cerevisiae, Drosophila melanogaster, Caenorhabditis elegans, Mus musculus and Homo Sapiens. IsoRank and IsoRankN (10,11) software was used to globally align PPI networks across multiple species and the results were then used to cluster proteins across the various species such that these clusters best represent proteins with conserved biological function. The software is efficient and automatically adjusts to the wide variation in sizes of the known species-specific networks. IsoBase will be continually updated as more PPI data become available for additional as well as currently supported species.
The IsoBase database may be browsed for functionally related proteins across two or more species. It may also be queried in various ways: based on accession numbers, species-specific identifiers (e.g. CG numbers), gene names or descriptions. IsoBase allows batch querying by uploading a file with multiple gene ids, names and/or keywords. The database can also be bulk-downloaded. The displayed results include mean normalized entropy scores for each cluster, allowing users to further filter the data by cluster consistency.
Compared with existing sequence-only approaches (Homologene (12), Inparanoid (6) and OrthoMCL-DB (7)), we showed previously (10,11) and further demonstrate in ‘Statistics’ on the IsoBase website that incorporating PPI data helps significantly in finding functionally related proteins. Compared with methods like OrthoMCL, which explicitly claim to evolutionary insights, our approach produces protein–protein correspondences (which we refer to as ‘isologs’) that better preserve Gene Ontology (GO) functional similarity within each cluster. Furthermore, our isology mappings outperform those based on local network alignment (10,11), such as NetworkBLAST-M (13) and Graemlin 2.0 (14).
DESIGN AND IMPLEMENTATION
Data
IsoBase is compiled from two forms of data: PPI networks and sequence similarity scores between pairs of proteins. PPI networks from five major eukaryotic model organisms (H. sapiens, M. musculus, D. melanogaster, C. elegans and S. cerevisiae) were constructed by combining data from the Database of Interacting Proteins (DIP) (15), BioGRID (16) and Human Protein Reference Database (HPRD) databases (17). In total, these PPI networks contained 48 120 proteins and 114 897 known interactions. As new PPI data become available and are released by DIP, BioGRID or HPRD, the IsoBase database will be updated; please see the website for the currently used version of the underlying data. Sequence similarity scores of pairs of proteins were obtained from Ensembl (18) and consisted of BLAST Bit-values of the sequences.
IsoRank and IsoRankN algorithm
We briefly describe the algorithm used in the database construction. For a fuller description, along with analysis and evaluations of the algorithms, please see (10,11).
The input to the algorithm consists of PPI and sequence data from multiple species. The algorithm first integrates sequence and PPI data to construct pairwise scores between the proteins in its input; it then uses these scores to cluster the proteins. Both the stages use spectral techniques. In the first stage, for every protein pair (i,j), where i and j are from different species, we compute the score Rij. We pose this computation as an eigenvalue problem, explicitly modeling the tradeoff between the twin objectives of high PPI network overlap and high sequence similarity between the protein pairs. Let R be the vector of scores Rij, normalized so that ΣRij = 1. We require
Here, α is a free parameter and E is the vector of sequence similarity scores Eij; we use the BLAST bit score. A is a matrix that encodes the PPI networks’ connectivity information. Its rows and columns correspond to protein pairs:
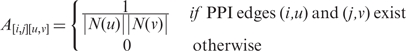 |
The eigenvalue equation above captures the following intuition: the score Rij for matching a protein pair (i,j) is a weighted sum of the sequence similarity score Eij and the total support provided to the match by each of the |N(i)||N(j)| possible matches between the neighbors of i and j. In return, each candidate pair of matching proteins (u,v) must distribute back its entire score Ruv equally among the |N(u)||N(v)| possible matches between its neighbors.
The scores Rij can be interpreted as a graph H, where each protein i corresponds to a node and an edge (i,j) exists with weight Rij, if Rij > 0. Given this graph, the second stage of our algorithm uses a spectral clustering approach. We choose an arbitrary species to start with and for each protein v in it, compute the subgraph Sv consisting of v and all nodes in H connected to it with a large weight. We then use spectral partitioning to identify 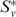 , a high-weight clique-like subset of Sv. If two clusters
, a high-weight clique-like subset of Sv. If two clusters 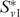 and
and 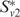 have edges with high weight between them, we merge them. We repeat the entire process until all the proteins have been assigned to clusters (please see (11) for more details).
have edges with high weight between them, we merge them. We repeat the entire process until all the proteins have been assigned to clusters (please see (11) for more details).
EVALUATION OF PREDICTIONS IN IsoBase
The key motivation behind IsoBase is the hypothesis that the combination of sequence and PPI data should enable better identification of functionally related proteins across species than just using sequence data. However, there is a lack of standardized techniques for benchmarking how well an orthology detection method captures functional similarity (19). To that end, we create an evaluation measure that can be used for benchmarking in an unbiased way and make it available for download on the IsoBase website.
To evaluate our predicted clustering, we measured the within-cluster consistency of GO (20) annotation of the predicted clusters. The intuition here is that each cluster should correspond to a set of genes with the same function. Thus, consistency measures the functional uniformity of genes in each cluster, represented by mean normalized entropies calculated for each predicted cluster over all proteins within the PPI networks used by IsoRankN. Clusters with greater consistency have lower entropy and, therefore, a greater indication of proteins sharing the same function. The entropy of a given cluster S*v is:
where pi is the fraction of S*v with GO term i, and d is the number of GO terms in each cluster. Mean entropy was then normalized by the number of distinct GO terms in a cluster so that 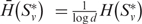 .
.
An important factor we considered when evaluating GO enrichment of clusters was the use of standardized sets of GO terms. It would not make sense to conclude that a group of genes are not functionally related if all that differs is the level of detail in their GO annotation; recall that GO terms are related to each other as part of a directed acyclic graph (DAG). The use of GO Slim sets has become popular for similar reasons (18). We created a standardized set by projecting GO terms to a common level of GO hierarchy. Details on the set of GO terms used and scripts for mapping GO terms to a common level in the GO hierarchy can be found on the ‘Download’ page of the IsoBase website.
Using the benchmark described above, we compared IsoRankN predictions to that of Homologene and OrthoMCL on five major eukaryotic networks (yeast, worm, fly, mouse and human). We did not compare to InParanoid, because it only provides pairwise orthology predictions, rather than multispecies groupings. Of 87 737 total proteins, IsoRankN clustered 48 120 (54.8%) proteins into 12 693 isologous groups. It outperformed the other methods in terms of within-cluster consistency of GO annotations. Across all predicted clusters, mean normalized entropy for IsoRankN (0.0586) was substantially lower than Homologene (0.255) and OrthoMCL (0.215) (Table 1). Additionally, mean normalized entropies for predictions on pairs of species produced similar results. Clusters consisting of only one protein were not considered in the entropy comparisons because these cases provide no information regarding functional relatedness between orthologs. Details on the entropy comparisons among IsoBase, Homologene and OrthoMCL can be found on the ‘Statistics’ page of the IsoBase website.
Table 1.
Comparative consistency on the five eukaryotic networks
| IsoRankN | Homologene | OrthoMCL | |
|---|---|---|---|
| Mean entropy | 0.0740 | 0.284 | 0.241 |
| Mean normalized entropy | 0.0586 | 0.255 | 0.215 |
| Exact cluster ratioa | 0.489 (6204/12 693) | 0.355 (4470/12 579) | 0.237 (1973/8326) |
| Exact protein ratiob | 0.539 (25 929/48 120) | 0.469 (13 134/27 988) | 0.364 (5796/15 940) |
Mean entropy and mean normalized entropy of predicted clusters. Note that the boldface numbers represent the best performance with respect to each measure.
aThe fraction of predicted clusters that are ‘exact’, that is all contained proteins have the same GO term.
bThe fraction of proteins in exact clusters.
We also measured the fraction of predicted clusters that are ‘exact’, i.e. all contained proteins have the same GO term. We find that IsoRankN predicts a higher fraction of exact clusters (0.489) than that for Homologene (0.355) and OrthoMCL (0.237) (Table 1).
In addition, we evaluated IsoRankN, Homologene and OrthoMCL predictions on human–fly orthologs in particular. Upon closer examination, we find that IsoRankN predicts a higher number of clusters (151) involving many fly genes mapped to one human gene than either Homologene (3) or OrthoMCL (1). For example, all methods predict fly gene CG8399 as an ortholog for human gene FRRS1. But IsoRankN also predicts CG14515 and CG7532 as orthologs. A closer look at these two fly genes reveals domain overlap with FRRS1. Another example shows all methods identifying fly homolog Dcr-1 for human DICER1, a ribonuclease that plays a key role in the RNA interference (RNAi) pathway; but IsoRankN solely identifies fly homolog Dcr-2 (with domain and GO overlap) as well. See the ‘Statistics’ page for further examples.
In our previous work, we showed that IsoRankN outperforms other related techniques for PPI network alignment (NetworkBLAST-M (13) and Græmlin2K (14)) in terms of number of clusters predicted, within-cluster consistency and GO/Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment. See Liao et al. (10) for details. We also showed that IsoRank, the basis of IsoRankN, compares favorable to InParanoid on pairs of species (11).
IsoBase WEB INTERFACE
IsoBase provides a variety of ways to access functionally related proteins through its web interface. Query options and search results detailing fully annotated orthologs are summarized in Figure 1. We provide an online ‘Help’ page at the website which describes possible query options, supported gene ids and interpretation of search results.
Figure 1.

Web interface and output of IsoBase. (A and B) Webserver entry page. (C) Example of an output page when choosing to browse through all ortholog clusters predicted over the PPI network alignment of two species, D. melanogaster and S. cerevisiae. Mean entropy scores normalized by the number of distinct GO terms for an ortholog cluster are displayed along with external sequence database links for each ortholog and associated KEGG and GO annotations.
Gene search
The user can search for isologs of their favorite protein based on gene name, gene symbol or a wide array of gene identifiers, including ‘ids’ from Ensembl, Entrez, GenBank, RefSeq, UniProtKB, Wormbase, Mouse Genome Informatics, FlyBase, Saccharomyces Genome Database, HPRD and DIP (Figure 1A). Upon submitting a query, IsoBase returns a cluster of functionally related proteins as well as a mean normalized entropy score computed for the cluster (Figure 1C). IsoBase annotates and interactively links each isolog to GO, KEGG and various genome databases. Batch querying is also supported, giving users the option to upload a list of query proteins or genes in any of the supported identifier formats. IsoBase then returns a cluster of isologs for each query gene or protein in its search results (Figure 1B).
Keyword search
In addition, users can search using a single keyword, such as a description or general function of a protein. IsoBase will retrieve all clusters having identifiers or descriptions containing the keyword. For example, a non-exact match for a keyword ‘YAL’ will retrieve an identifier ‘YAL027W’, while an exact match would not.
Browse
IsoBase can be browsed in its entirety. Users can filter through the entire set of clusters by selecting which eukaryotic PPI networks are included in the PPI network alignment. For instance, if three species are selected, IsoBase returns clusters that include proteins from only those three species. Entropy score cut-offs can also be lowered to increase the consistency of GO and KEGG annotations within each cluster, with an entropy of 0 indicating maximum consistency. In the ‘Statistics’ page of the website, we discuss the evaluation of our results using mean normalized entropy and how entropy is computed.
Data availability
Although isolog predictions are accessible through query and browse functions from the IsoBase web interface, predictions are also freely available via bulk download. In addition, the website contains the set of clusters for all species, mean normalized entropy scores associated with each cluster and KEGG/GO annotations for each predicted isolog. IsoBase also provides mappings between IsoBase internal identifiers and identifiers from a variety of external genome databases. We further provide the GO information used in entropy calculations, the GO hierarchy (represented as a DAG) and scripts to generate DAGs and identify all the GO terms at a given level. PPI networks for all eukaryotic species (fly, yeast, mouse, worm and human) and BLAST data have been made available in addition to the executables for running IsoRank and IsoRankN algorithms. The initial database covers the five species for which significant amount of PPI data are available; in the future, we anticipate that more PPI data may enable us to support additional species as well as better support the current species. We plan to update IsoBase on a semi-yearly basis.
DISCUSSION
We have presented IsoBase, a database that contains groups of proteins predicted to be functionally related. Unlike much of the existing work in sequence-based orthology detection, IsoBase is primarily designed to provide function-oriented ortholog detection. This focus on functional relationships is of significant practical value (2). Although our approach is not based on phylogenetic considerations, the phylogenetic and functional interpretations of orthology are closely related. In keeping with this intuition, sequence similarity information provides a large part of the signal used by our prediction algorithm, and our predictions broadly agree with existing sequence-based orthology predictions. The key contribution of IsoBase is the simultaneous use of PPI and sequence data in the prediction process. With the rapid growth of PPI data, the functional information provided by such data can be valuable in identifying functionally related proteins across species. The integrative approach used here allows us to make predictions where the within-cluster GO annotation similarity is better than in the predictions from sequence-only approaches.
In future work, we intend to explore synergies between our approach and existing sequence-only approaches. For example, using our method as a post-processing step after these approaches may help identify orthologs for proteins outside the existing methods’ coverage. Also, in cases where existing methods produce multiple matches, our method may be used to rank them in the order of functional similarity. We also intend to expand the number of species available in our database. Finally, as more PPI data become available, we will update the database with improved predictions.
ACKNOWLEDGEMENTS
We thank Claire (Yanhui) Hu, Stephanie Mohr and Norbert Perrimon for most helpful discussions, comparative analysis to other methods, and feedback on the database.
FUNDING
This publication was made possible by Grant Number 1R01GM081871; from the National Institute of General Medical Sciences. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or NIGMS. The Fannie and John Hertz Foundation and the NSF MSPRF (to M.B., in part); the National Science Council (NSC99-2218-E-007-010 to C.-S.L.). Funding for open access charge: National Institutes of Health (1R01GM081871).
Conflict of interest statement. None declared.
REFERENCES
- 1.Koonin E.V. Orthologs, paralogs, and evolutionary genomics. Ann. Rev. Genet. 2005;39:309–338. doi: 10.1146/annurev.genet.39.073003.114725. [DOI] [PubMed] [Google Scholar]
- 2.Fang G., Bhardwaj N., Robilotto R., Gerstein M.B. Getting started in gene orthology and functional analysis. PLoS Comp. Biol. 2010;6:e1000703. doi: 10.1371/journal.pcbi.1000703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tatusov R.L., Galperin M.Y., Natale D.A., Koonin E.V. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schneider A., Dessimoz C., Gonnet G.H. OMA Browser—exploring orthologous relations across 352 complete genomes. Bioinformatics. 2007;23:2180–2182. doi: 10.1093/bioinformatics/btm295. [DOI] [PubMed] [Google Scholar]
- 5.DeLuca T.F., Wu I.H., Pu J., Monaghan T., Peshkin L., Singh S., Wall D.P. Roundup: a multi-genome repository of orthologs and evolutionary distances. Bioinformatics. 2006;22:2044–2046. doi: 10.1093/bioinformatics/btl286. [DOI] [PubMed] [Google Scholar]
- 6.Chen F., Mackey A.J., Stoeckert C.J., Jr, Roos D.S. OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 2006;34:D363–D368. doi: 10.1093/nar/gkj123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Brien K.P., Remm M., Sonnhammer E.L. Inparanoid: a comprehensive database of eukaryotic orthologs. Nucleic Acids Res. 2005;33:D476–D480. doi: 10.1093/nar/gki107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Przytycka T.M., Singh M., Slonim D.K. Toward the dynamic interactome: it’s about time. Brief Bioinform. 2010;11:15–29. doi: 10.1093/bib/bbp057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharan R., Ulitsky I., Shamir R. Network-based prediction of protein function. Mol. Syst. Biol. 2007;3:88. doi: 10.1038/msb4100129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liao C.S., Lu K., Baym M., Singh R., Berger B. IsoRankN: spectral methods for global alignment of multiple protein networks. Bioinformatics. 2009;25:i253–i258. doi: 10.1093/bioinformatics/btp203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh R., Xu J., Berger B. Global alignment of multiple protein interaction networks with application to functional orthology detection. Proc. Natl Acad. Sci. USA. 2008;105:12763–12768. doi: 10.1073/pnas.0806627105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sayers E.W., Barrett T., Benson D.A., Bolton E., Bryant S.H., Canese K., Chetvernin V., Church D.M., DiCuccio M., Federhen S., et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2010;38:D5–D16. doi: 10.1093/nar/gkp967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalaev M., Smoot M., Ideker T., Sharan R. NetworkBLAST: comparative analysis of protein networks. Bioinformatics. 2008;24:594–596. doi: 10.1093/bioinformatics/btm630. [DOI] [PubMed] [Google Scholar]
- 14.Flannick J., Novak A., Srinivasan B.S., McAdams H.H., Batzoglou S. Graemlin: general and robust alignment of multiple large interaction networks. Genome Res. 2006;16:1169–1181. doi: 10.1101/gr.5235706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salwinski L., Miller C.S., Smith A.J., Pettit F.K., Bowie J.U., Eisenberg D. The Database of Interacting Proteins: 2004 update. Nucleic Acids Res. 2004;32:D449–D451. doi: 10.1093/nar/gkh086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Breitkreutz B.J., Stark C., Reguly T., Boucher L., Breitkreutz A., Livstone M., Oughtred R., Lackner D.H., Bähler J., Wood V., et al. The BioGRID Interaction Database: 2008 update. Nucleic Acids Res. 2008;36:D637–D640. doi: 10.1093/nar/gkm1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keshava Prasad T.S., Goel R., Kandasamy K., Keerthikumar S., Kumar S., Mathivanan S., Telikicherla D., Raju R., Shafreen B., Venugopal A., et al. Human Protein Reference Database—2009 update. Nucleic Acids Res. 2009;37:D767–D772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hubbard T.J., Aken B.L., Ayling S., Ballester B., Beal K., Bragin E., Brent S., Chen Y., Clapham P., Clarke L., et al. Ensembl 2009. Nucleic Acids Res. 2009;37:D690–D697. doi: 10.1093/nar/gkn828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gabaldón T., Dessimoz C., Huxley-Jones J., Vilella A.J., Sonnhammer E.L., Lewis S. Joining forces in the quest for orthologs. Genome Biol. 2009;10:403. doi: 10.1186/gb-2009-10-9-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000; 25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Although isolog predictions are accessible through query and browse functions from the IsoBase web interface, predictions are also freely available via bulk download. In addition, the website contains the set of clusters for all species, mean normalized entropy scores associated with each cluster and KEGG/GO annotations for each predicted isolog. IsoBase also provides mappings between IsoBase internal identifiers and identifiers from a variety of external genome databases. We further provide the GO information used in entropy calculations, the GO hierarchy (represented as a DAG) and scripts to generate DAGs and identify all the GO terms at a given level. PPI networks for all eukaryotic species (fly, yeast, mouse, worm and human) and BLAST data have been made available in addition to the executables for running IsoRank and IsoRankN algorithms. The initial database covers the five species for which significant amount of PPI data are available; in the future, we anticipate that more PPI data may enable us to support additional species as well as better support the current species. We plan to update IsoBase on a semi-yearly basis.