


Abstract
We describe the Biomolecule Stretching Data Base that has been recently set up at http://www.ifpan.edu.pl/BSDB/. It provides information about mechanostability of proteins. Its core is based on simulations of stretching of 17 134 proteins within a structure-based model. The primary information is about the heights of the maximal force peaks, the force–displacement patterns, and the sequencing of the contact-rupturing events. We also summarize the possible types of the mechanical clamps, i.e. the motifs which are responsible for a protein's resistance to stretching.
INTRODUCTION
Despite more than a decade of experiments on single biomolecule manipulation, mechanical properties of only several scores of proteins have been measured. A characteristic scale of the force of resistance to stretching, Fmax, has been found to range between ∼10 and 480 pN. The Biomolecule Stretching Data Base (BSDB) described here provides information about expected values of Fmax for, currently, 17 134 proteins. The values and other characteristics of the unfolding process, including the nature of identified mechanical clamps, are available at http://www.ifpan.edu.pl/BSDB/. They have been obtained through simulations within a structure-based model which correlates satisfactorily with the available experimental data on stretching. BSDB also lists experimental data and results of the existing all atom simulations. It is intended to be updated internally, through users input, and by making requests for needed calculations. The database offers a Protein-Data-Bank-wide guide to mechano-stability of proteins.
Functioning of a biological cell involves conversion of chemical energy into conformational changes of proteins, nucleic acids and their complexes. Understanding of mechanical processes in the cell requires information about mechanical properties of the constituting biomolecules. One way to obtain it is through single-molecule manipulation such as stretching by a tip of an atomic force microscope (1,2). The scope of such studies is very limited compared to the number of the kinds of the molecules that are present in the cell. There is then a need to have estimates of mechanical parameters for a much larger set of molecules. These estimates are necessary to provide guidance when selecting targets for experimental studies and when making theoretical models.
Recently, we have provided such estimates for 17 134 proteins (3) by performing molecular dynamics simulations within a coarse-grained, structure-based model. We have considered all proteins comprising up to 250 amino acids and having structures deposited in the Protein Data Bank (PDB) (4). The cutoff size well exceeds a typical single domain size which is of order 100–150 residues. This data set includes also proteins with knots of type 31 and one slipknotted protein (5,6). Reference (3) presents a summary of the results and lists proteins with particularly large values of Fmax. One of these proteins, scaffoldin c7A with the PDB structure code 1aoh, has been confirmed experimentally to be highly resistant to stretching. Its measured is equal to 480 pN (7). Here, we describe the Biomolecule Stretching Database (BSDB) in which results for all studied proteins are deposited. Initially, it is based on the results derived for ref. (3) but it is intended to grow. A synthetic discussion of the results is contained in (3). It includes presenting probability distributions of the values of Fmax for the whole set and for various structural categories of proteins. In particular, it identifies categories which are likely to yield large forces. It also discusses the nature of possible mechanical clamps—structural regions which generate the most significant resistance to stretching. Usually, resistance arises through shear between parallel or antiparallel β-strands (8–10). However, it should attain its top strength if formation of the cysteine slipknot motif is operational dynamically (3). This novel mechanism of strength has been discovered as a result of making the PDB-wide survey. It involves pulling a piece of the backbone through a cysteine knot as explained in the Discussion section where we also provide classification of all types of mechanical clamps found so far.
MATERIALS AND METHODS
The theoretical model used in the survey
An input to structure-based models of proteins comes from the PDB. Such models are defined by requirement that the ground state of the system coincides with the native structure. They usually come with an implicit solvent and are coarse-grained. The simplest choice is to represent residues by Cα atoms. There is no unique prescription for a good structure-based model. A total of 62 possible variants have been analyzed in (11). Their mechanical and folding properties differ substantially. Several of them are optimal and we use the simplest of them as described in references (10,12–14).
An attractive native contact between Cα atoms in residues i and j (at distance rij) is declared to arise if van der Waals spheres (enlarged to account for attraction) associated with the heavy atoms overlap (15). Non-native contacts are considered repulsive. In the first survey (10), all native contacts were included in the energy function. Here, we remove the i, i + 2 contacts as they are usually dispersive and weak (16).
The potential energy of the system of N amino acids is given by 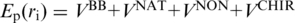 . The harmonic
. The harmonic 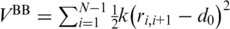 tethers consecutive beads at the equilibrium bond length and k = 100 ε/Å−2, where ε is defined below. A similar potential is used to describe the disulphide bonds. The native contacts are described by the Lennard-Jones potentials with the uniform energy parameter ε:
tethers consecutive beads at the equilibrium bond length and k = 100 ε/Å−2, where ε is defined below. A similar potential is used to describe the disulphide bonds. The native contacts are described by the Lennard-Jones potentials with the uniform energy parameter ε:
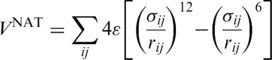 |
(1) |
The length parameters, σij, are selected so that the minima of the potentials agree with the native distances between the Cα atoms in the contact. VNON, the potential in the non-native contacts, is similar to VNON but it has purely repulsive terms.
The model also contains a four-body chirality term that favors the native sense of chirality (17), 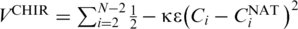 where
where 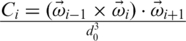 and CiNAT is the chirality of residue i in the native conformation and κ = 1. Here,
and CiNAT is the chirality of residue i in the native conformation and κ = 1. Here, 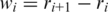 . A positive Ci corresponds to right-handed chirality. VCHIR favors native values of the dihedral angles (11). The model considered here has similar properties (18) to the model with the 10–12 contact potentials of Clementi et al. (19).
. A positive Ci corresponds to right-handed chirality. VCHIR favors native values of the dihedral angles (11). The model considered here has similar properties (18) to the model with the 10–12 contact potentials of Clementi et al. (19).
In our stretching simulations, both termini are attached to harmonic springs of elastic constant k = 0.12ε/Å. The choice of k affects mostly the location of the force peaks. One end of the spring is pulled at a constant speed, vp, along the initial end-to-end position vector. We consider vp = 0.005 Å/τ, where τ is of order 1 ns since this is a characteristic time to cover a typical distance of 5 Å through diffusion in the implicit solvent. The experimental results are obtained at various speeds that are all at least two orders of magnitude slower (<104 nm/s). Using one speed for all proteins facilitates making comparisons. We monitor F as a function of d and record Fmax.
The value of ε should be within 800–2300 K since it averages over of all non-covalent interactions in proteins. Our previous simulations of folding (13,20) were optimal with the dimensionless temperature 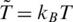 of order 0.3 which corresponds to room temperature if ε is ∼900 K (kB is the Boltzmann constant and T is temperature). Our simulations are performed at
of order 0.3 which corresponds to room temperature if ε is ∼900 K (kB is the Boltzmann constant and T is temperature). Our simulations are performed at 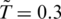 . Our latest estimate of ε is about 110 pNÅ or 1.5 kcal/mole (3).
. Our latest estimate of ε is about 110 pNÅ or 1.5 kcal/mole (3).
Thermal fluctuations are introduced by random Gaussian forces together with a velocity dependent damping. This noise mimics the random effects of the solvent and provides thermostating. The equation of motion for each Cα reads 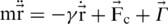 where FC is the net force due to the molecular potentials. The damping constant Γ is taken to be equal to 2m/τ and the dispersion of the random forces is equal to
where FC is the net force due to the molecular potentials. The damping constant Γ is taken to be equal to 2m/τ and the dispersion of the random forces is equal to 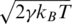 . This choice of Γ corresponds to a situation in which the inertial effects are negligible (13) but the damping action is not yet as strong as in water. Larger values of γ have only a minor effect on the F–d curves. The equations of motion are solved by a fifth order Gear predictor-corrector scheme (21). Due to the overdamping, our results are equivalent to those obtained by the Brownian dynamics algorithm (22). The F–d curves are averaged over a pulling distance of 0.5 Å.
. This choice of Γ corresponds to a situation in which the inertial effects are negligible (13) but the damping action is not yet as strong as in water. Larger values of γ have only a minor effect on the F–d curves. The equations of motion are solved by a fifth order Gear predictor-corrector scheme (21). Due to the overdamping, our results are equivalent to those obtained by the Brownian dynamics algorithm (22). The F–d curves are averaged over a pulling distance of 0.5 Å.
Proteins with non-trivial topology
The BSDB includes proteins with knots which were recognized by the KMT algorithm (23) as implemented in (24,25). These proteins were stretched to a maximal distance defined as the end-to-end distance along the backbone from which the sequential size of the maximally tightened knot (of order 12 residues for the trefoil knot) is subtracted. Currently, the BSDB does not include other types of knots since the corresponding proteins contain more than the cutoff value of 250 residues. BSDB includes one slip knotted protein which was recognized by the technique described in (26).
The force peak recognition
An automated detection of Fmax poses difficulties due to existence of fluctuations and a need to discard the final indefinite growth in F when the resistance to pulling is provided only by the unbreakable couplings. There is no absolute prescription as to when it starts. The strongest proteins often give rise to isolated force peaks on such ‘final’ slopes. In our procedure, a trajectory is split into 100 parts in which we determine the maximum 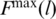 and time averaged
and time averaged 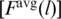 values of F. The maximum arises in the l’th partition provided
values of F. The maximum arises in the l’th partition provided 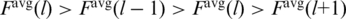 . The value of Fmax is then read off as
. The value of Fmax is then read off as 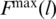 in this partition. Mistaken calls are due to large fluctuations in the final ascent and they occur with a probability of ∼1%. Thus peaks signaled as arising toward the end of the process are examined separately. If no isolated force peak is detected, Fmax is declared to be 0.
in this partition. Mistaken calls are due to large fluctuations in the final ascent and they occur with a probability of ∼1%. Thus peaks signaled as arising toward the end of the process are examined separately. If no isolated force peak is detected, Fmax is declared to be 0.
For each molecule, at least two trajectories have been studied to get a measure of errors. If the two trajectories are significantly distinct, the statistics is raised to 10 trajectories to identify various pathways (e.g. for 1qu0). The Fmax specified in the data base is then given by the weighted average over the pathways. The experimentally studied 1aoh has small pieces of its contact map incomplete (due to missing locations of some atoms in the side chains). This contact map was completed (7) by calculating the Cα – Cα distances in the affected regions and accepting distances smaller that 7.5 Å as corresponding to the missing native contacts.
RESULTS
Stretching protocol
The stretching protocol used in the survey involves anchoring one terminus to an immobile substrate and pulling another terminus by an elastic spring which moves at a constant speed. There is a huge number of other possibilities to choose pairs of amino acids as attachment points. Some of the available experimental data correspond to such non-terminal manipulation. This fact has been taken into account when comparing experimental to theoretical values of Fmax to relate the effective unit of the calculated force to measurements. This unit is equal to ε/Å, where the energy parameter ε denotes the depth of the attractive potential well in native contacts between the Cα atoms. We estimate ε/Å to be equal to 110 ± 30 pN (3). The estimation procedure involves making extrapolations to the experimental pulling speeds used. The existing studies (9,27–32) indicate that it is only for protein GFP (33) that non-terminal pulling may result in a larger Fmax than the terminal pulling. It is thus expected that the values of Fmax provided by the BSDB are usually the upper bounds for the various choices of the manipulation. The data for non-terminal pulling can be calculated upon request.
The BSDB also provides other information about stretching. In particular, it displays two calculated force, F, versus displacement, d, curves. If the two curves display noticeable differences then at least two distinct unfolding pathways are possible. The corresponding values of Fmax usually do not differ much. Larger statistics of trajectories were obtained only for a small number of proteins. The F – d curves can be used, for example, to identify mechanically stable intermediates along the unfolding pathway. These intermediates correspond to the force peaks and the associated conformations can be determined upon request. It has been shown (34) that such intermediates in FN-III are responsible for exposure of cryptic binding sites that allow for fibrillogenesis.
The BSDB also presents findings obtained through literature search. Specifically, it lists available experimental data on Fmax, together with the pulling speed used, and theoretical results that have been obtained by other methods, especially by all-atom simulations. It should be emphasized, however, that the literature data refer only to several tens of proteins.
Selection of structures from the PDB
PDB entries include proteins, nucleic acids, carbohydrates, and their complexes. We have downloaded all 54 807 structures that were available on 17 December 2008 by establishing a local mirror of the ftp://ftp.rcsb.org/pdb/ site and utilizing GNU wget program with an option preserving the database tree structure. We have restricted the survey to proteins of not more than 250 amino acids. We have used a script that eliminates entries containing words like DNA DUPLEX, RIBOSYME, OLIGONUCLEOTIDE, etc. in their HEADER and TITLE sections. Other keywords are detailed in the Database description presented online. However, special attention has been paid to files containing keywords like DNA as they may still refer to proteins, like the DNA ribonuclease.
Despite the thorough validation of structures in the PDB, not all structure files are readily available for molecular dynamics simulations, as they may have either missing residues or insufficiently resolved segments. Generally, such gap containing structures have been eliminated from our studies. We have also rejected structures in which some distances between consecutive Cα atoms are outside of the (3.6–3.95) Å range. This rule does not apply if one of the amino acids is proline as the corresponding distances fall then in the range (2.8–3.85) Å. In case of proteins that occur in multimeric or multi-protein complexes, we examine only the first chain and neglect any disulphide bridges to other units. We do not eliminate structures with artificial or modified amino acids. If there are multiple structures assigned to a protein (especially for structures determined through NMR), we take the first one. If there are alternative local placements, we take the first one if the distances between the Cα atoms are proper, otherwise we try the alternative. However, we reject files with ambiguous definitions of alternative local placements.
The disulfide bonds in our model cannot break and thus behave differently than contacts due to hydrogen bonds or ionic bridges. We detect such bonds using the information contained in the ‘SSBOND’ section of the PDB file.
Databases used in BSDB
The BSDB relates to four external databases: PDB (4), CATH (35), SCOP (36) and Gene Ontology database (37). The first of these is the source of structures for which the calculations were made. The next two assign a symbol of structure classification to a protein. The assignment based on CATH is algorithmic whereas the one on SCOP relies on human judgment. The fourth database provides description of biological function. The very strongest protein is currently predicted to be the extracellular bone morphogenic protein 1 bmp for which Fmax could be around 1 nN. Its mechanical clamp is that of the cysteine slipknot. It should be noted that there is a website, www.p-found.org, which allows for making and depositing all-atom simulation of protein stretching (38).
The Structural Classification of Proteins (SCOP) (36), accessible at http://scop.mrc-lmb.cam.ac.uk/scop, is a hierarchical scheme that classifies proteins according to features that relate to both structural and sequential features. The scheme is accessible either by name of a hierarchical level (Domain, Family, etc.), or it's ascension number (39) in the form of [a–k].xx.yy.zz where a–k stands for class of proteins, and xx, yy, zz for lower levels of hierarchy. The numbers are not immutable and change between releases of the SCOP, thus they are presented along with the corresponding names.
The Class, Architecture, Topology, Homology (CATH) (35,14) scheme is also hierarchical, but it considers four classes (α, β, α–β and no-structure). Each entry in our database is presented with the corresponding ascension number and name.
One should note, that a given PDB code may come with none, one or more SCOP or CATH entries, since a protein may be unclassified or contain two or more domains. When studying correlations between the dynamics and structure, we use classified structures and take the first assignment. In the 1.73 version of SCOP (11), there are 92 972 entries that relate to 34 495 pdb structures. These entries are divided into 3464 families, 1777 superfamilies and 1086 unique folds. The 3.2 version of CATH contains 114 215 domains, 2178 homologous superfamilies and 1110-fold groups (12).
Information provided
The structure-based data are presented in four lists: one ranked by the value of Fmax, another by the number of amino acids in the structure, the remaining two by the structure classification codes that come with the CATH and SCOP schemes. One can also access the results by providing the PDB code of a protein. An example of data available for a given protein is shown in Figure 1 for the scaffoldin 1aoh. They start with the chain used, value of Fmax in ε/Å together with the corresponding standard deviation, ΔFmax, based on several trajectories, and the value of Fmax converted to pN. They also specify value of the tip displacement, Dmax, at which the maximal peak force arises for vp = 0.005 Å/τ, where τ is of order 1 ns, and the corresponding end-to-end distance, Lmax. The force peak position is also described in terms of the dimensionless parameter λ defined as (Lmax − Ln)/(Lf − Ln), where Ln is the native end-to-end distance and Lf is the full backbone length. Small values of λ indicate that the force peak arises at the beginning of the pulling process. Lf is close to the end-to-end distance at full extension only provided there are no disulphide bridges that require significantly larger forces to get ruptured. The number of the disulphide bridges is given by the parameter nSS. In this example, there are no such bridges.
Figure 1.

Example of a screenshot from the BSDB for protein 1aoh.
The entry also specifies the related structure codes and corresponding names, if available, and all GO numbers that specify molecular function, biological process, and cellular component. In addition, it shows examples of two stretching trajectories in the F–d plots. Such plots indicate whether the force peaks are multiple or not. The arrow locates the displacement at which the biggest force maximum arises.
A simultaneous display of data for several proteins is implemented through the ‘add this protein to compare’ hyperlink and then using ‘compare’. It allows for an analysis of data with a common characteristic such as the system size, the value of Fmax or function. The F–d curves for the selected proteins are displayed in a column, where they can be inspected visually. Results can be sorted according to various properties, for example by the CATH or SCOP number. In this way, the user can inspect changes in extension curves within the same family, or between proteins with relatively low homology, allowing one to correlate the peak pattern with a degree of relationship.
An important feature of the BSDB is that it provides information about the microscopic nature of the unravelling process. It is contained in the so called scenario diagrams (14) which specify the pulling distances at which particular native contacts are seen as operational for the last time (i.e. distances between amino acids i and j do not exceed a threshold value). For each protein, the BSDB gives a listing of the breaking distances, du, for each native contact. A scenario diagram can be obtained if |j − i| is plotted against du as in (14). We provide examples of such diagrams for 1aoh (one of the strongest proteins) and 1j85 (a protein with a knot).
We provide hyperlinks to several other databases, such as CSU (16), that facilitate understanding of contacts and of identification of secondary structures to interpret the scenario diagrams.
DISCUSSION
The information contained in the scenario diagrams can be used to determine the nature of the relevant mechanical clamp. Accumulation of rupture events around a specific value of du signifies occurrence of a force peak. Some of the rupture events belonging to a peak are crucial dynamically—removal of the corresponding contacts reduces Fmax significantly. Such contacts identify the mechanical clamp. The remaining rupture events are just concurrent. It is hard to identify the mechanical clamps in an automated way since one needs to determine structurally relevant groups of contacts and then redo the simulation with various groups removed. We have accomplished this task for selected proteins, including the 64 strongest (results are listed in BSDB). Taking it together with discussions in the literature we can identify typical motifs which are associated with mechanical clamps. We divide these motifs into two groups: (i) involving strain in localized regions and (ii) involving a larger motion of a loop or two loops that are made of segments of the backbone.
Figure 2 shows examples of mechanical clamps belonging to the first group. The β-strands are indicated as black arrows in the figure. The simplest motifs are denoted by S, SA and Z. The first of these corresponds to shearing between parallel β-strands, similar to what takes place in a stretched titin (8,9). The longer the strand, the bigger the number of bonds that are sheared simultaneously and then the bigger the value of Fmax. It should be noted that Fmax is also affected by the direction of the stretching force relative to the orientation of the clamp. The second of these corresponds to shearing between antiparallel strands (10). For a given length of the strand, SA yields a smaller Fmax than S. The third motif is a zipper (40) which is the most fragile of them all since the contacts get ruptured one at a time. The elementary motifs S and Z can also arise with helices (in which case the arrows in Figure 2 would represent helices) but the corresponding values of Fmax then are expected to be smaller than for the β strands of a similar length. Another possibility is U—an unstructured clamp seen in 1qp1 and 1tum (10). It is similar to S except that two nearby β-strands are replaced by unstructured segments of the backbone (non-typical strands).
Figure 2.

Mechanical clamps involving shear or zipping in localized regions. Except for the zipper motif, all other motifs involve shear between secondary structures. The black arrows indicate β-strands. More generically, however, they may also depict α-helices. The lighter arrows indicate the sense of motion that is induced by pulling. The ‘delocalized’ clamp consists of multiple local shearing regions. Unlike the motifs shown in Figure 3, its operation does not rely on motion of any backbone loops. This is the reason why motif D is lamped together with the clamps displayed in the current Figure 3.
The elementary motifs S and SA can combine to form shear composite motifs shown in the middle line of the panels in Figure 2. Examples of these include disconnected shear motifs SD1 and SD2 and a supported shear motif SS. In SD1, discussed in (10) and observed in 1aoh (7), one S motif is followed by another S motif, separated by an essentially shear free region. SD2 is similar but it involves two SA motifs in a row. The panel illustrating SD2 is drawn in a particular fashion that indicates the geometry of fibronectin (8) in a schematic way. For this protein, the constituting SA motifs correspond to the β-hairpin. The SD2 motif is also found in proteins L and G (41). One can also consider a variant, SD3, in which S is combined with SA. It arises in GPF when pulled by residues 3 and 212 (33).
In SS characterizing 1cp4 (10), the main S motif is flanked by neighboring β-strands which stabilize it. Shearing the main S results also in a shear with the flanking β-strands. We have found a few examples of still other possibilities in group I. One of them is SB—a shear box in protein 1pav as discussed in (10). It involves a sheared two-strand β-sheet that is placed below a sheared two-helix ‘sheet’ so that a shear in one strand induces a shear on the helix above (not shown). Still another possibility is D—a delocalized clamp with multiple elementary clamps exerting comparable resistances to pulling. This happens, e.g. in 1lsl. Its schematic representation shown at the bottom of Figure 2 indicates that some of the contributing strands may be unstructured.
Figure 2 shows one more shearing motif denoted by T for torsion. It has been argued to be operational in polyankyrin (42). It combines shear in multiple S-like motifs with undoing of the overall horseshoe shape of the protein. This shape seems to result from a combination of steric stiffness and some contacts in the loop-like linkers. The reported experimental value of Fmax for 12 domains is larger than that of 1aoh (42), however for six domains is much smaller (∼50 ± 20 pN) (43). Our simulations for 12 and 3 domains have yielded Fmax of only of order 140 and 60 pN respectively (J.I. Sułkowska, unpublished data). A small force for a single domain has been also derived through all-atom simulations (44) [see also ref. (45)].
Figure 3 illustrates another category of mechanical clamps belonging to the second group in which topological loops matter. In four of these, CK, CL1, CL2 and CSK, the loops arise due to the presence of between one and three relevant disulphide bonds (indicated by short solid lines) between pairs of cysteines. The cysteine knot, CK, corresponds to shearing taking place inside a cysteine knot (3)—a loop that is created by two disulphide bonds.
Figure 3.

Types of the force clamps that involve backbone loops.
CL1 and CL2 are cysteine loops. In CL1 shearing results because one branch of § is pulled by a cysteine loop (3). CL2 is similar but the motion of the cysteine loop also drags another piece of the backbone that transmits shear to the other branch of the S motif (3). CSK is the cysteine slipknot motif (3), it involves three disulfide bridges which generate two loops: knot-loop (which is an example of a cysteine knot) and a slip-loop. The mechanical resistance to pulling comes from pulling the slip-loop through the knot-loop. The fifth motif shown in Figure 3 is SK—the slipknot motif (26) that is observed, for instance, in protein 2j85. It is created by two interacting loops, the slip-loop and the knot-loop, that move simultaneously on pulling. If the knot loops shrinks faster than the knot loop then the slipknot gets tightened temporarily and a ‘catch bond’ (46) is formed. This intermediate and metastable configuration eventually gets untied upon further stretching. The existence of such a metastable conformation where the slipknot is jammed (26) is responsible for lengthening of unravelling times at a higher velocity or force.
When we rank order the proteins according to the value of Fmax, as calculated in our model, and ask in what order particular mechanical clamps are seen for the first time on reducing Fmax, then their strengths can be rank ordered in the following succession: CSK, SS, SK, S, SD1, CL1, CL2, SD2 and Z. However, the strengths of various types of clamps intermix when going down the ranking ladder because they are influenced by dynamical details such as the length of a strand. In the case of CSK and SK, Fmax depends on the relationship between sizes of the cysteine- and knot-loops in a strong way.
We plan to expand and improve the database. In particular, we intend to implement a possibility of self-submitted calculations for desired pulling directions and speeds. Currently, the requests for data should be sent to bsdb@ifpan.edu.pl. In particular a structure file which is not in the PDB can also be submitted. The resulting data are added to the database (which may affect ranking of the proteins).
FUNDING
Funding for open access charge: Grant N N202 0852 33 from the Ministry of Science and Higher Education in Poland; EC FUNMOL project under FP7-NMP-2007-SMALL-1; European Union within European Regional Development Fund, through grant Innovative Economy (POIG.01.01.02-00-008/08); Center for Theoretical Biological Physics sponsored by the NSF (Grant PHY-0822283); NSF-MCB-0543906 (to J.I.S.).
Conflict of interest statement. None declared.
REFERENCES
- 1.Carrion-Vazquez M, Cieplak M, Oberhauser AF. Protein mechanics at the single-molecule level. In: Meyers RA, editor. Encyclopedia of Complexity and Systems Science. NY: Springer; 2009. pp. 7026–7050. [Google Scholar]
- 2.Galera-Prat A, Gomez-Sicilia A, Oberhauser AF, Cieplak M, Carrion-Vazquez M. Understanding biology by stretching proteins: recent progress. Curr. Opin. Struct. Biol. 2010;20:63–69. doi: 10.1016/j.sbi.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sikora M, Sułkowska JI, Cieplak M. Mechanical strength of 17 134 model proteins and cysteine slipknots. PLoS Comp. Biol. 2009;5:e1000547. doi: 10.1371/journal.pcbi.1000547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berman H, Henrick K, Nakamura H, Markley JL. The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007;35:D301–D303. doi: 10.1093/nar/gkl971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yeates TO, Norcross TS, King NP. Knotted and topologically complex proteins as models for studying folding and stability. Curr. Opin. Chem. Biol. 2007;11:596. doi: 10.1016/j.cbpa.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bolinger D, Sułkowska JI, Hsu H-P, Mirny LA, Kardar M, Onuchic JN, Virnau PA. Stevedore's protein knot. PLoS Comp. Biol. 2010;6:e1000731. doi: 10.1371/journal.pcbi.1000731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Valbuena A, Oroz J, Hervás R, Vera AM, Rodríguez D, Menéndez M, Sulkowska JI, Cieplak M, Carrión-Vázquez M. On the remarkable mechanostability of scaffoldins and the mechanical clamp motif. Proc. Natl Acad. Sci. USA. 2009;106:13791–13796. doi: 10.1073/pnas.0813093106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu H, Schulten K. Steered molecular dynamics simulation of conformational changes of immunoglobulin domain I27 interpret atomic force microscopy observations. J. Chem. Phys. 1999;247:141–153. [Google Scholar]
- 9.Brockwell DJ, Paci E, Zinober RC, Beddard G, Olmsted PD, Smith DA, Perham RN, Radford SE. Pulling geometry defines mechanical resistance of β-sheet protein. Nat. Struct. Biol. 2003;10:731–737. doi: 10.1038/nsb968. [DOI] [PubMed] [Google Scholar]
- 10.Sułkowska JI, Cieplak M. Mechanical stretching of proteins—a theoretical survey of the Protein Data Bank. J. Phys. Cond. Mat. 2007;19:283201. [Google Scholar]
- 11.Sułkowska JI, Cieplak M. Selection of optimal variants of Go-like models of proteins through studies of stretching. Biophys. J. 2008;95:3174–3191. doi: 10.1529/biophysj.107.127233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoang TX, Cieplak M. Molecular dynamics of folding of secondary structures in Go-like models of proteins. J. Chem. Phys. 2000;112:6851–6862. [Google Scholar]
- 13.Cieplak M, Hoang TX. Universality classes in folding times of proteins. Biophys. J. 2003;84:475–488. doi: 10.1016/S0006-3495(03)74867-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cieplak M, Hoang TX, Robbins MO. Thermal effects in stretching of Go-like models of titin and secondary structures. Proteins Struct. Funct. Bio. 2004;56:285–297. doi: 10.1002/prot.20081. [DOI] [PubMed] [Google Scholar]
- 15.Tsai J, Taylor R, Chothia C, Gerstein M. The packing density in proteins: standard radii and volumes. J. Mol. Biol. 1999;290:253–266. doi: 10.1006/jmbi.1999.2829. [DOI] [PubMed] [Google Scholar]
- 16.Sobolev V, Sorokine A, Prilusky J, Abola EE, Edelman M. Automated analysis of interatomic contacts in proteins. Bioinformatics. 1999;15:327–332. doi: 10.1093/bioinformatics/15.4.327. [DOI] [PubMed] [Google Scholar]
- 17.Kwiecinska JI, Cieplak M. Chirality and protein folding. J. Phys. Cond. Mat. 2005;17:S1565–S1580. [Google Scholar]
- 18.Cieplak M, Hoang TX. Folding of proteins in Go modles with angular interactions. Physica A. 2003;330:195–205. [Google Scholar]
- 19.Clementi C, Nymeyer H, Onuchic JN. Topological and energetic factors: what determines the structural details of the transition state ensemble and “on-route” intermediates for protein folding? An investigation for small globular proteins. J. Mol. Biol. 2000;298:937–953. doi: 10.1006/jmbi.2000.3693. [DOI] [PubMed] [Google Scholar]
- 20.Cieplak M, Hoang TX. Kinetic non-optimality and vibrational stability of proteins. Proteins Struct. Funct. Genet. 2001;44:20–25. doi: 10.1002/prot.1067. [DOI] [PubMed] [Google Scholar]
- 21.Gear WC. Numerical Initial Value Problems in Ordinary Differential equations. Upper Saddle River, NJ, USA: Prentice-Hall, Inc.; 1978. [Google Scholar]
- 22.Ermak DL, McCammon JA. Brownian dynamics with hydrodynamic interactions. J. Chem. Phys. 1978;69:1352–1360. [Google Scholar]
- 23.Koniaris K, Muthukumar M. Knottedness in ring polymers. Phys. Rev. Lett. 1991;66:2211–2214. doi: 10.1103/PhysRevLett.66.2211. [DOI] [PubMed] [Google Scholar]
- 24.Sułkowska JI, Sułkowski P, Szymczak P, Cieplak M. Tightening of knots in the proteins. Phys. Rev. Lett. 2008;100:058106. doi: 10.1103/PhysRevLett.100.058106. [DOI] [PubMed] [Google Scholar]
- 25.Sułkowska JI, Sułkowski P, Szymczak P, Cieplak M. Stabilizing effect of knots on proteins—how knots influence properties of proteins. Proc. Natl Acad. Sci. USA. 2008;105:19714–19719. doi: 10.1073/pnas.0805468105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sułkowska JI, Sułkowski P, Onuchic JN. Jamming proteins with slipknots and their free energy landscapes. Phys. Rev. Let. 2009;103:268103. doi: 10.1103/PhysRevLett.103.268103. [DOI] [PubMed] [Google Scholar]
- 27.Carrion-Vazquez M, Li H, Lu H, Marszalek PE, Oberhauser AF, Fernandez JM. The mechanical stability of ubiquitin is linkage dependent. Nat. Struct. Biol. 2003;10:738. doi: 10.1038/nsb965. [DOI] [PubMed] [Google Scholar]
- 28.Cieplak M, Marszalek PE. Mechanical unfolding of ubiquitin molecules. J. Chem. Phys. 2005;123:194903. doi: 10.1063/1.2046609. [DOI] [PubMed] [Google Scholar]
- 29.Nome R, Zhao J, Hoff W, Scherer N. Axis-dependent anisotropy in protein unfolding from integrated nonequilibrium response spectroscopy. Proc. Natl Acad. Sci. USA. 2007;104:20799–20804. doi: 10.1073/pnas.0701281105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Best R, Hummer G. Protein folding kinetics under force from molecular simulation. J. Am. Chem. Soc. 2008;130:3706. doi: 10.1021/ja0762691. [DOI] [PubMed] [Google Scholar]
- 31.Kumar S, Giri D. Does changing the pulling direction give better insight of biomolecules. Phys. Rev. Lett. 2007;98:048101. doi: 10.1103/PhysRevLett.98.048101. [DOI] [PubMed] [Google Scholar]
- 32.Matouschek A, Bustamante C. Finding a protein's Achilles heel. Nat. Struct. Biol. 2003;10:674–676. doi: 10.1038/nsb0903-674. [DOI] [PubMed] [Google Scholar]
- 33.Dietz H, Berkemeier F, Bertz M, Rief M. Anisotropic deformation response of single protein molecules. Proc. Natl Acad. Sci. USA. 2006;103:12724–12728. doi: 10.1073/pnas.0602995103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao M, Craig D, Lequin O, Campbell ID, Vogel V, Schulten K. Structure and functional significance of mechanically unfolded fibronectin type III1 intermediates. Proc. Natl Acad. Sci. USA. 2003;100:14784–14789. doi: 10.1073/pnas.2334390100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cuff AL, Sillitoe I, Lewis T, Redfern OC, Garratt R, Thornton J, Orengo CA. The CATH classification revisited–architectures reviewed and new ways to characterize structural divergence in superfamilies. Nucleic Acids Res. 2009;37:D310–D314. doi: 10.1093/nar/gkn877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andreeva A, Howorth D, Chandonia JM, Brenner SE, Hubbard TJ, Chothia C, Murzin AG. Data growth and its impact on the SCOP database: new developments. Nucleic Acids Res. 2008;36:D419–D425. doi: 10.1093/nar/gkm993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.The Gene Ontology Consortium. Gene ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brito RMM, Dubitzky W, Rodrigues JR. Protein folding and unfolding simulations: a new challenge for data mining. OMICS J. Integr. Biol. 2004;8:153–166. doi: 10.1089/1536231041388311. [DOI] [PubMed] [Google Scholar]
- 39.Lo Conte L, Brenner SE, Hubbard TJP, Chothia C, Murzin AG. SCOP database in 2002: refinements accommodate structural genomics. Nucleic Acids Res. 2002;30:264–267. doi: 10.1093/nar/30.1.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carrion-Vazquez M, Oberhauser AF, Fisher TE, Marszalek PE, Li H, Fernandez JM. Mechanical design of proteins studied by single-molecule force spectroscopy and protein engineering. Prog. Biophys. Mol. Biol. 2000;74:63–91. doi: 10.1016/s0079-6107(00)00017-1. [DOI] [PubMed] [Google Scholar]
- 41.Brockwell DJ, Beddard GS, Paci E, West DK, Olmsted PD, Smith DA, Radford SE. Mechanically unfolding the small topologically simple protein L. Biophys. J. 2005;89:506–519. doi: 10.1529/biophysj.105.061465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee G, Abdi K, Jiang Y, Michaely P, Bennett V, Marszalek PE. Nanospring behavior of ankyrin repeats. Nature. 2006;440:246–249. doi: 10.1038/nature04437. [DOI] [PubMed] [Google Scholar]
- 43.Li L, Wetzel S, Pluckthun A, Fernandez JM. Stepwise unfolding of ankyrin repeats in a single protein revealed by atomic force microscopy. Biophys. J. 2006;90:L30–L32. doi: 10.1529/biophysj.105.078436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sotomayor M, Corey DP, Schulten K. In search of the hair-cell gating spring elastic properties of ankyrin and cadherin repeats. Structure. 2005;13:669–682. doi: 10.1016/j.str.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 45.Makarov DE. A theoretical model for the mechanical unfolding of repeat proteins. Biophys. J. 2009;96:2160–2167. doi: 10.1016/j.bpj.2008.12.3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dembo M, Torney DC, Saxman K, Hammer D. The reaction-limited kinetics of membrane-to-surface adhesion and detachment. Proc. R. Soc. Lond. B. Biol. Sci. 1988:55–83. doi: 10.1098/rspb.1988.0038. [DOI] [PubMed] [Google Scholar]