


Abstract
Protein phosphorylation catalyzed by kinases plays crucial regulatory roles in intracellular signal transduction. With the increasing number of experimental phosphorylation sites that has been identified by mass spectrometry-based proteomics, the desire to explore the networks of protein kinases and substrates is motivated. Manning et al. have identified 518 human kinase genes, which provide a starting point for comprehensive analysis of protein phosphorylation networks. In this study, a knowledgebase is developed to integrate experimentally verified protein phosphorylation data and protein–protein interaction data for constructing the protein kinase–substrate phosphorylation networks in human. A total of 21 110 experimental verified phosphorylation sites within 5092 human proteins are collected. However, only 4138 phosphorylation sites (∼20%) have the annotation of catalytic kinases from public domain. In order to fully investigate how protein kinases regulate the intracellular processes, a published kinase-specific phosphorylation site prediction tool, named KinasePhos is incorporated for assigning the potential kinase. The web-based system, RegPhos, can let users input a group of human proteins; consequently, the phosphorylation network associated with the protein subcellular localization can be explored. Additionally, time-coursed microarray expression data is subsequently used to represent the degree of similarity in the expression profiles of network members. A case study demonstrates that the proposed scheme not only identify the correct network of insulin signaling but also detect a novel signaling pathway that may cross-talk with insulin signaling network. This effective system is now freely available at http://RegPhos.mbc.nctu.edu.tw.
INTRODUCTION
Protein phosphorylation is the most widespread and well-studied post-translational modification in eukaryotic cells. It has been estimated that one-third to one-half of all proteins in a eukaryotic cell are phosphorylated (1). Phosphorylation can regulate almost every property of a protein and is involved in all fundamental cellular processes. In addition, protein phosphorylation catalyzed by kinase plays crucial regulatory roles in intracellular signal transduction. The networks of proteins and small molecules that transmit information from the cell surface to the nucleus, where they ultimately effect transcriptional changes (2). Thus, a full understanding of the mechanism of intracellular signal transduction remains a major challenge in cellular biology. Mass spectrometry (MS)-based proteomics have enabled the large-scale mapping of in vivo phosphorylation sites (3). There are several databases storing experimentally verified phosphorylation sites with catalytic kinases, such as Phospho.ELM (4), PhosphoSite (5), UniProtKB/Swiss-Prot (6), Phosphorylation Site Database (7) and PHOSIDA (8). PhosPhAt (9) is a database of phosphorylation sites in Arabidopsis thaliana. PhosphoPOINT (10) provides robust annotation for kinases, their down-stream substrates and their interaction (phospho)-proteins and this should accelerate the functional characterization of kinome-mediated signaling.
Manning et al. (11) have identified 518 human kinase genes, the so-called ‘kinome’, that provides a starting point for comprehensive analysis of protein phosphorylation networks. To explore the protein kinase–substrate phosphorylation networks, the experimentally verified kinase-specific phosphorylation sites can be collected from the public resources. However, only 20% of the experimentally verified phosphorylation sites have the annotation of catalytic kinases. Recently, with exponential increase in protein phosphorylation sites identified by MS, many researches are undertaken to identify the kinase-specific phosphorylation sites, including NetPhosK (12), Scansite 2.0 (13), GPS (14,15), PPSP (16) and KinasePhos (17–19). The summary of the previously developed phosphorylation site prediction methods is listed in Supplementary Table S1. Particularly, Linding et al. (20) have proposed an excellent method, namely NetworKIN, that augments motif-based predictions with the network context of kinases and phosphoproteins.
Although the proposed resources can be utilized to construct the phosphorylation network between kinase and substrate proteins, the experimental data need to be combined by systems biology analysis, which translates the separate, large-scale datasets into signaling networks (21). Many studies have been proposed to model signaling networks using various approaches (22–26). Additionally, Steffen et al. (2) have developed a computational approach for generating static models of signal transduction networks. It utilizes protein-interaction maps generated from large-scale two-hybrid screens and DNA microarrays expression profiles. However, it is still insufficient to discover signaling networks in a gene group that have similar microarray expression profiles. To fully investigate how protein kinases regulate the intracellular processes, it is necessary to accurately identify the catalytic kinases for phosphoproteins. In this study, a knowledgebase named RegPhos is developed to integrate experimentally verified protein phosphorylation data and protein–protein interaction data for constructing the protein kinase–substrate phosphorylation networks in human. A graph searching algorithm, Breadth-first search (BFS) (27), is applied to explore the intracellular phosphorylation network starting from receptor kinases to transcription factors, associated with the information of protein subcellular localization. Supplementary Figure S1 demonstrates the concept of RegPhos. This effective system can let users input a group of human proteins; consequently, the phosphorylation network associated with the protein subcellular localization can be explored.
For the phosphoproteins without the annotation of catalytic kinases, KinasePhos (17–19) is incorporated with protein association for assigning the potential kinase. A case study is demonstrated that RegPhos not only identify the correct network of insulin signaling but also detect a novel signaling pathway that may cross-talk with insulin signaling network. Additionally, time-coursed microarray expression data is subsequently used to represent the degree of similarity in the expression profiles of network members.
MATERIALS AND METHODS
The system flow of RegPhos is shown in Figure 1, mainly including the collection of experimentally verified phosphorylation sites, identification of experimentally confirmed kinase–substrate interactions and construction of intracellular phosphorylation networks. To fully investigate how protein kinases regulate the intracellular processes, a published method, KinasePhos (17–19), is combined with protein associations for identifying kinase-specific phosphorylation sites. Time-coursed microarray expression data is then used to validate the degree of similarity in the expression profiles of network members.
Figure 1.

System flow of RegPhos.
Collection of experimentally verified phosphorylation sites
The experimental verified phosphorylation sites are extracted from dbPTM (28) which has integrated version 8.0 of Phospho.ELM (4), release 55.0 of UniProtKB/Swiss-Prot (29) and version 1.0 of PHOSIDA (8). As shown in Table 1, Phospho.ELM, Swiss-Prot and PHOSIDA contains 21 542, 24 628 and 6600 experimental verified phosphorylation sites within 6520, 8606 and 2244 phosphoproteins, respectively. Additionally, Human Protein Reference Database (HPRD) (30), which integrates a wealth of information relevant to the function of human proteins in health and disease, is integrated in this work. In release 7.0 of HPRD, there are totally 16 972 PTMs within 2830 protein entries, of 7438 PTMs are phosphorylation sites within 1774 proteins. Furthermore, data pertaining to thousands of protein–protein interactions, posttranslational modifications, enzyme/substrate relationships, disease associations, tissue expression and subcellular localization were extracted from the literature for a non-redundant set of 25 661 human proteins. We are prompted to construct human phosphorylation network in this study, the collected phosphorylation sites in human proteins are separately represented in Table 1. After removing the redundant data among these databases, the number of human phosphorylation sites and phosphoproteins are 21 110 and 5092, respectively.
Table 1.
Statistics of the public phosphorylation databases integrated in RegPhos
| Database | Version | All species |
Human |
||
|---|---|---|---|---|---|
| Number of phosphoprotein | Number of phosphosite | Number of phosphoprotein | Number of phosphosite | ||
| Phospho.ELM | 8.0 | 6520 | 21 542 | 4067 | 13 833 |
| UniProtKB/Swiss-Prot | 55.0 | 8606 | 24 328 | 3746 | 11 862 |
| PHOSIDA | 1.0 | N/A | N/A | 2212 | 8969 |
| HPRD | 7.0 | – | – | 1774 | 7438 |
| Combined (NRa) | – | – | – | 5092 | 21 110 |
aNR, non-redundant.
Identification of experimentally confirmed kinase–substrate interactions
The human kinase annotations extracted from KinBase (11) are used to unify the kinase names among the external phosphorylation site databases which contain various names for a kinase. To unify the heterogeneous data of kinases and phosphoproteins, the kinase names in KinBase and phosphoproteins in public resources are both mapped to the UniProtKB/Swiss-Prot ID and accession number. Due to the classification of kinase identified by Manning et al. (11), 518 kinases are categorized by their annotated family or subfamily, including totally 221 kinase families. The 518 kinases are major nodes in the construction of human phosphorylation networks. Several representative kinase families are listed in Supplementary Table S2; for instance, the family of protein kinase B (PKB) consists of three kinase members such as AKT1, AKT2 and AKT3. With the integration of experimental phosphorylation sites from public resources, totally 89 phosphorylation sites of 63 human phosphoproteins are catalyzed by PKB kinase family. The knowledgebase contains 21 110 experimentally verified phosphorylation sites within 5092 human proteins, of 4138 phosphorylation sites (∼20%) have the annotation of catalytic kinases. According to the annotations of 4138 experimentally confirmed kinase-specific phosphorylation sites, a total of 1306 experimentally kinase–substrate interactions are identified.
Construction of intracellular phosphorylation networks
Manning et al. (11) have identified 518 human kinase genes, that provides a starting point for investigating protein phosphorylation networks. With the identification of experimentally confirmed kinase–substrate interactions, the intracellular phosphorylation networks can be reconstructed. A graph-based method is adopted to formalize the construction of intracellular phosphorylation network to a path search problem in graph theory. The intracellular protein phosphorylation networks are visualized as an directed graph G = (V, E), where x, y ∈ V and (x, y) ∈ E. Let x and y represent kinase and substrate proteins, respectively and (x, y) ∈ E represent a phosphorylation interaction when kinase x phosphorylates substrate y. However, the intracellular phosphorylation networks (signaling networks) contain not only the kinase cascades or kinase–substrate interactions, but also protein–protein interactions or protein complex, such as insulin signaling network (31). To make the construction of signaling networks feasible, the experimental protein–protein interactions or protein complexes in human are integrated from DIP (32,33), MINT (34), IntAct (35) and HPRD (30), as shown in Supplementary Table S3. In this work, V refers to all human proteins in UniProtKB (36) and E refers to all experimental interactions in knowledgebase including experimentally verified kinase–substrate interactions and experimental protein–protein interactions.
Moreover, the cellular localization of proteins is used to constrain the search of phosphorylation network. Supplementary Table S4 lists the public databases of protein subcellular localization, including LOCATE (37), DBSubLoc (38), Organelle DB (39) and PSORTdb (40). Due to the annotation of cellular localization databases, there are 84 cell membrane-associated kinases being the start points of the phosphorylation networks. With the annotation of TRANSFAC version 11.0 (41), there are 1364 transcription factors in human. To identify the phosphorylation networks starting from membrane receptor to transcription factor in nucleus, the graph-based definition can be refined as follows: given a directed weighted graph G = (V, E) with n nodes, m edges, a set S of start nodes (receptor) and a set T of end nodes (TF). As shown in Supplementary Figure S2, for each node s in S, a acyclic path p = (s, c1, … , ck, t) with length k that starts from S and ends at t within T, passed through cytoplasmic proteins c1, … , ck is found. A graph searching algorithm, BFS (27), is applied to explore the intracellular phosphorylation network associated with the information of protein subcellular localization. BFS is one of the basic schemes for searching a subgraph or a path in a graph. Given a graph G = (V, E) where V represents the set of proteins and E is the set of physical interactions between proteins and a distinguished source vertex s, BFS systematically explores the edges of G to discover every vertex that is reachable from s. We restrict attention to simple paths that was constrained the order of occurrence of proteins in a defined path length 8 (2).
Systematically exploring the intracellular phosphorylation networks, starting from membrane receptor to transcription factor in nucleus, may produce a lot of false positive networks. Clustering genes with similar profiles into a group is a proven method for grouping functionally related genes (21). Therefore, the identified signaling networks are further examined the degree of similarity in the expression profiles of network members. The time-coursed gene expression samples from Affymetrix GeneChip Human Genome U133 Array Set HG-U133A platform (GPL96) (42), which consists of 22 283 probe set for 12 678 genes, is used to explore the co-expression of kinase and substrate genes. Gene expression data, including Esophageal cell response to low pH (GSE2144), Lung cancer cell line response to motexafin gadolinium (GSE2189), Cyanobacterial metabolite apratoxin A cytotoxic effect on colon adenocarcinoma cells (GSE2742), Interleukin 13 effect on bronchial cell line (GSE3183), Endotoxin effect on leukocytes (GSE3284), Blood response to various beverages (GSE3846) Androgen receptor modulator effect (GSE4636), Glucocorticoid receptor activation effect on breast cancer cells (GSE4917) and Epidermal growth factor effect on cervical carcinoma cell line (GSE6783), were quantified by Robust Multichip Average (RMA) algorithm (43). RMA quantification was performed by the justRMA function of Bioconductor Affy package in R program language using raw data (Affymatrix CEL file). Then, Pearson correlation coefficient is used to measure the trends of two expression profiles.
Computational identification of kinase–substrate interactions
With the integration of public phosphorylation resources, most of the experimentally verified phosphorylation sites (∼80%) do not have the annotation of catalytic kinases. To fully investigate how protein kinases regulate the intracellular processes, it is necessary to accurately link the experimental phosphorylation sites to catalytic kinases. With reference to the approach of NetworKIN (20), a published kinase-specific phosphorylation site prediction tool, named KinasePhos (17–19), is incorporated with protein association for assigning the potential kinase. The association context for each kinase–substrate pair is investigated by the information of protein–protein interactions, functional associations (physical protein interactions, curated pathway, co-occurrence in literature abstracts, mRNA co-expression studies and genomic context) and cellular co-localization. A public SVM library, namely LibSVM (44), is adopted to train the kinase-specific predictive models, including more than 100 kinase families, with the encoded amino acid sequences and structural features, such as secondary structure (SS), accessible surface area (ASA) and disorder region (DIS). Radial basis function (RBF) 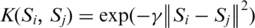 is selected as the kernel function of SVM. Each model is evaluated the discriminatory power between phosphorylated and non-phosphorylated sites, based on five-fold cross-validation.
is selected as the kernel function of SVM. Each model is evaluated the discriminatory power between phosphorylated and non-phosphorylated sites, based on five-fold cross-validation.
To investigate the possibility of using association context to enhance the identification of kinase-specific substrates, the constructed SVM models are combined with protein associations including protein–protein interactions, functional associations and subcellular localization. This work extract human protein–protein interactions from DIP (32,33), MINT (34), IntAct (35) and HPRD (30), as shown in Supplementary Table S3. Moreover, to capture the complete biological context of a substrate, the functional associations extracted from the STRING database (45) are integrated. In order to identify the direct and indirect connection between kinase and substrate, a graph searching algorithm, BFS, is also adopted.
The eukaryotic cell is a composite system internally subdivided into membrane-enveloped compartments that perform particular functions (46). The proteins, which are involved in similar biological functions, are closely located in the same subcellular localization. Therefore, knowing the localization of every protein is important for elucidating its interactions with other molecules and for understanding its biological function. In order to accurately identify the interaction of kinase–substrate phosphorylation, the information of subcellular localization is used to evaluate the co-localization between kinases and phosphoproteins. Supplementary Table S4 shows the list of integrated databases of protein subcellular localization, including LOCATE (37), DBSubLoc (38), Organelle DB (39) and PSORTdb (40).
Logistic regression has been adopted to evaluate the confidence value of protein–protein (kinase–substrate) interaction (25). In this study, a modified version of the Sharan et al. (47) method was utilized to evaluate the confidence values of the discovered kinase–substrate interactions (see Supplementary Figure S3). In the logistic regression model, we incorporate four sets of variables for a given interaction set, including (i) the prediction score of the kinase-specific SVM model, (ii) the depth of interaction between kinase and substrate was observed, (iii) the confidence score of the STRING functional association and (iv) the binary (0/1) protein subcellular localization data of interacting pairs. The computationally identified kinase–substrate interactions can be considered into the construction of intracellular phosphorylation networks, which may make the discovered network more feasible. Since exploring the protein phosphorylation networks, each edge has the weighted score from 0 to 1, 1 for the experimentally verified kinase–substrate interaction and logistic regression probability value for the computationally identified kinase–substrate interaction.
RESULTS AND DISCUSSIONS
The aim of this work is to develop an effective system, namely RegPhos, for exploring the protein kinase–substrate phosphorylation networks in human. The information of subcellular localization is utilized to construct the intracellular phosphorylation network starting from membrane receptor to transcription factor in nucleus. In order to enhance the identification of kinase–substrate interactions, the protein associations (protein–protein interaction, functional association and subcellular localization) between kinases and phosphoproteins are carefully investigated.
Investigation of association context among kinases, phosphoproteins and interacting proteins
With the annotations of 4138 experimentally confirmed kinase-specific phosphorylation sites in human, a total of 1306 experimental kinase–substrate interactions are identified; as presented in Supplementary Figure S4, 1039 kinase–substrate pairs of which have been annotated as protein–protein interactions, based on the collection of protein interactions from DIP (32,33), MINT (34), IntAct (35) and HPRD (30) databases. According to annotations in the four integrated interaction databases, a total of 1801 phosphoproteins have the direct interaction to 430 human kinases. Furthermore, the indirect links between kinases and their substrates are also taken into account. Those unobvious relationships would be very difficult to predict by manually inspecting the available sequence motifs. To investigate the interacting distance of indirect connection between kinases and substrates, the number of substrates interacting to a specific kinase family is observed in different interacting distance. As shown in Table 2, the numbers of interacting substrates in PKA, PKC, CK2, CDK, Src, EGFR and INSR families are listed with various interacting distance. For instance, PKA family, consisting of PKACa, PKACb and PKACg kinases, has 123 (63%) directly interacting substrates. About 37% of PKA-specific substrates are indirect connection to PKA kinases. Base on the statistics of interacting distance between kinases and their substrates, most of the substrates (∼95%) are connecting to kinases within the distance of three interacting nodes (proteins). Both direct and indirect protein associations are adopted to help the identification of kinase–substrate interactions.
Table 2.
The interacting distance between kinases and their substrates
| Kinase family | Kinase members | Number of substrates | Number of substrates in a specific interacting distance |
|||
|---|---|---|---|---|---|---|
| Distance = 1 (direct interaction) | Distance = 2 (indirect interaction) | Distance = 3 (indirect interaction) | Distance > 4 (indirect interaction) | |||
| PKA | PKACa, PKACb, PKACg | 194 | 123 | 39 | 25 | 7 |
| PKC | PKCh, PKCa, PKCb, PKCd, PKCe, PKCg, PKCi, PKCt, PKCz | 231 | 175 | 41 | 6 | 9 |
| CK2 | CK2a1, CK2a2, CK2b, CK2al-rs | 158 | 120 | 28 | 9 | 1 |
| CDK | CDC2, CDK2, CDK3, CDK4, CDK5, CDK6, CDK7, CDK8, CDK9, CDK10, CDK11, | 157 | 135 | 15 | 2 | 5 |
| Src | Src | 92 | 68 | 19 | 3 | 2 |
| EGFR | EGFR | 27 | 25 | 0 | 1 | 1 |
| InsR | InsR | 14 | 12 | 0 | 1 | 1 |
Investigation of cellular co-localization between kinases and substrates
To easily categorize the subcellular localization for kinases and substrates, the localization of substrates is mainly classified into nuclear and cytoplasmic substrates. We mapped localizations from UniProtKB/Swiss-Prot to the kinase-specific substrates, which resulted in 3863 phosphoproteins that are described as localizing to either the cytoplasm or the nucleus. The statistics of substrate localization preference of kinase families is listed in Table 3. The statistically significant (P < 0.05) localization preference of kinase family is marked in bold. Based on the statistics, we found 33 kinase groups that show a statistically significant preference for either cytoplasmic or nuclear substrates. For the kinase groups that are primarily localized in the nucleus (ATM, DNAPK, RSK, CK2, CDK, CDC2 and Aurora), their preference were about two-fold more nuclear than cytoplasmic targets. However, GRK, ROCK, BARK, CaMK2 and CK1 have strong preference for cytoplasmic substrates. PKA, PKC, PKB, Abl, IKK and MAP2K families are both fairly pleiotropic kinases, which in the phosphorylation network show a slight preference for cytoplasmic substrates. In the case of membrane-associated kinase families, EGFR, INSR, JAK, Src, FYN, LCK, LYN and SYK have the high preference of cytoplasmic substrates.
Table 3.
Cellular co-localization of human kinases and their substrates
| Kinase family | Cellular localization of kinases | All substrates | Cytoplasmic substrates | Nuclear substrates | Cytoplasmic and nuclear substrates |
|---|---|---|---|---|---|
| PKA | Cytoplasm, nucleus | 151 | 96 | 74 | 21 |
| PKC | Cytoplasm, nucleus | 168 | 105 | 81 | 26 |
| PKB | Cell membrane, cytoplasm, nucleus | 63 | 49 | 32 | 19 |
| GRK | Cytoplasm | 19 | 18 | 2 | 2 |
| ROCK | Cytoplasm | 15 | 15 | 1 | 1 |
| BARK | Cytoplasm | 14 | 14 | 1 | 1 |
| CaMK2 | Cytoplasm | 36 | 29 | 11 | 6 |
| CaMK1 | Cytoplasm, nucleus | 14 | 5 | 8 | 2 |
| CK1 | Cytoplasm | 33 | 29 | 14 | 10 |
| ATM | Nucleus | 34 | 11 | 32 | 9 |
| DNAPK | Nucleus | 13 | 3 | 12 | 2 |
| RSK | Nucleus | 31 | 15 | 25 | 9 |
| CK2 | Nucleus | 123 | 46 | 91 | 17 |
| CDK | Nucleus | 121 | 34 | 79 | 30 |
| CDC2 | Nucleus | 95 | 37 | 66 | 17 |
| GSK | Nucleus | 34 | 15 | 23 | 9 |
| MAPK | Cytoplasm, nucleus | 140 | 59 | 91 | 29 |
| JNK | Cytoplasm, nucleus | 27 | 13 | 22 | 9 |
| P38 | Cytoplasm, nucleus | 35 | 15 | 22 | 4 |
| ERK | Nucleus | 88 | 41 | 63 | 18 |
| Aurora | Nucleus | 19 | 8 | 14 | 4 |
| IKK | Cytoplasm, nucleus | 12 | 10 | 8 | 6 |
| PAK | Cytoplasm | 25 | 19 | 6 | 1 |
| MAP2K | Cytoplasm, nucleus | 13 | 9 | 6 | 2 |
| Abl | Cytoplasm, nucleus | 26 | 18 | 13 | 5 |
| EGFR | Cell membrane, nucleus | 22 | 18 | 0 | 4 |
| InsR | Cell membrane | 9 | 9 | 0 | 0 |
| JAK | Membrane associated | 17 | 17 | 6 | 6 |
| Src | Membrane associated | 68 | 61 | 22 | 16 |
| FYN | Membrane associated | 21 | 16 | 9 | 5 |
| LCK | Membrane associated | 25 | 22 | 1 | 1 |
| LYN | Membrane associated | 20 | 17 | 3 | 3 |
| SYK | Membrane associated | 17 | 15 | 1 | 1 |
| Total | 3863 | 1661 | 2195 | 612 |
The bold value means P-value < 0.05.
Predictive performance of computationally identifying kinase–substrate interactions
To fully investigate how protein kinases regulate the intracellular processes, this work proposes a computational model for assigning the potential kinase for each experimental phosphorylation sites without the annotation of catalytic kinase. With reference to NetworKIN (20), that has augmented motif-based predictions with the functional association context of kinases and phosphoproteins, we adopt the similar data set to evaluate the performance of the proposed method. Using only SVM-based model (KinasePhos), the predictive accuracies are 84, 89.6, 91.5 and 81.9% in PKC, CDK, PIKK and INSR, respectively (Supplementary Table S5). The cross classifying specificity among PKC, CDK, PIKK and INSR families are listed in Supplementary Table S6. The specificity (Sp) of CDK, PIKK and INSR sets corresponding to the PKC model are 81.9, 89.1 and 83.3%, respectively. Similarly, the cross specificity values among PKC, CDK, PIKK and INSR are generally higher than 80%. However, the specificity of INSR model is slightly weak when differentiating PKC substrates from INSR substrates. The higher specificity in the cross-validation, the less incorrect prediction of the phosphorylation sites in other groups. By incorporating contextual information of protein association, the prediction accuracy improves to 84.1, 91.6, 91.9 and 91.9% in PKC, CDK, PIKK and INSR, respectively, because of the improvement of specificity (Supplementary Figure S5). However, there are slight drops in predictive sensitivity. These results highlight the importance of including contextual information in identifying kinase–substrate relationships for experimentally verified phosphorylation sites without annotated catalytic kinases. The computationally identified kinase–substrate interactions can make the construction of intracellular phosphorylation networks more feasible.
A case study of identifying catalytic kinases for insulin receptor substrate 1
Insulin receptor substrate 1 (IRS1), which mediate the control of various cellular processes by insulin (48), were used to present the effectiveness of computational identification of kinase-specific phosphorylation sites. With the annotation of Phospho.ELM (4) and UniProtKB/Swiss-Prot (29), IRS1 has totally 32 experimentally verified phosphorylation sites. However, some of the experimental phosphorylation sites do not have the annotation of catalytic kinases. Based on the trained threshold of logistic regression probability score in each kinase group, these phosphorylation sites were annotated the potential catalytic kinases. As illustrated in Figure 2, seven kinase-specific phosphorylation sites with their protein associations are identified. For instance, the tyrosine phosphorylation sites ‘Y612’ and ‘Y632’ were potentially catalyzed by Janus kinase 1 (JAK1), with the indirect protein–protein interaction which was linked by v-erb-b2 erythroblastic leukemia viral oncogene homolog 2 (ErbB2). The tyrosine phosphorylation sites ‘Y46’ and ‘Y896’ were catalyzed by Insulin-like Growth Factor I Receptor (IGF1R), with the directly functional association annotated by STRING database. Phosphoserine ‘S636’ was catalyzed by Mitogen-Activated Protein Kinase (MAPK) group and a functional association shows that Mitogen-Activated Protein Kinase 1 (MAPK1 or Erk2) was directly link to IRS1. Phosphotyrosine ‘Y1229’ was catalyzed by insulin receptor (InsR) with the direct protein–protein interaction (DIP:429E) of DIP database. Some phosphorylation sites were identified by more than two kinases, for example phosphoserine ‘S1145’ was potentially catalyzed by v-akt murine thymoma viral oncogene homolog 1 (Akt1) with directly functional association or was potentially catalyzed by protein kinase C epsilon (PKCe) with indirect link in distance of three protein–protein interactions, passing through Stratifin (SFN) and B-Raf proto-oncogene serine/threonine-protein kinase (BRAF).
Figure 2.

Case study of computationally identified kinase-specific phosphorylation sites in Insulin Receptor Substrate 1 (IRS1).
Web interface of exploring protein phosphorylation networks
To facilitate the investigation of protein kinase and their substrate, a web-based system, named RegPhos, was implemented for users to efficiently browse the protein kinases and their substrate proteins in a user-friendly manner. Three major functions, including browsing kinase or substrate (see Supplementary Figure S7), constructing phosphorylation network and microarray expression analysis (see Supplementary Figure S8), are provided in the proposed system. The JMol viewer (49) is adopted for the visualization of PDB (50) structures of kinases and substrates. The proposed system can let users input a group of gene/protein names; the phosphorylation network associated with protein subcellular localization can be automatically constructed. To fully investigate how protein kinase control the intracellular processes, the experimentally verified kinase–substrate phosphorylations and the computationally discovered kinase–substrate interactions are incorporated to explore the phosphorylation networks starting from receptor kinases associated with membrane to transcription factors located in nucleus. However, the phosphorylation-driven signal transduction pathway is not always the phosphorylation cascade. Some protein–protein interactions are involved in the signal transduction pathway, such as IRS1–GRB2 interaction, GRB2–SOS1 interaction, SOS1–HRAS interaction and HRAS–RAF1 interaction in insulin signaling pathway (31). Supplementary Figure S9 shows an example of insulin signaling network in the construction of phosphorylation network. A group of proteins associated with insulin signaling pathway are inputted to construct the network from membrane-associated proteins to nuclear proteins.
A case study of the discovered networks associated with insulin signaling pathway
To demonstrate the effectiveness of the proposed method, the discovered phosphorylation networks associated with the insulin signaling pathway are represented in Figure 3. Insulin regulates both metabolism and gene expression; the insulin signal transduction passes from the plasma membrane receptor to insulin-sensitive metabolic enzymes and finally to the nucleus, where it stimulates the transcription of specific genes (31). The well-known insulin signaling pathway, INSR → IRS1 — Grb2 — SOS1 — RAS — Raf1 → MEK → ERK1 → Elk1, can be successfully identified by the presented graph-based phosphorylation network searching method (‘→’ stands for phosphorylation and ‘—’ stands for protein–protein interaction). Due to the protein–protein interactions, which are allowed in the network searching, numerous insulin receptor (INSR) related signaling pathways have been discovered, which contain about 2000 pathways with length of eight proteins. After the validation of time-coursed microarray data, the discovered INSR-related phosphorylation networks can be decreased to about 50 pathways. Some of the well-known signaling networks are discovered and marked with red lines in Figure 3. RegPhos not only identify the correct network of insulin signaling but also detect a potentially novel signaling pathway that may cross-talk with insulin signaling network. For instance, Qin-induced kinase (QIK) posphorylates ‘Ser-794’ of IRS1 in insulin-stimulated adipocytes, potentially modulating the efficiency of insulin signal transduction (51); SHC-transforming protein 1 (SHC1) is a signaling adapter that couples activated growth factor receptors to signaling pathway (48); GRB2-associated-binding protein 1 (GAB1) probably involved in EGF and insulin receptor signaling (52). The phosphoregulators, such as QIK, IRS1, SHC1 and GAB1, are considerably involved in cross-talk between signaling cascades (53).
Figure 3.

Case study of the discovered phosphorylation networks associated with insulin signaling pathway.
Investigation of co-expressed kinases and substrates
To investigate the statistically significant syn-expressed pair of kinase and substrate genes, all the pairs of genes are calculated for background correlation. However, it is time-expensive for calculating all pairs of genes. Therefore, the random sampling is adopted to extract 100 000 gene pairs as the background set for estimating the distribution of Pearson correlation coefficients of background gene pairs (see Supplementary Figure S10). The distribution of Pearson correlation coefficient of pairs of specific kinases and their substrates is also investigated. Supplementary Figure S11 shows the distribution of correlation coefficient of PKA-substrate pairs, CDC2-substrate pairs and EGFR-substrate pairs, based on 98 microarray series. Most of the PKA-substrate pairs (40%) belong to the low positive correlation (0 < r < 0.4), with the average correlation coefficient 0.08. In particular, about 65% of CDC2-substrate pairs have the positive correlation, with ∼20% high positive correlation (r > 0.7). The average correlation coefficient of CDC2-substrate pairs is 0.14. In the case of EGFR-substrate pairs, the distribution of correlation coefficient is similar to the distribution of all kinase–substrate pairs. The average correlation coefficient of EGFR-substrate pairs is 0.028.
Moreover, the distribution of Pearson correlation coefficient of pairs of specific kinases and their substrates is investigated based on time-coursed microarray data. Supplementary Figure S11 shows the distribution of correlation coefficient of PKA-substrate pairs, CDC2-substrate pairs and EGFR-substrate pairs based on nine time-coursed microarray series (described in ‘Materials and methods’ section). The average correlation coefficient of PKA-substrate pairs is up to 0.12. The proportion of PKA-substrate pairs belonged to the low positive correlation (0 < r < 0.4) is increased from 40 to 45%. In the case of EGFR-substrate pairs, the average correlation coefficient of EGFR-substrate pairs is raised from 0.028 to 0.08. The proportion of EGFR-substrate pairs belonged to high positive correlation (r > 0.6) is approaching 16%. However, based on time-coursed microarray data, the average correlation coefficient of CDC2-substrate pairs is decreased to 0.10. Generally, the experimentally confirmed kinase–substrate pairs have higher value of Pearson correlation coefficient based on time-coursed microarray expression data. Thus, the time-coursed microarray data of Affymetrix GeneChip Human Genome U133 Array Set HG-U133A platform (GPL96) are used to test the degree of similarity in the expression profiles of network members.
CONCLUSION
With the increasing number of in vivo phosphorylation sites, which have been identified, the desire of mapping the network of protein kinase and substrate is motivated. The experimental kinase-specific substrates, ultimately, need to be combined by systems biology analysis, which translates the separate, large-scale datasets into signaling networks. Therefore, this study has incorporated the experimentally verified kinase–substrate interactions with experimental protein–protein interactions to construct the intracellular phosphorylation network starting from receptor kinases to transcription factors, associated with the information of subcellular localization. With the integration of public phosphorylation resources, most of the experimentally verified phosphorylation sites (∼80%) do not have the annotation of catalytic kinases. A published kinase-specific phosphorylation site prediction tool, KinasePhos (17–19), is incorporated with protein association (protein–protein interaction, functional association and protein subcellular localization) for assigning the potential kinase. After the evaluation, the proposed method improves the predictive power and highlights the importance of kinase–substrate interactions in the specificity of protein phosphorylation within cells. Moreover, the experimental expression evidence, such as gene microarray data, was adopted to validate the syn-expression of the discovered phosphorylation network with statistical significance. To facilitate the investigation of protein kinases and their substrates, a web-based system, named RegPhos, was implemented for users to efficiently browse the protein kinases and their substrate proteins in a user-friendly manner. A case study demonstrates that RegPhos not only identify the correct network of insulin signaling but also detect a novel signaling pathway that may cross-talk with insulin signaling network. In prospective works, protein phosphatase, act as oppositive function to protein kinases, is needed to be considered in construction of protein phosphorylation network. Protein kinases and phosphatases can regulate the phosphorylation status of the protein complement of a cell and in turn, regulate the activity of their target phosphoproteins in cellular processes. Defining the entire complement of these proteins gives us an opportunity to view the system as a whole.
AVAILABILITY
The RegPhos database will be continuously maintained and updated. All the experimentally verified data on protein phosphorylation and protein–protein interaction will be updated quarterly. The time-coursed microarray expression data collected from Gene Expression Omnibus (GEO) will also be updated quarterly. The resource is now freely available at http://RegPhos.mbc.nctu.edu.tw.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Council of the Republic of China under (Contract Numbers of NSC 98-2627-B-009-005, NSC 99-2320-B-155-001, NSC 99-2627-B-009-003, NSC 98-2311-B-009-004-MY3, NSC 99-2621-B-006-001-MY2 and NSC 99-2628-B-006-016-MY3); National Research Program for Genomic Medicine (NRPGM), Taiwan.
Conflict of interest statement. None declared.
REFERENCES
- 1.Hubbard MJ, Cohen P. On target with a new mechanism for the regulation of protein phosphorylation. Trends Biochem. Sci. 1993;18:172–177. doi: 10.1016/0968-0004(93)90109-z. [DOI] [PubMed] [Google Scholar]
- 2.Steffen M, Petti A, Aach J, D'Haeseleer P, Church G. Automated modelling of signal transduction networks. BMC Bioinformatics. 2002;3:34. doi: 10.1186/1471-2105-3-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 4.Diella F, Cameron S, Gemund C, Linding R, Via A, Kuster B, Sicheritz-Ponten T, Blom N, Gibson TJ. Phospho.ELM: a database of experimentally verified phosphorylation sites in eukaryotic proteins. BMC Bioinformatics. 2004;5:79. doi: 10.1186/1471-2105-5-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hornbeck PV, Chabra I, Kornhauser JM, Skrzypek E, Zhang B. PhosphoSite: A bioinformatics resource dedicated to physiological protein phosphorylation. Proteomics. 2004;4:1551–1561. doi: 10.1002/pmic.200300772. [DOI] [PubMed] [Google Scholar]
- 6.Boeckmann B, Bairoch A, Apweiler R, Blatter MC, Estreicher A, Gasteiger E, Martin MJ, Michoud K, O'Donovan C, Phan I, et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wurgler-Murphy SM, King DM, Kennelly PJ. The Phosphorylation Site Database: A guide to the serine-, threonine-, and/or tyrosine-phosphorylated proteins in prokaryotic organisms. Proteomics. 2004;4:1562–1570. doi: 10.1002/pmic.200300711. [DOI] [PubMed] [Google Scholar]
- 8.Gnad F, Ren S, Cox J, Olsen JV, Macek B, Oroshi M, Mann M. PHOSIDA (phosphorylation site database): management, structural and evolutionary investigation, and prediction of phosphosites. Genome Biol. 2007;8:R250. doi: 10.1186/gb-2007-8-11-r250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heazlewood JL, Durek P, Hummel J, Selbig J, Weckwerth W, Walther D, Schulze WX. PhosPhAt: a database of phosphorylation sites in Arabidopsis thaliana and a plant-specific phosphorylation site predictor. Nucleic Acids Res. 2008;36:D1015–D1021. doi: 10.1093/nar/gkm812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang C-T, Chang C-H, Yu Y-L, Emma Lin T-C, Lee S-A, Yen C-C, Yang J-M, Lai J-M, Hong Y-R, Tseng T-L, et al. PhosphoPOINT: a comprehensive human kinase interactome and phospho-protein database. Bioinformatics. 2008;24:14–20. doi: 10.1093/bioinformatics/btn297. [DOI] [PubMed] [Google Scholar]
- 11.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 12.Blom N, Sicheritz-Ponten T, Gupta R, Gammeltoft S, Brunak S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics. 2004;4:1633–1649. doi: 10.1002/pmic.200300771. [DOI] [PubMed] [Google Scholar]
- 13.Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xue Y, Zhou F, Zhu M, Ahmed K, Chen G, Yao X. GPS: a comprehensive www server for phosphorylation sites prediction. Nucleic Acids Res. 2005;33:W184–W187. doi: 10.1093/nar/gki393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou FF, Xue Y, Chen GL, Yao X. GPS: a novel group-based phosphorylation predicting and scoring method. Biochem. Biophys. Res. Commun. 2004;325:1443–1448. doi: 10.1016/j.bbrc.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 16.Xue Y, Li A, Wang L, Feng H, Yao X. PPSP: prediction of PK-specific phosphorylation site with Bayesian decision theory. BMC Bioinformatics. 2006;7:163. doi: 10.1186/1471-2105-7-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang HD, Lee TY, Tzeng SW, Horng JT. KinasePhos: a web tool for identifying protein kinase-specific phosphorylation sites. Nucleic Acids Res. 2005;33:W226–W229. doi: 10.1093/nar/gki471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang HD, Lee TY, Tzeng SW, Wu LC, Horng JT, Tsou AP, Huang KT. Incorporating hidden Markov models for identifying protein kinase-specific phosphorylation sites. J. Comput. Chem. 2005;26:1032–1041. doi: 10.1002/jcc.20235. [DOI] [PubMed] [Google Scholar]
- 19.Wong YH, Lee TY, Liang HK, Huang CM, Wang TY, Yang YH, Chu CH, Huang HD, Ko MT, Hwang JK. KinasePhos 2.0: a web server for identifying protein kinase-specific phosphorylation sites based on sequences and coupling patterns. Nucleic Acids Res. 2007;35:W588–W594. doi: 10.1093/nar/gkm322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Linding R, Jensen LJ, Ostheimer GJ, van Vugt MA, Jorgensen C, Miron IM, Diella F, Colwill K, Taylor L, Elder K, et al. Systematic discovery of in vivo phosphorylation networks. Cell. 2007;129:1415–1426. doi: 10.1016/j.cell.2007.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Janes KA, Yaffe MB. Data-driven modelling of signal-transduction networks. Nat. Rev. Mol. Cell Biol. 2006;7:820–828. doi: 10.1038/nrm2041. [DOI] [PubMed] [Google Scholar]
- 22.Neves SR, Iyengar R. Modeling of signaling networks. Bioessays. 2002;24:1110–1117. doi: 10.1002/bies.1154. [DOI] [PubMed] [Google Scholar]
- 23.Choi C, Crass T, Kel A, Kel-Margoulis O, Krull M, Pistor S, Potapov A, Voss N, Wingender E. Consistent re-modeling of signaling pathways and its implementation in the TRANSPATH database. Genome Inform. 2004;15:244–254. [PubMed] [Google Scholar]
- 24.Sachs K, Perez O, Pe'er D, Lauffenburger DA, Nolan GP. Causal protein-signaling networks derived from multiparameter single-cell data. Science. 2005;308:523–529. doi: 10.1126/science.1105809. [DOI] [PubMed] [Google Scholar]
- 25.Bebek G, Yang J. PathFinder: mining signal transduction pathway segments from protein-protein interaction networks. BMC Bioinformatics. 2007;8:335. doi: 10.1186/1471-2105-8-335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eungdamrong NJ, Iyengar R. Modeling cell signaling networks. Biol. Cell. 2004;96:355–362. doi: 10.1016/j.biolcel.2004.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Knuth DE. The Art of Computer Programming. 3rd edn. Boston: Addison-Wesley; 1997. [Google Scholar]
- 28.Lee TY, Huang HD, Hung JH, Huang HY, Yang YS, Wang TH. dbPTM: an information repository of protein post-translational modification. Nucleic Acids Res. 2006;34:D622–D627. doi: 10.1093/nar/gkj083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Farriol-Mathis N, Garavelli JS, Boeckmann B, Duvaud S, Gasteiger E, Gateau A, Veuthey AL, Bairoch A. Annotation of post-translational modifications in the Swiss-Prot knowledge base. Proteomics. 2004;4:1537–1550. doi: 10.1002/pmic.200300764. [DOI] [PubMed] [Google Scholar]
- 30.Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R, Shafreen B, Venugopal A, et al. Human Protein Reference Database–2009 update. Nucleic Acids Res. 2009;37:D767–D772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lehninger AL, Nelson DL, Cox MM. Lehninger Principles of Biochemistry. 2005. 4th edn., W. H. Freeman, Worth Publisher, USA. [Google Scholar]
- 32.Xenarios I, Salwinski L, Duan XJ, Higney P, Kim SM, Eisenberg D. DIP, the Database of Interacting Proteins: a research tool for studying cellular networks of protein interactions. Nucleic Acids Res. 2002;30:303–305. doi: 10.1093/nar/30.1.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xenarios I, Fernandez E, Salwinski L, Duan XJ, Thompson MJ, Marcotte EM, Eisenberg D. DIP: The Database of Interacting Proteins: 2001 update. Nucleic Acids Res. 2001;29:239–241. doi: 10.1093/nar/29.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chatr-Aryamontri A, Ceol A, Palazzi LM, Nardelli G, Schneider MV, Castagnoli L, Cesareni G. MINT: the Molecular INTeraction database. Nucleic Acids Res. 2007;35:D572–D574. doi: 10.1093/nar/gkl950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kerrien S, Alam-Faruque Y, Aranda B, Bancarz I, Bridge A, Derow C, Dimmer E, Feuermann M, Friedrichsen A, Huntley R, et al. IntAct–open source resource for molecular interaction data. Nucleic Acids Res. 2007;35:D561–D565. doi: 10.1093/nar/gkl958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boutet E, Lieberherr D, Tognolli M, Schneider M, Bairoch A. UniProtKB/Swiss-Prot: The Manually Annotated Section of the UniProt KnowledgeBase. Methods Mol. Biol. 2007;406:89–112. [Google Scholar]
- 37.Sprenger J, Lynn Fink J, Karunaratne S, Hanson K, Hamilton NA, Teasdale RD. LOCATE: a mammalian protein subcellular localization database. Nucleic Acids Res. 2008;36:D230–D233. doi: 10.1093/nar/gkm950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo T, Hua S, Ji X, Sun Z. DBSubLoc: database of protein subcellular localization. Nucleic Acids Res. 2004;32:D122–D124. doi: 10.1093/nar/gkh109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wiwatwattana N, Landau CM, Cope GJ, Harp GA, Kumar A. Organelle DB: an updated resource of eukaryotic protein localization and function. Nucleic Acids Res. 2007;35:D810–D814. doi: 10.1093/nar/gkl1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rey S, Acab M, Gardy JL, Laird MR, deFays K, Lambert C, Brinkman FS. PSORTdb: a protein subcellular localization database for bacteria. Nucleic Acids Res. 2005;33:D164–D168. doi: 10.1093/nar/gki027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wingender E, Karas H, Knüppel R. TRANSFAC database as a bridge between sequence data libraries and biological function. In: Altman RB, Dunker AK, Hunter L, Klein TE, editors. Pacific Symposium on Biocomputing '97 (PSB'97) Singapore: World Scientific; 1997. pp. 477–485. [PubMed] [Google Scholar]
- 42.Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Edgar R. NCBI GEO: mining tens of millions of expression profiles–database and tools update. Nucleic Acids Res. 2007;35:D760–D765. doi: 10.1093/nar/gkl887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hochreiter S, Clevert DA, Obermayer K. A new summarization method for Affymetrix probe level data. Bioinformatics. 2006;22:943–949. doi: 10.1093/bioinformatics/btl033. [DOI] [PubMed] [Google Scholar]
- 44.Chang C-C, Lin C-J. LIBSVM: a library for support vector machines. 2001 Software available at http://www.csie.ntu.edu.tw/∼cjlin/libsvm (date last accessed, 30 September 2009) [Google Scholar]
- 45.von Mering C, Jensen LJ, Kuhn M, Chaffron S, Doerks T, Kruger B, Snel B, Bork P. STRING 7–recent developments in the integration and prediction of protein interactions. Nucleic Acids Res. 2007;35:D358–D362. doi: 10.1093/nar/gkl825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pierleoni A, Martelli PL, Fariselli P, Casadio R. BaCelLo: a balanced subcellular localization predictor. Bioinformatics. 2006;22:e408–e416. doi: 10.1093/bioinformatics/btl222. [DOI] [PubMed] [Google Scholar]
- 47.Sharan R, Suthram S, Kelley RM, Kuhn T, McCuine S, Uetz P, Sittler T, Karp RM, Ideker T. Conserved patterns of protein interaction in multiple species. Proc. Natl Acad. Sci. USA. 2005;102:1974–1979. doi: 10.1073/pnas.0409522102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Craparo A, O'Neill TJ, Gustafson TA. Non-SH2 domains within insulin receptor substrate-1 and SHC mediate their phosphotyrosine-dependent interaction with the NPEY motif of the insulin-like growth factor I receptor. J. Biol. Chem. 1995;270:15639–15643. doi: 10.1074/jbc.270.26.15639. [DOI] [PubMed] [Google Scholar]
- 49. Jmol: an open-source Java viewer for chemical structures in 3D. http://www.jmol.org/ (date last accessed, 30 September 2009)
- 50.Deshpande N, Addess KJ, Bluhm WF, Merino-Ott JC, Townsend-Merino W, Zhang Q, Knezevich C, Xie L, Chen L, Feng Z, et al. The RCSB Protein Data Bank: a redesigned query system and relational database based on the mmCIF schema. Nucleic Acids Res. 2005;33:D233–D237. doi: 10.1093/nar/gki057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Horike N, Takemori H, Katoh Y, Doi J, Min L, Asano T, Sun XJ, Yamamoto H, Kasayama S, Muraoka M, et al. Adipose-specific expression, phosphorylation of Ser794 in insulin receptor substrate-1, and activation in diabetic animals of salt-inducible kinase-2. J. Biol. Chem. 2003;278:18440–18447. doi: 10.1074/jbc.M211770200. [DOI] [PubMed] [Google Scholar]
- 52.Holgado-Madruga M, Emlet DR, Moscatello DK, Godwin AK, Wong AJ. A Grb2-associated docking protein in EGF- and insulin-receptor signalling. Nature. 1996;379:560–564. doi: 10.1038/379560a0. [DOI] [PubMed] [Google Scholar]
- 53.Forrest AR, Ravasi T, Taylor D, Huber T, Hume DA, Grimmond S. Phosphoregulators: protein kinases and protein phosphatases of mouse. Genome Res. 2003;13:1443–1454. doi: 10.1101/gr.954803. [DOI] [PMC free article] [PubMed] [Google Scholar]