

Abstract
Air pollution is associated with significant adverse health effects, including increased cardiovascular morbidity and mortality. Exposure to particulate matter with an aerodynamic diameter of <2.5 μm (PM2.5) increases ischemic cardiovascular events and promotes atherosclerosis. Moreover, there is increasing evidence that the smallest pollutant particles pose the greatest danger because of their high content of organic chemicals and prooxidative potential. To test this hypothesis, we compared the proatherogenic effects of ambient particles of <0.18 μm (ultrafine particles) with particles of <2.5 μm in genetically susceptible (apolipoprotein E–deficient) mice. These animals were exposed to concentrated ultrafine particles, concentrated particles of <2.5 μm, or filtered air in a mobile animal facility close to a Los Angeles freeway. Ultrafine particle–exposed mice exhibited significantly larger early atherosclerotic lesions than mice exposed to PM2.5 or filtered air. Exposure to ultrafine particles also resulted in an inhibition of the antiinflammatory capacity of plasma high-density lipoprotein and greater systemic oxidative stress as evidenced by a significant increase in hepatic malondialdehyde levels and upregulation of Nrf2-regulated antioxidant genes. We conclude that ultrafine particles concentrate the proatherogenic effects of ambient PM and may constitute a significant cardiovascular risk factor.
Keywords: air pollution, ultrafine particles, atherosclerosis, oxidative stress, HDL
It is increasingly being recognized that exposure to ambient particulate matter (PM) contributes to significant adverse health effects and is a risk factor for the development of ischemic cardiovascular events via exacerbation of atherosclerosis, coronary artery disease, and the triggering of myocardial infarctions.1 Although this association has been documented for PM with a mean aerodynamic diameter of < 10 μm (PM10), there is increasing evidence that smaller particles may pose an even greater health risk. A growing literature indicates that fine particles (FPs) with an average aerodynamic diameter of <2.5 μm (PM2.5) exert adverse health effects of greater magnitude. For example, the “Women’s Health Initiative study demonstrated a 24% increase in the incidence of cardiovascular events and a 76% increase in cardiovascular mortality for every 10 μg/m3 increase in the annual average PM2.5 level.2 It appears that the smallest particles that exist in the urban environment are the most dangerous.3 Ambient ultrafine particles (UFPs) that have an aerodynamic diameter of <0.18 μm are by far the most abundant particles by number in urban environments such as Los Angeles. Because these particles are emitted mainly by vehicular emissions and other combustion sources, they contain a high content of redox-cycling organic chemicals that could be released deep into the lungs or could even spill over into the systemic circulation. Thus, UFPs may be particularly relevant from the perspective of cardiovascular injury.3
In spite of the epidemiological evidence indicating that ambient PM can promote cardiovascular injury and atherosclerosis, the mechanisms of the cardiovascular injury and proatherogenic effects are not clear. However, experimental studies in susceptible animal models have shed some light on disease pathogenesis. For instance, intratracheal administration of ambient PM10 in Watanabe rabbits4 or long-term exposure of apolipoprotein (apo)E-null mice to PM2.55,6 enhanced atherosclerotic plaque growth. Moreover, a cross-sectional exposure study in humans showed a 5.9% increase in carotid intima–medial thickness for every 10 μg/m3 rise in PM2.5 levels,7 and a prospective cohort study supported an association between long-term residential exposure to high-traffic levels of PM2.5 and coronary atherosclerosis, as assessed by coronary artery calcification scores,8 demonstrating that the proatherogenic effects of PM are clinically relevant.7,8 Air pollution has also been linked to the triggering of acute coronary ischemic events in humans, including myocardial infarction.9
We have demonstrated that ambient PM exerts proinflammatory effects in target cells such as endothelial cells,10 macrophages,11 and epithelial cells12 through the generation of reactive oxygen species (ROS) and oxidative stress.11,12 These prooxidative effects are mediated, in part, by redox-cycling organic chemicals and transition metals that are present on the particle surface.11 Ambient PM can synergize with oxidized phospholipids in the induction of a wide array of genes involved in vascular inflammatory processes such as atherosclerosis.10 Moreover, when comparing concentrated ambient particles (CAPs) of various sizes in the Los Angeles basin, UFPs were shown to have the highest content of redox cycling chemicals and therefore displayed the largest prooxidant potential, both abiotically and biotically.13 We hypothesized, therefore, that UFPs may concentrate some of the PM proatherogenic effects by promoting prooxidant and proinflammatory effects. We used the particle concentrator technology available in the Southern California Particle Center to evaluate the atherogenic potential of concentrated UFPs versus concentrated PM2.5 in apoE-null mice. In addition, we evaluated the effects of particle exposures on the plasma high-density lipoprotein (HDL) antiinflammatory activity as well as markers of systemic oxidative stress. Our data show that UFPs are more proatherogenic, exert the strongest prooxidative effects, and are associated with the largest decrease in HDL protective activity. These data are of considerable significance from a regulatory perspective.
Materials and Methods
Detailed methods about histology, immunohistochemistry, blood chemistry, monocyte chemotactic assays, lipid peroxidation assay, RNA extraction, and real-time RT-PCR can be found in the online data supplement at http://circres.ahajournals.org.
Animals and Diet
The Animal Research Committee at The University of California at Los Angeles (UCLA) approved all animal protocols. ApoE−/− (C57BL/6J background) male mice were obtained from The Jackson Laboratory (Bar Harbor, Me). Animals were brought to the UCLA animal facility at 4 weeks of age. Mice were fed a regular chow diet (NIH-31 modified 6% diet; Harlan Teklad, Madison, Wis). Both water and food were administered ad libitum. Animals were randomly assigned to 3 groups (n=17/group) that were sent to a mobile inhalation toxicology laboratory located 300 meters from the 110 Freeway. This freeway carries a high volume of gasoline and diesel motor vehicle transit, resulting in high levels of PM2.5 mass and UFP counts at the exposure site (Table). The mobile research laboratory (AirCARE 1) is owned by Michigan State University.14 Mice were subjected to CAP exposures starting at 6 weeks of age over a 40-day period. One mouse in the FP group and 2 in the UFP group died during the course of the study. Animals were euthanized 24 to 48 hours after completion of the last CAP exposure, and aortas and various organs were harvested. Between exposures, mice were housed in a Hazelton chamber15 that was ventilated with air from which 99.9% of the incident particles were removed by a HEPA filter.
Table.
Characteristics of Experimental Exposure Protocol of ApoE-Null Mice Fed a Normal Chow Diet
| Experimental Parameter | |
|---|---|
| Groups | FA, FP, UFP |
| Exposure time (dates) | 11/03/2005 to 12/12/2005 |
| Exposure time (hours) | 75 |
| Total ambient particle no. (particles/cm3) | 3.42 (±0.96)×104 |
| No. concentration in FA chamber (particles/cm3) | <5000 |
| No. concentration in FP chamber (particles/cm3) | 4.56 (±1.06)×105 |
| Calculated UFP no. concentration in the FP chamber (particles/cm3) | 3.88 (±1.06)×105 |
| No. concentration in UFP chamber (particles/cm3) | 5.59 (±1.23)×105 |
| Ratio of UFP in the FP vs the UFP chamber* | 1:1.44 |
| FP chamber particle enrichment factor | 13.35 (±1.6) |
| UFP chamber particle enrichment factor | 16.4 (±1.8) |
| Mass in FP exposure chamber (μg/m3) | 438.29 |
| Mass in UFP exposure chamber (μg/m3) | 112.61 |
| PM2.5 mass in ambient air (μg/m3) | 26.78 |
| UFP mass in ambient air (μg/m3) | 8.43 |
FA, FP, and UFP groups were exposed in a mobile laboratory located in downtown Los Angeles. Values shown are means(±SD).
This ratio was obtained by reducing the particle no. in the FP chamber by 15%, which represents the contribution of particles in the 0.18 –2.5 μm range. This also translates into an ≈2-fold increase in surface area if a spherical particle shape is assumed.
CAP Exposures and Chemical Characterization
Whole-body exposures were performed simultaneously in sessions of 5 hours per day, 3 days per week, for a combined total of 75 hours. Particle concentrator technology was used to deliver the CAP exposures. Three animal groups were simultaneously exposed to atmospheres containing concentrated particles of <2.5 μm (FPs), particles of <0.18 μm (UFPs), and filtered air (FA). Briefly, ambient air was drawn through an aluminum duct into the VACES (Versatile Aerosol Concentration Enrichment System)16,17 and delivered to whole-body exposure chambers.18,19 The FP and UFP aerosol concentrators delivered 0.01- to 2.5-μm and 0.01- to 0.18-μm aerosols, respectively (Table). The FP atmosphere included sub-18 μm particles that were ≈40% fewer particles than in the UFP chamber. Temperature and airflow were controlled to ensure adequate ventilation and minimize buildup of animal-generated dander, ammonia, CO2, and thermal stress. Mobilization of mice between the Hazelton chamber and the exposures chambers was performed over the shortest time period possible to limit the exposure to ambient air PM in the trailer. CAP number concentrations were measured with a TSI 3022 Condensation Particle Counter, and particle mass concentration was assessed with a DataRAM Model DK-2000.
Particle mass concentration and elemental CAP composition were measured by particle collection on 37-mm Teflon filters (PTFE 2-μm pore, Gelman Science, Ann Arbor, Mich). Concentrations of inorganic ions (sulfate and nitrate), elemental carbon, organic carbon (OC), polycyclic aromatic hydrocarbon (PAH) content, and particle-bound trace elements and metals was performed as previously described.16–18
Statistical Analysis
All data were expressed as means±SEM unless indicated otherwise. Differences between experimental groups were analyzed by 1-way ANOVA with a 1-tailed Fisher protected least-significance difference (PLSD) post hoc analysis test. Differences were considered statistically significant at P<0.05.
Results
UFP Exposures Are Enriched in OC Substances Such As PAHs
Six-week-old male apoE-null mice were exposed in a mobile inhalation toxicology laboratory in downtown Los Angeles to CAPs in the size range of <2.5 μm (FP exposures) or <0.18 μm (UFP exposures). Controls consisted of mice exposed to FA (Figure 1A). Animals were simultaneously exposed to UFPs, FPs, and FA for a total of 75 hours over a 40-day time period while being kept on a chow diet. The atmospheric conditions and particle characteristics in the FP and UFP chambers are summarized in the Table. Because the FP atmosphere included particles of <0.18 μm (UFPs) that accounted for up to 85% of the total particle number, the actual number of these sub-0.18 μm particles was ≈44% greater in the UFP chamber (Table), despite a total UFP mass that was approximately one-quarter of the FP mass. Assuming a roughly spherical shape for the particles, this 44% increase in sub-0.18 μm particle numbers in the UFP chamber translates into an ≈2-fold increase in the particle surface area. This was also accompanied by an ≈2-fold increase in fractional OC content (Figure 1B and 1C), which is theoretically more bioavailable than the smaller organic fraction on FPs (Figure 1B). Thus, the increased particle number, greater surface area, and higher fractional carbon composition could combine to deliver a much higher biological effective dose of the injurious components in the UFP compared with the FP chamber. In fact, measurement of a set of signature PAHs in filter samples that were collected concurrently with the CAP exposures, demonstrated that the PAH content of the UFPs was roughly twice as high as the FP content when corrected for a per mass basis (Figure 2). Although there is no definitive evidence that PAHs are those responsible for adverse cardiovascular effects, we have previously demonstrated that their abundance is a good proxy for the prooxidant potential of PM.13
Figure 1.

CAP exposures. A, Experimental protocol. Three groups (n=17) of 6-week-old male apoE-null mice were exposed to FA, PM2.5, and PM of <0.18 μm (UFPs) for 40 days. B and C, Chemical composition of CAPs. UFP air had a greater content of organic and elemental carbon than FP air. Particle chemical composition of the FP (B) and UFP (C) chambers was performed as described in Materials and Methods.
Figure 2.

OC composition. Mass concentration fraction of PAHs in the FP (gray) and UFP (black) chambers. Data are shown as nanogram per milligram of PM mass and represent the average of composition analysis performed on filter samples collected for 2 experiments. PAH analysis was performed by means of gas chromatography–mass spectroscopy as described.16–18
UFP Exposure Promotes Atherosclerosis
Exposure to the UFP atmosphere for 75 hours over a 40-day interval resulted in 55% greater aortic atherosclerotic lesion development (33 011±3741, n=15) as compared with FA controls (21 362±2864, n=14; P=0.002) (Figure 3). Exposure to the FP atmosphere resulted in a similar trend but of lesser magnitude (P=0.1). Interestingly, UFP mice exhibited a 25% increase in atherosclerotic lesions in comparison with FP mice (26 361±2275, n=16, P=0.04), which suggests that the smallest particles are indeed more proatherogenic.
Figure 3.

UFP is the most proatherogenic PM fraction. Atherosclerotic lesions were quantitatively analyzed in serial aortic root sections and stained with oil red O. Lesional area was scored as square micrometer per section, averaged ≥25 sections per animal. Group averages are indicated by straight horizontal bars. One FA mouse was an obvious outlier in its group and removed from the atherosclerotic lesion analysis. However, its inclusion did not modify the overall significance. FA mice are represented by dotted circles (n=14), FPs by stripped circles (n=16), and UFPs by filled circles (n=15).
Histological analysis revealed that lesions were predominantly comprised of macrophage infiltration with intracellular lipid accumulation (foam cells) (Figure 4). These cells contributed, on average, >85% of the total lesional area in all the groups (supplemental Table I). UFP-exposed animals developed more extensive as well as thicker atherosclerotic plaques that showed the same relative abundance of macrophages and smooth muscle cells, as determined by MOMA-2 and α-actin immunohistochemical staining (Figure 4 and supplemental Table I).
Figure 4.
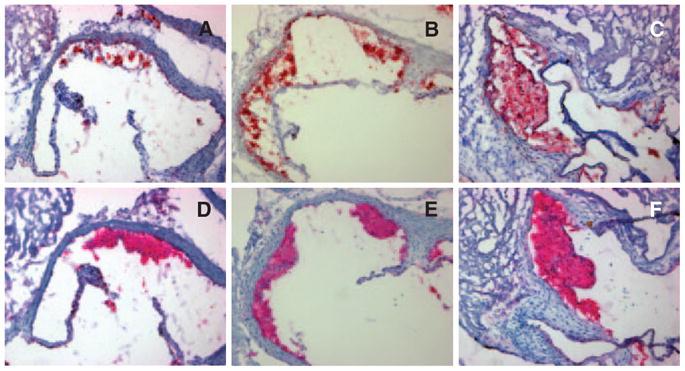
Representative histological photomicrographs. A through C, Oil red O staining for neutral lipids in representative aortic root sections of FA (A), FP (B), and UFP (C) mice. D through F, MOMA-2 immunohistochemical staining in adjacent aortic root sections to those shown in the top row, corresponding to the same FA (D), FP (E), and UFP (F) mice. Both oil red O and MOMA-2 staining yielded red-stained areas. UFP mice exhibited more extensive atherosclerotic plaques (C and F) than FP (B and E) or FA animals (A and D), all consisting primarily of foam cells and macrophages (fatty streaks). Original magnification, ×100.
Exposure to Ambient CAPs Results in Loss of HDL Antiinflammatory Properties
FP but not UFP exposures resulted in a small but significant increase in plasma total cholesterol in comparison to other groups (supplemental Table II). Although all animals displayed similar levels of plasma HDL cholesterol (supplemental Table II), we did observe a change in HDL antiinflammatory properties. This was demonstrated by comparing the antiinflammatory protective capacity of HDL against LDL-induced chemotaxis (Figure 5). Plasma HDL from both FP and UFP animals exhibited significantly less protective effect than HDL from the FA group (Figure 5). Moreover, the antiinflammatory effect of HDL from the UFP group was significantly decreased compared with the FP group. These results are in good agreement with the extent of vascular lesions in the different animal groups, suggesting that a PM-induced decrease in the HDL antiinflammatory protective capacity could contribute to atherogenesis.
Figure 5.

PM exposure leads to a loss of HDL antiinflammatory properties. Pooled plasma HDL from FA (n=16), FP (n=16) and UFP mice (n=15) was added to cocultures of human artery wall cells in the presence of standard (Std) human LDL, as described in Materials and Methods. Values are expressed as means±SEM of the number of migrated monocytes in 9 fields. Statistical analysis was performed by 1-way ANOVA (Fisher PLSD).
UFP Exposure Leads to the Expression of Systemic Biomarkers of Oxidative Stress and Activation of the Unfolded Protein Response
One of the major mechanistic hypotheses regarding PM injury is the ability of the particles to induce ROS production and oxidative stress. To probe for the presence of oxidative stress, we explored whether CAP exposure could result in lipid peroxidation in the liver. We observed statistically significant increases in the hepatic malondialdehyde (MDA) levels in the UFP compared with the FA group (P=0.02) (Figure 6). FP mice also demonstrated increases in lipid peroxidation compared with the FA group (P=0.03). These data suggest that CAP exposure leads to systemic oxidative stress.
Figure 6.

UFP exposure increases liver lipid peroxidation. MDA was assessed in liver homogenates as described in Materials and Methods. Values are expressed as the means±SEM of MDA (nmol/g) in animals from the FA (n=16), FP (n=15), and UFP (n=14) groups. Statistical analysis was performed by 1-way ANOVA (1-tailed Fisher PLSD).
We also explored whether differences in lipid peroxidation were accompanied by phase II antioxidant responses that are mediated via the p45-NFE2–related transcription factor 2, Nrf2.11 This constitutes one of the most sensitive oxidative stress effects that can be traced to prooxidative PM in vitro and in vivo.11,20 UFP mice exhibited a significant increase in the expression of Nrf2 as well as genes that are secondarily regulated by this transcription factor (Figure 7). Indeed, UFP mice displayed Nrf2 mRNA levels that were 68% greater than FA and FP mice (P=0.01). Likewise, as compared with the FA group, UFP mice displayed significantly greater levels of catalase (3.7-fold), glutathione S-transferase Ya (5.3-fold), NAD(P)H-quinone oxidoreductase 1 (1.8-fold), and superoxide dismutase 2 (1.4-fold) (Figure 7). Interestingly, increased tissue oxidative stress was also accompanied by the activation of the unfolded protein response in the liver because the UFP-exposed mice displayed 41% and 37% greater expression of activating transcription factor 4 than FP mice (P=0.01) and FA controls (P=0.02) (Figure 7).
Figure 7.

UFP exposure leads to upregulation of antioxidant genes in the liver. mRNA levels for antioxidant genes were determined by quantitative PCR in the livers of chow-fed mice exposed to CAPs for 40 days. Values are expressed as the means±SEM of mRNA levels normalized by β-actin mRNA. Ten samples per group were assayed in duplicate. Statistical analysis was performed by 1-way ANOVA (1-tailed Fisher PLSD; *P<0.05). ATF4 indicates activating transcription factor 4; GST-Ya, glutathione S-transferase Ya; NQO-1, NAD(P)H-quinone oxidoreductase 1; SOD2, superoxide dismutase 2.
Discussion
We demonstrate that atherosclerotic plaque formation in apoE-null male mice is enhanced by exposure to sub-0.18 μm particles. Mice exposed to UFPs alone exhibited greater and more advanced lesions compared with FA- or FP-exposed animals. UFP mice also showed a comparatively greater decline in the antiinflammatory capacity of plasma HDL as well as increased phase II enzyme mRNA expression in the liver. These results support the hypothesis that exposure to UFPs may enhance atherosclerosis via the promotion of systemic prooxidant and proinflammatory effects.
Our study significantly extends previous data showing that PM potentiates atherosclerotic lesion development in animals.4–6 The fact that FP mice displayed a nonstatistically significant trend to develop more atherosclerotic lesions than FA controls could be attributable to the relatively short duration of our exposure (40 days), which stands in contrast to the 5- to 6-month exposure period that was previously used to demonstrate a 45% to 58% increment in atherosclerotic lesion development during PM2.5 exposure.5,6 Of interest, our UFP animals exhibited a similar 55% increment over FA controls despite an exposure duration that was 4 to 5 times shorter, indicating the greater proatherogenic toxicity of sub-0.18 μm particles. This supports the notion that the adverse cardiovascular effects of PM are exaggerated by a small particle size.
A number of injury mechanisms have been proposed to explain the adverse health effects of PM, including its ability to stimulate oxidative stress and inflammation, alter blood clotting, stimulate autonomic nervous system activity, or act as a carrier for endotoxin.1 A key injury mechanism appears to be the generation of inflammation as a direct consequence of the ability of ambient particles and their adsorbed chemicals to induce ROS and oxidative stress.21 Oxidative stress initiates proinflammatory signaling cascades, including the Jun kinase and nuclear factor κB cascades16,22,23 that are relevant to atherogenesis. According to the hierarchical oxidative stress hypothesis, the induction of Nrf2-induced phase II enzyme expression is an integral oxidative stress protective pathway that acts as a sensitive marker for oxidative stress.24 Indeed, important cytoprotective, antiinflammatory and antioxidant phase II enzymes including catalase, superoxide dismutase 2, glutathione S-transferase Ya, and NAD(P)H-quinone oxidoreductase 1 were all significantly upregulated in the liver of UFP mice (Figure 7) and, together with Nrf2 upregulation, suggest the triggering of a Nrf2-driven antioxidant response.
Our results support the notion that the generation of systemic oxidative stress is responsible for the observed vascular effects. Possible explanations for these systemic effects are: First, inhaled particles may release organic chemicals and transition metals from the lung to the systemic circulation. Second, pulmonary inflammation could lead to the release of ROS, cytokines and chemokines to the systemic circulation. Although we did not observe any major increase in inflammatory cells during the performance of bronchoalveolar lavage in these mice, future studies will need to address whether any subtle proinflammatory effects in the lung could play a role. Third, UFPs could gain access to the systemic circulation by directly penetrating the alveolar/capillary barrier.25 However, this possibility is still controversial. Although reports of the systemic translocation of 99mTc-labeled ultrafine carbon particles25 or albumin nanocolloid particles of <80 nm26 have appeared in the literature, skepticism has been expressed about the stability of the labeling procedures. Moreover, the same has not been demonstrated for ambient air “nanoparticles.”
The particles or their chemicals may generate ROS systemically via a number of different pathways, including redox cycling of quinones, metabolism and functionalization of PAHs, activation of leukocyte NADPH oxidase and myeloperoxidase, or interference in 1-electron transfers in the mitochondrial inner membrane.27 It is also possible that the particles themselves or their chemical components may synergize with oxidized LDL in promoting endothelial cell dysfunction. Indeed, we have shown that ambient PM can synergize with oxidized phospholipids in the induction of a large number of genes in a human microvascular endothelial cell line, many of which belong to antioxidant, proinflammatory, unfolded protein response, or proapoptotic pathways.10 ROS generation and antioxidant responses constitute a dynamic equilibrium. The greater prooxidant stimulus delivered by the UFPs could be more prone to overwhelm the concomitant generation of a protective antioxidant response. On the other hand, it is interesting that no differences were noted between the FP and UFP exposures in the MDA assay. Although the methodology used is sensitive and specific for the determination of MDA,28 there are several limitations in this assay in reflecting the degree of lipid peroxidation, as reviewed by Janero et al,29, such as: (1) MDA yield as a result of lipid peroxidation varies with the nature of the polyunsaturated fatty acids peroxidized (especially its degree of unsaturation) and the peroxidation stimulus; (2) only certain lipid oxidation products decompose to yield MDA; (3) MDA is only one of several (aldehydic) end products of fatty peroxide formation and decomposition; (4) the peroxidation environment influences both the formation of lipid-derived MDA precursors and their decomposition to MDA; (5) MDA itself is a reactive substance that can be oxidatively and metabolically degraded; (6) oxidative injury to nonlipid biomolecules has the potential to generate MDA. Thus, if FP and UFP exposures impacted these factors in a different extent, it may explain a greater degree of lipid peroxidation not reflected by the MDA measurements.
PM-induced systemic inflammation and oxidative stress could also adversely affect lipoprotein function, including interfering in the beneficiary effects of HDL on reverse cholesterol transport30 and the antiinflammatory31 effects of this lipoprotein fraction. Indeed, both FP and UFP mice exhibited the development of dysfunctional HDL, which was more severe in the latter group in terms of its proinflammatory potential (Figure 5). Such proinflammatory effects were also supported by the greater expression of activating transcription factor 4 in liver, an unfolded protein response component that we have shown to exert proinflammatory effects in endothelial cells by inducing the expression of interleukin-6, interleukin-8, and monocyte chemotactic protein 1.32 Likewise, we have also shown that prooxidative diesel exhaust particle chemicals induce an unfolded protein response in bronchial epithelial cells.33 Changes in HDL function were observed in the absence of changes on HDL quantitative levels. On the other hand, FP exposures did result in greater total cholesterol levels in the FP versus FA mice, whereas UFP levels were unaffected. These higher cholesterol levels in the FP mice may have resulted in narrowing of the differences in atherosclerosis in between FP and UFP mice that otherwise could have been larger than the 23% observed difference. Consistent with our results, it has been reported that the HDL antiinflammatory profile can be hampered by environmental factors such as the exposure to prooxidative chemicals present in cigarette smoke.34 For example, mice exposed to second-hand smoke develop dysfunctional HDL.35 A possible mechanism could be interference with paraoxonase and lecithin cholesterol acyltransferase activities by redox-active chemical compounds. In particular, prooxidative PM chemicals may affect critical thiol groups that are responsible for the catalytic activity of paraoxonase, leading to increased susceptibility to atherosclerosis.36
The fact that the FP atmosphere contains both UFPs and particles of >0.18 μm makes interpretation of those data complex. However, we have shown that the 25% difference in atherosclerotic lesion scores could be explained by the 44% increase in UFP particle number (Table and Figure 3). Total particle mass was clearly not a determining factor because the FP atmosphere had ≈3.9-fold greater mass than the UFP aerosol. What is likely significant is that UFPs have an ≈2-fold increase in the OC and PAH content on a per mass basis (Figures 1 and 2). It is possible that these prooxidative components could be delivered from a surface area that is twice as big in particles associated with the UFP atmosphere. Although we cannot claim that the PAHs are actually responsible for the lesion development, these organic chemical compounds are a good proxy for the prooxidative potential of UFPs.13
How do our experimental atmospheres relate to real life exposures? The particle numbers in our study were 2- to 6-fold higher than the in-vehicle exposures that commuters may encounter while traveling on Los Angeles freeways.37 It was not logistically feasible to perform detailed dose– and time–response studies; this type of data will be important to obtain in future studies. Although it would clearly be advantageous to know the minimum exposure that is required for proatherogenic effects, previous epidemiological studies have shown that cardiovascular morbidity and mortality increase linearly without a threshold effect.38,39 Differences in the physiology of genetically susceptible animals and humans also have to be taken into consideration when extrapolating this work to cardiovascular disease in humans.
In conclusion, we demonstrate that UFP exposures have a higher proatherogenic potential than FP exposures. These effects could be linked to a greater propensity of UFPs to generate systemic oxidative stress and to interfere with the antiinflammatory capacity of plasma HDL. Our findings are important in explaining how ambient PM may contribute to daily total and cardiovascular mortality.40 Although such an association has been established previously for PM10 and PM2.5,2,41,42 we demonstrate that UFP exposure could be of even greater relevance. Further epidemiological and experimental data collection are required to determine the critical physicochemical and toxicological properties of UFPs in humans.
Supplementary Material
Acknowledgments
We thank Larry Castellani for the plasma lipoprotein determinations.
Sources of Funding
This work was supported by National Institute of Environmental Health Sciences grant RO1 ES13432 (to A.N.); National Institute of Allergy and Infectious Diseases grant U19 AI070453 (to A.N.); a U.S. Environmental Protection Agency ENERGY STAR Award to the Southern California Particle Center; a Robert Wood Johnson Foundation Harold Amos Medical Faculty Development Program Award (to J.A.A.); and National Heart, Blood, and Lung Institute grant HL30568 (to A.J.L.).
Footnotes
Disclosures
None.
References
- 1.Nel A. Atmosphere. Air pollution-related illness: effects of particles. Science. 2005;308:804–806. doi: 10.1126/science.1108752. [DOI] [PubMed] [Google Scholar]
- 2.Miller KA, Siscovick DS, Sheppard L, Shepherd K, Sullivan JH, Anderson GL, Kaufman JD. Long-term exposure to air pollution and incidence of cardiovascular events in women. N Engl J Med. 2007;356:447–458. doi: 10.1056/NEJMoa054409. [DOI] [PubMed] [Google Scholar]
- 3.Utell MJ, Frampton MW. Acute health effects of ambient air pollution: the ultrafine particle hypothesis. J Aerosol Med. 2000;13:355–359. doi: 10.1089/jam.2000.13.355. [DOI] [PubMed] [Google Scholar]
- 4.Suwa T, Hogg JC, Quinlan KB, Ohgami A, Vincent R, van Eeden SF. Particulate air pollution induces progression of atherosclerosis. J Am Coll Cardiol. 2002;39:935–942. doi: 10.1016/s0735-1097(02)01715-1. [DOI] [PubMed] [Google Scholar]
- 5.Sun Q, Wang A, Jin X, Natanzon A, Duquaine D, Brook RD, Aguinaldo JG, Fayad ZA, Fuster V, Lippmann M, Chen LC, Rajagopalan S. Long-term air pollution exposure and acceleration of atherosclerosis and vascular inflammation in an animal model. JAMA. 2005;294:3003–3010. doi: 10.1001/jama.294.23.3003. [DOI] [PubMed] [Google Scholar]
- 6.Chen LC, Nadziejko C. Effects of subchronic exposures to concentrated ambient particles (CAPs) in mice. V. CAPs exacerbate aortic plaque development in hyperlipidemic mice. Inhal Toxicol. 2005;17:217–224. doi: 10.1080/08958370590912815. [DOI] [PubMed] [Google Scholar]
- 7.Künzli N, Jerrett M, Mack WJ, Beckerman B, LaBree L, Gilliland F, Thomas D, Peters J, Hodis HN. Ambient air pollution and atherosclerosis in Los Angeles. Environ Health Perspect. 2005;113:201–206. doi: 10.1289/ehp.7523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoffmann B, Moebus S, Mohlenkamp S, Stang A, Lehmann N, Dragano N, Schmermund A, Memmesheimer M, Mann K, Erbel R, Jockel KH. Residential exposure to traffic is associated with coronary atherosclerosis. Circulation. 2007;116:489–496. doi: 10.1161/CIRCULATIONAHA.107.693622. [DOI] [PubMed] [Google Scholar]
- 9.Peters A, Dockery DW, Muller JE, Mittleman MA. Increased particulate air pollution and the triggering of myocardial infarction. Circulation. 2001;103:2810–2815. doi: 10.1161/01.cir.103.23.2810. [DOI] [PubMed] [Google Scholar]
- 10.Gong KW, Zhao W, Li N, Barajas B, Kleinman M, Sioutas C, Horvath S, Lusis AJ, Nel A, Araujo JA. Air-pollutant chemicals and oxidized lipids exhibit genome-wide synergistic effects on endothelial cells. Genome Biol. 2007;8:R149. doi: 10.1186/gb-2007-8-7-r149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li N, Alam J, Venkatesan MI, Eiguren-Fernandez A, Schmitz D, Di Stefano E, Slaughter N, Killeen E, Wang X, Huang A, Wang M, Miguel AH, Cho A, Sioutas C, Nel AE. Nrf2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals. J Immunol. 2004;173:3467–3481. doi: 10.4049/jimmunol.173.5.3467. [DOI] [PubMed] [Google Scholar]
- 12.Hiura TS, Kaszubowski MP, Li N, Nel AE. Chemicals in diesel exhaust particles generate reactive oxygen radicals and induce apoptosis in macrophages. J Immunol. 1999;163:5582–5591. [PubMed] [Google Scholar]
- 13.Li N, Sioutas C, Cho A, Schmitz D, Misra C, Sempf J, Wang M, Oberley T, Froines J, Nel A. Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environ Health Perspect. 2003;111:455–460. doi: 10.1289/ehp.6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harkema JR, Keeler G, Wagner J, Morishita M, Timm E, Hotchkiss J, Marsik F, Dvonch T, Kaminski N, Barr E. Effects of concentrated ambient particles on normal and hypersecretory airways in rats. Res Rep Health Eff Inst. 2004:1–68. [PubMed] [Google Scholar]
- 15.Brown MG, Moss OR. An inhalation exposure chamber designed for animal handling. Lab Anim Sci. 1981;31:717–720. [PubMed] [Google Scholar]
- 16.Kim S, Jaques PA, Chang MC, Barone T, Xiong C, Friedlander SK, Sioutas C. Versatile aerosol concentration enrichment system (VACES) for simultaneous in vivo and in vitro evaluation of toxic effects of ultrafine, fine and coarse ambient particles. Part II: field evaluation. J Aerosol Sci. 2001;32:1299–1314. [Google Scholar]
- 17.Kim S, Jaques PA, Chang MC, Froines JR, Sioutas C. Versatile aerosol concentration enrichment system (VACES) for simultaneous in vivo and in vitro evaluation of toxic effects of ultrafine, fine and coarse ambient particles. Part I: development and laboratory characterization. J Aerosol Sci. 2001;32:1281–1297. [Google Scholar]
- 18.Kleinman MT, Hamade A, Meacher D, Oldham M, Sioutas C, Chakrabarti B, Stram D, Froines JR, Cho AK. Inhalation of concentrated ambient particulate matter near a heavily trafficked road stimulates antigen-induced airway responses in mice. J Air Waste Manag Assoc. 2005;55:1277–1288. doi: 10.1080/10473289.2005.10464727. [DOI] [PubMed] [Google Scholar]
- 19.Oldham MJ, Phalen RF, Robinson RJ, Kleinman MT. Performance of a portable whole-body mouse exposure system. Inhal Toxicol. 2004;16:657–662. doi: 10.1080/08958370490464670. [DOI] [PubMed] [Google Scholar]
- 20.Li N, Nel AE. Role of the Nrf2-mediated signaling pathway as a negative regulator of inflammation: implications for the impact of particulate pollutants on asthma. Antioxid Redox Signal. 2006;8:88–98. doi: 10.1089/ars.2006.8.88. [DOI] [PubMed] [Google Scholar]
- 21.Nel A, Xia T, Madler L, Li N. Toxic potential of materials at the nanolevel. Science. 2006;311:622–627. doi: 10.1126/science.1114397. [DOI] [PubMed] [Google Scholar]
- 22.Nel AE, Diaz-Sanchez D, Ng D, Hiura T, Saxon A. Enhancement of allergic inflammation by the interaction between diesel exhaust particles and the immune system. J Allergy Clin Immunol. 1998;102:539–554. doi: 10.1016/s0091-6749(98)70269-6. [DOI] [PubMed] [Google Scholar]
- 23.Gius D, Botero A, Shah S, Curry HA. Intracellular oxidation/reduction status in the regulation of transcription factors NF-kappaB and AP-1. Toxicol Lett. 1999;106:93–106. doi: 10.1016/s0378-4274(99)00024-7. [DOI] [PubMed] [Google Scholar]
- 24.Xiao GG, Wang M, Li N, Loo JA, Nel AE. Use of proteomics to demonstrate a hierarchical oxidative stress response to diesel exhaust particle chemicals in a macrophage cell line. J Biol Chem. 2003;278:50781–50790. doi: 10.1074/jbc.M306423200. [DOI] [PubMed] [Google Scholar]
- 25.Nemmar A, Hoet PH, Vanquickenborne B, Dinsdale D, Thomeer M, Hoylaerts MF, Vanbilloen H, Mortelmans L, Nemery B. Passage of inhaled particles into the blood circulation in humans. Circulation. 2002;105:411–414. doi: 10.1161/hc0402.104118. [DOI] [PubMed] [Google Scholar]
- 26.Nemmar A, Vanbilloen H, Hoylaerts MF, Hoet PH, Verbruggen A, Nemery B. Passage of intratracheally instilled ultrafine particles from the lung into the systemic circulation in hamster. Am J Respir Crit Care Med. 2001;164:1665–1668. doi: 10.1164/ajrccm.164.9.2101036. [DOI] [PubMed] [Google Scholar]
- 27.Xia T, Korge P, Weiss JN, Li N, Venkatesen MI, Sioutas C, Nel A. Quinones and aromatic chemical compounds in particulate matter induce mitochondrial dysfunction: implications for ultrafine particle toxicity. Environ Health Perspect. 2004;112:1347–1358. doi: 10.1289/ehp.7167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Erdelmeier I, Gerard-Monnier D, Yadan JC, Chaudiere J. Reactions of N-methyl-2-phenylindole with malondialdehyde and 4-hydroxyalkenals. Mechanistic aspects of the colorimetric assay of lipid peroxidation. Chem Res Toxicol. 1998;11:1184–1194. doi: 10.1021/tx970180z. [DOI] [PubMed] [Google Scholar]
- 29.Janero DR. Malondialdehyde and thiobarbituric acid-reactivity as diagnostic indices of lipid peroxidation and peroxidative tissue injury. Free Radic Biol Med. 1990;9:515–540. doi: 10.1016/0891-5849(90)90131-2. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, Zanotti I, Reilly MP, Glick JM, Rothblat GH, Rader DJ. Overexpression of apolipoprotein A-I promotes reverse transport of cholesterol from macrophages to feces in vivo. Circulation. 2003;108:661–663. doi: 10.1161/01.CIR.0000086981.09834.E0. [DOI] [PubMed] [Google Scholar]
- 31.Barter PJ, Nicholls S, Rye KA, Anantharamaiah GM, Navab M, Fogelman AM. Antiinflammatory properties of HDL. Circ Res. 2004;95:764–772. doi: 10.1161/01.RES.0000146094.59640.13. [DOI] [PubMed] [Google Scholar]
- 32.Gargalovic PS, Gharavi NM, Clark MJ, Pagnon J, Yang WP, He A, Truong A, Baruch-Oren T, Berliner JA, Kirchgessner TG, Lusis AJ. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:2490–2496. doi: 10.1161/01.ATV.0000242903.41158.a1. [DOI] [PubMed] [Google Scholar]
- 33.Jung EJ, Avliyakulov NK, Boontheung P, Loo JA, Nel AE. Pro-oxidative DEP chemicals induce heat shock proteins and an unfolding protein response in a bronchial epithelial cell line as determined by DIGE analysis. Proteomics. 2007;7:3906–3918. doi: 10.1002/pmic.200700377. [DOI] [PubMed] [Google Scholar]
- 34.Erguder IB, Erguder T, Ozkan C, Bozkurt N, Soylu K, Devrim E, Durak I. Short-term effects of smoking cessation on blood antioxidant parameters and paraoxonase activity in healthy asymptomatic long-term cigarette smokers. Inhal Toxicol. 2006;18:575–579. doi: 10.1080/08958370600686325. [DOI] [PubMed] [Google Scholar]
- 35.Navab M, Hama SY, Hough GP, Subbanagounder G, Reddy ST, Fogelman AM. A cell-free assay for detecting HDL that is dysfunctional in preventing the formation of or inactivating oxidized phospholipids. J Lipid Res. 2001;42:1308–1317. [PubMed] [Google Scholar]
- 36.Gur M, Aslan M, Yildiz A, Demirbag R, Yilmaz R, Selek S, Erel O, Ozdogru I. Paraoxonase and arylesterase activities in coronary artery disease. Eur J Clin Invest. 2006;36:779–787. doi: 10.1111/j.1365-2362.2006.01727.x. [DOI] [PubMed] [Google Scholar]
- 37.Westerdahl D, Fruin S, Sax T, Fine PM, Sioutas C. Mobile platform measurements of ultrafine particles and associated pollutant concentrations on freeways and residential streets in Los Angeles. Atmos Environ. 2005;39:3597–3610. [Google Scholar]
- 38.Pope CA, 3rd, Burnett RT, Thun MJ, Calle EE, Krewski D, Ito K, Thurston GD. Lung cancer, cardiopulmonary mortality, and long-term exposure to fine particulate air pollution. JAMA. 2002;287:1132–1141. doi: 10.1001/jama.287.9.1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Daniels MJ, Dominici F, Samet JM, Zeger SL. Estimating particulate matter-mortality dose-response curves and threshold levels: an analysis of daily time-series for the 20 largest US cities. Am J Epidemiol. 2000;152:397–406. doi: 10.1093/aje/152.5.397. [DOI] [PubMed] [Google Scholar]
- 40.Dominici F, McDermott A, Daniels D. Special Report: Revised Analyses of Time-Series Studies of Air Pollution and Health. Boston, Mass: Health Effects Institute; 2003. Mortality among residents of 90 cities. [Google Scholar]
- 41.Samet JM, Dominici F, Curriero FC, Coursac I, Zeger SL. Fine particulate air pollution and mortality in 20 U.S. cities, 1987–1994. N Engl J Med. 2000;343:1742–1749. doi: 10.1056/NEJM200012143432401. [DOI] [PubMed] [Google Scholar]
- 42.Pope CA, 3rd, Burnett RT, Thurston GD, Thun MJ, Calle EE, Krewski D, Godleski JJ. Cardiovascular mortality and long-term exposure to particulate air pollution: epidemiological evidence of general pathophysiological pathways of disease. Circulation. 2004;109:71–77. doi: 10.1161/01.CIR.0000108927.80044.7F. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.