

Abstract
AIMS
The role of CYP pharmacogenetics in the bioactivation of cyclophosphamide is still controversial. Recent clinical studies have suggested a role for either CYP2C19 or CYP2B6. The aim of this study was to clarify the role of these pharmacogenes.
METHODS
We used a combined in vitro–in vivo approach to determine the role of these pharmacogenes in the bioactivation of the prodrug to 4-hydroxy cyclophosphamide (4-OHCP). Cyclophosphamide metabolism was determined in a human liver biobank (n = 14) and in patients receiving the drug for treatment of lupus nephritis (n = 16)
RESULTS
In livers of known CYP2C19 and CYP2B6 genotype and protein expression we observed that there was a combined role for both CYP2C19 and CYP2B6 in the bioactivation of cyclophosphamide in vitro. The presence of at least one loss of function (LoF) allele at either CYP2C19 or CYP2B6 resulted in a significant decrease in both Vmax (P = 0.028) and CLint (P = 0.0017) compared with livers with no LoF alleles. This dual genotype relationship was also observed in a preliminary clinical study, with patients who had ≥1 LoF allele at either CYP2C19 or CYP2B6 also displaying significantly (P = 0.0316) lower bioactivation of cyclophosphamide. The mean 4-OHCP : CP bioactivation ratio was 0.0014 (95% CI 0.0007, 0.002) compared with 0.0071 (95% CI 0.0001, 0.014) in patients with no LoF alleles at either of these genes.
CONCLUSIONS
The presence of ≥1 LoF allele(s) at either CYP2B6 or CYP2C19 appeared to result in decreased bioactivation of cyclophosphamide both in vitro and in patients. Further clinical studies to confirm this relationship are warranted.
Keywords: CYP2C19, CYP2B6, cyclophosphamide, lupus
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
The prodrug cyclophosphamide requires bioactivation by liver CYP enzymes.
Controversy exists about which CYP isoforms are important in the in vitro bioactivation of this drug.
Recent clinical studies have highlighted a role for either CYP2C19 or CYP2B6 in the therapeutic response to cyclophosphamide in lupus patients.
However, the role of these isoforms in the bioactivation of cyclophosphamide in lupus patients has not been previously demonstrated.
WHAT THIS STUDY ADDS
Low bioactivation of cyclophosphamide by human liver appears to be dependent on a combination of both CYP2C19 and CYP2B6 loss of function variants.
In a preliminary study of lupus patients poor bioactivation of cyclophosphamide was also observed in those individuals who had at least one loss of function allele at either CYP2C19 or CYP2B6.
Introduction
Cyclophosphamide (CP) is a cytotoxic prodrug that requires hepatic bioactivation via 4-hydroxy cyclophosphamide (4-OH CP), to the ultimate cytotoxin, phosphoramide mustard [1]. It has long been recognized that the initial hydroxylation step is catalysed by cytochrome P450 (CYP) enzymes, whilst the subsequent conversion of 4-OH CP to phosphoramide mustard is thought to be non-enzymatic.
Many CYP isozymes have been shown to catalyse the conversion of cyclophosphamide to 4-OH CP, including: CYP2B6 [2–5], CYP2C19 [6, 7], CYP2C9 and CYP3A4/5[8]. Many of these CYP isozymes have genetic variants which result in either loss of function (LoF) - CYP2C19*2, CYP2C19*3, CYP2B6*5[9, 10] or increased activity - CYP2C19*17[11, 12] and CYP2B6*6[3, 13] although CYP2B6*6 is also reported to result in lower expression levels [10]. Considerable inter-individual variation in 4-OH CP plasma concentrations has also been demonstrated but the role of pharmacogenetics is still controversial and which CYP genes are important has not been clearly resolved.
Preliminary in vitro evidence [7] indicated a possible role for CYP2C19 in the variable bioactivation of cyclophosphamide whilst a role for polymorphic CYP2B6 in the 4-hydroxylation of cyclophosphamide has also been demonstrated [3]. However, both these studies only measured bioactivation at limited (0.25 and 1 mm) cyclophosphamide concentrations rather than determining intrinsic clearance or catalytic efficiency via this route.
A number of recent studies have also tried to identify the pharmacogenes associated with this clinical variability. Some studies have suggested a role for CYP2B6[13, 14], while other studies have reported a role for variant CYP2C19 alleles in the decreased clearance of cyclophosphamide [15, 16]. In addition, a lack of therapeutic response in patients homozygous for CYP2B6*5 or CYP2C19*2 has been reported in lupus nephritis patients [17]. A lower risk of premature ovarian failure in those heterozygous or homozygous for CYP2C19*2 was also seen and this finding was confirmed by Singh et al. [18]. None of these clinical studies found any association with CYP3A4/5, CYP2C9, GST or ALDH.
Our aim was to clarify the role of CYP2B6 and CYP2C19 pharmacogenetics in the variable bioactivation of cyclophosphamide. We investigated the enzyme kinetics of 4-OH CP formation in a human liver biobank that had been fully characterized for CYP2C19 and CYP2B6 genotype and protein expression. In addition, we also investigated the relationship between cyclophosphamide 4-hydroxylation and CYP2C19 and CYP2B6 genotype in patients receiving the drug for treatment of lupus nephritis.
Methods
Cyclophosphamide bioactivation in vitro
Human liver microsomes were prepared from 14 donors (five male and nine female donors aged 29–70 years, details reported previously [19]. The livers were genotyped for CYP2C19 plus CYP2B6 and protein expression determined (see below). CYP2B6 and CYP2C19 supersomes were obtained from BD Biosciences (New Jersey, USA). Microsomes (0.5 mg) or supersomes (2 pmol CYP 450) were incubated with cyclophosphamide (0.25–30 mm) and 10 mm NADPH in 67 mm phosphate buffer (pH 7.4) in triplicate for 10 min at 37°C. The reaction was terminated with ice-cold acetonitrile (250 µl) and the samples centrifuged (13 000 g for 5 min). The supernatant was removed and 4-OH CP derivatized as described previously [20]. After centrifugation (13 000 g for 5 min) the supernatant was diluted with 10 mm sodium phosphate buffer (pH 3.5, 350 µl) and analysed by HPLC.
HPLC analysis was carried out using an Agilent 1200 system with fluorescence detection on a Zorbax Extend C18 5 µm (4.6 × 150 mm) column (Agilent technologies, USA). The mobile phase consisted of 10 mm sodium phosphate buffer (pH 3.5) : acetonitrile (66:34) at a flow rate of 2 ml min−1. Fluorescence detection was at excitation 350 nm and emission 550 nm (PMT 12, gate 0.03 ms) and the limit of quantification was 0.2 µm with a coefficient of variation of <10% [20]. Quantification of 4-OH CP was determined using a calibration curve of 4-OH CP (10–100 µm) prepared fresh by incubation of 4-hydroperoxy cyclophosphamide (kind gift from Dr P. Kestell, University of Auckland) with sodium thiosulphate (25 mm) as described previously [21]. Non-linear regression analysis of the rate of 4-OH CP formation (nmol min−1 mg−1) was determined and the parameters of the Michaelis-Menten equation were fitted to the data using GraphPad Prism version 5.02.
Cyclophosphamide bioactivation in vivo
Ethics approval for the clinical study was obtained from the New Zealand Health and Disability Northern X Regional Ethics Committee. Following written informed consent, sixteen patients with lupus nephritis (systemic lupus erythematosus with renal disease) were recruited and received a 1 h intravenous infusion of cyclophosphamide with doses ranging from 0.29–1.16 g m−2. A blood sample was taken 15 min after completion of the infusion, immediately centrifuged (13 000 g for 5 min), with the plasma sample divided into two and processed as follows. Plasma (1 ml) was transferred to a tube containing 1 ml ice cold acetonitrile, vortex mixed and centrifuged (13 000 g for 5 min). The deproteinized supernatant was then removed and transferred to a fresh tube and immediately analysed for 4-OH CP as described above. The remainder of the plasma sample was stored at −80°C until analysis for CP. Briefly, plasma (0.4 ml) was spiked with ifosfamide as internal standard and diluted to 2 ml with Milli Q water. Following solid phase extraction (C18 cartridges, Alltech associates, USA) the eluate was evaporated to dryness and the residue reconstituted in 100 µl mobile phase prior to LC/MS analysis as previously described [22]. The limit of quantification was 0.1 µm with a coefficient of variation of <10%.The bioactivation ratio was calculated as the ratio of 4-OH CP concentration divided by the cyclophosphamide concentration (4-OHCP : CP) at 15 min post-infusion which is approximately tmax and is directly related to the ratio of total drug and metabolite exposure (AUC) [14].
A further blood sample (8.5 ml) was collected into PAXgene™ blood tubes (Qiagen, Hilden, Germany) for CYP2C19 and CYP2B6 genotyping.
CYP2C19 and CYP2B6 genotyping
Genomic DNA was extracted from human liver tissue using a DNeasy tissue kit or from whole blood using a PAXgene™ blood DNA kit according to manufacturer's instructions (Qiagen, Hilden, Germany). The CYP2C19 genotype of individual human livers was determined by PCR-RFLP analysis of the two major LoF allelic variants (CYP2C19*2 and CYP2C19*3) and the gain of function variant (CYP2C19*17) using previously published methods [9, 11]. CYP2B6 genotype was determined by PCR-RFLP analysis of the G516T, A785G and C1459T SNPs which allows determination of the, *4 (A785G), *5 (C1459T), *6 (G516T, A785G), *7 (G516T, A785G and C1459T) and *9 (G516T) alleles as described previously [23].
CYP2C19 and CYP2B6 protein expression
Expression of CYP2C19 and CYP2B6 protein in human liver microsomes was determined by immunoblotting using standard techniques and commercially available primary antibodies (BD Biosciences, New Jersey, USA and Abcam Inc., Cambridge, UK, respectively). Immunoreactive proteins were visualized by enhanced chemiluminescence detection (Pierce Biotechnology, Illinois, USA) and the relative intensity of the signal analysed with the Scion imaging analysis program (Scion Corporation, Maryland, USA).
Statistical analysis
The relationship between CYP protein expression and cyclophosphamide bioactivation was analysed using Spearman rank order correlation. Student's t-test (two-tailed) was used to determine the statistical difference between groups using GraphPad Prism version 5.02. Values of P < 0.05 were considered to be statistically significant. Patients on long-term treatment with known inducers of CYP were excluded from the analysis between genotype and bioactivation. Data are expressed where appropriate as the mean and 95% confidence interval (95% CI).
Results
Cyclophosphamide bioactivation in vitro
Variability in the level of CP bioactivation was observed across the human liver biobank (n = 14), with examples of livers having low, intermediate and high activity (Figure 1). The kinetics of 4-OH CP formation were fitted with a single enzyme Michaelis-Menten model (confirmed by linear Eadie-Hofstee plots) and the parameters, Km and Vmax, were used to calculate the intrinsic clearance (CLint) of cyclophosphamide (Table 1). The rate of 4-OH CP metabolite formation (Vmax) ranged from 0.72 to 13.37 (mean 4.54, 95% CI 2.28, 6.81) nmol min−1 mg−1. The intrinsic clearance of cyclophosphamide via 4-hydroxylation varied more than 13-fold with a range of values between 0.12 and 1.65 µl min−1 mg−1 (mean 0.74, 95% CI 0.48, 1.00).
Figure 1.
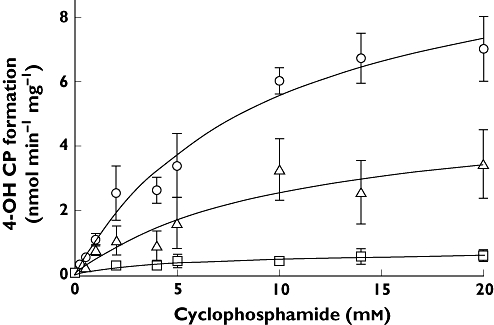
Formation of 4-hydroxy cyclophosphamide (4-OH CP) from cyclophosphamide (0.25–20 mm) by human liver microsomes in vitro. Examples of livers with high (circles), intermediate (triangles) and low (squares) metabolic capacity are shown. Data shown as the mean ± SD of triplicate determinations
Table 1.
The relationship between CYP2C19 and CYP2B6 genotype and the in vitro bioactivation (rate Vmax, affinity Km and intrinsic clearance: CLint) of cyclophosphamide by human liver
| Human liver | CYP2C19 | CYP2B6 | Vmax (nmol min−1 mg−1) | Km (mm) | CLint (µl min−1 mg−1) |
|---|---|---|---|---|---|
| 5 | *1/*1† | *1/*5 | 0.72 (0.43, 0.99) | 5.8 (0.06, 11.5) | 0.12 |
| 18 | *1/*2 | *1/*5 | 1.69 (1.25, 2.12) | 6.0 (2.15, 9.88) | 0.28 |
| 11 | *17/*17 | *4/*5 | 1.08 (0.83, 1.33) | 2.7 (0.67, 4.76) | 0.39 |
| 15 | *1/*1 | *5/*6 | 3.09 (2.29, 3.88) | 7.7 (3.15, 12.25) | 0.4 |
| 8 | *1/*2 | *1/*6 | 5.11 (2.38, 7.85) | 10.2 (0.0, 21.55) | 0.5 |
| 12 | *2/*2 | *1/*1 | 1.99 (1.58, 2.45) | 3.9 (1.24, 6.66) | 0.5 |
| 6 | *1/*1 | *1/*1 | 4.09 (2.66, 5.53) | 7.6 (0.15, 14.9) | 0.54 |
| 7 | *1/*1 | *1/*5 | 2.64 (1.78, 3.49) | 3.9 (0.22, 7.55) | 0.68 |
| 14 | *1/*1 | *1/*1 | 2.47 (1.54, 3.39) | 3.3 (0.0, 7.15) | 0.75 |
| 13 | *1/*17 | *1/*1 | 13.37 (0.24, 26.5) | 17.2 (0.0, 51.6) | 0.78 |
| 16 | *1/*1 | *1/*1 | 10.89 (8.13, 13.47) | 9.4 (4.46, 14.4) | 1.15 |
| 9 | *1/*17 | *1/*6 | 9.34 (6.16, 12.5) | 7.4 (0.32, 14.6) | 1.25 |
| 4 | *1/*1 | *1/*6 | 2.01 (1.58, 2.44) | 1.4 (0.0, 2.93) | 1.39 |
| 10 | *1/*1 | *1/*1 | 5.15 (3.89, 6.4) | 3.1 (0.22, 6.02) | 1.65 |
Data are shown as mean (95% CI). †Human liver 5 has null CYP2C19 protein expression and null CYP2C19 activity (as determined using both proguanil and omeprazole as substrates).
Two livers were heterozygous variant for *2 and one liver was homozygous variant (*2/*2). No CYP2C19*3 variant alleles were detected (Table 1). The liver bank was also genotyped for the CYP2C19*17 variant which is associated with an ultra-rapid metabolizer phenotype. The incidence of this allelic variant in the present study was 0.14, with two livers heterozygote *17 carriers and one liver (HL11) homozygous for this gain of function allele. Hence the frequency of the CYP2C19*1, *2 and *17 alleles was 0.71, 0.14 and 0.14, respectively.
The CYP2B6 genotype of the livers was also determined (Table 1). Six livers were homozygous wild type (*1/*1). Three livers were *1/*5 and three livers were *1/*6. One liver had the *4/*5 genotype and one liver was*5/*6. The frequency of the CYP2B6*1, *5 and *6 alleles was 0.64, 0.18 and 0.14, respectively.
Variable expression was observed for both CYP2C19 and CYP2B6 protein with a >23-fold variation and 52-fold variation for CYP2C19 and CYP2B6, respectively (Figure 2A,B). The expression of CYP2C19 protein was related to LoF genotype as no detectable CYP2C19 protein expression was observed for the homozygote variant (*2/*2) liver (HL 12). One liver (HL 5) also had no detectable immunoreactive protein and the lack of CYP2C19 protein expression in this liver was confirmed by null functional CYP2C19 activity as determined using both proguanil and omeprazole as substrates (data not shown). This liver was classified as homozygous wild-type but null protein may also be due to the presence of an unidentified variant allele.
Figure 2.
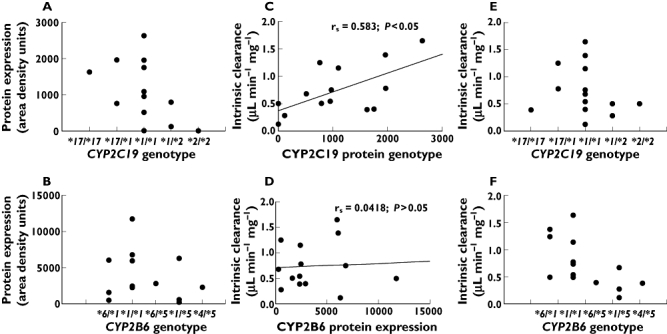
The relationship between CYP2C19 and CYP2B6 genotype, protein expression and cyclophosphamide bioactivation (intrinsic clearance) in the Auckland human liver bank. Spearman rank order correlation (rs) of protein expression vs. intrinsic clearance is shown (C) and (D)
There was no simple relationship between the CYP2B6 protein expression and CYP2B6 variant alleles (Figure 2B). Protein expression in the wild-type livers was 5729.33 (95% CI 1355, 9203) area density units (range 2316–11749). Livers heterozygous for the LoF CYP2B6*5 allele had relatively low, but highly variable, protein expression (range 225–6289 area density units). Of the three livers which were *1/*6 genotype, two had relatively low CYP2B6 expression (487 and 1616 area density units). No livers had null CYP2B6 protein expression.
There was no correlation between CYP2B6 protein concentrations and cyclophosphamide bioactivation (rs = 0.042, P = 0.88, Figure 2D), but there was a relationship between CYP2B6*5 genotype and decreased cyclophosphamide bioactivation in vitro (Table 1). The presence of one variant CYP2B6*5 allele resulted in a significantly decreased CLint compared with livers that were homozygous wild-type CYP2B6*1/*1 (P = 0.037, t = 2.44, d.f. = 9) (Figure 2F). In contrast to CYP2B6, there was a statistically significant correlation between CYP2C19 protein expression and cyclophosphamide CLint (rs = 0.58, P = 0.03, Figure 2C), also indicating a clear role for CYP2C19 protein in the bioactivation of cyclophosphamide in vitro. Both Vmax and CLint were lower in livers (Table 1) with one (CYP2C19*1/*2) or two (CYP2C19*2/*2) variant alleles (mean Vmax = 2.93, 95% CI 1.77, 7.63 nmol min−1 mg−1 and mean CLint = 0.43, 95% CI 0.11, 0.74 µl min−1 mg−1), compared with CYP2C19*1/*1 wild-type livers (mean Vmax = 4.98, 95% CI 2.11, 7.86 nmol min−1 mg−1 and CLint = 0.82, 95% CI 0.51, 1.14 µL min−1 mg−1). Nonetheless, the two livers with no detectable CYP2C19 protein expression (i.e. HL12, CYP2C19*2/*2 and HL5, CYP2C19*1/*1) had detectable 4-hydroxylase activity (Table 1).
Using supersomes®, CYP2B6 had a high catalytic efficiency (Vmax = 47.8, 95% CI 38.0, 57.7) nmol min−1 nmol−1 P450, Km = 0.84, 95% CI 0.24, 1.45 mm) for cyclophosphamide bioactivation. CYP2C19 also hydroxylated cyclophosphamide (Vmax = 4.8, 95% CI 4.02, 5.6 nmol min−1 nmol−1 P450) with a similar Km (Km = 0.96, 95% CI 0.54, 1.41 mm) for the substrate.
Examining the CYP2B6 and CYP2C19 genotypes together, it can be seen that combined genotype is important for the intrinsic clearance in vitro (Figure 3). The presence in either CYP2C19 or CYP2B6 of at least one LoF variant allele (i.e. CYP2C19*2 or CYP2B6*5) resulted in a significant decrease in both Vmax (P = 0.0276, t = 2.50, d.f. 12) and CLint (P = 0.0017, t = 4.03, d.f. 12, Figure 4A) compared with livers which were homozygous wild-type or heterozygous for CYP2C19*17 or CYP2B6*6. No livers were homozygous LoF variant at both loci (e.g. CYP2C19*2/*2 and CYP2B6*5/*5). HL5 which was heterozygous for CYP2B6*5 also had no detectable CYP2C19 protein expression, despite being CYP2C19*1/*1 genotype. Overall this liver had the lowest CLint (0.124 µl min−1 mg−1) of the livers studied (Figure 3). From these data it would be predicted that only a liver that was homozygous variant at both genes would be a null cyclophosphamide bioactivator in vitro.
Figure 3.
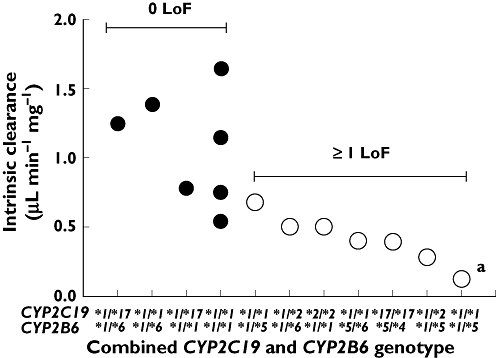
The role of the combined CYP2C19 and CYP2B6 genotype in the bioactivation of cyclophosphamide in vitro by human liver. aThis liver has no functional CYP2C19 activity
Figure 4.
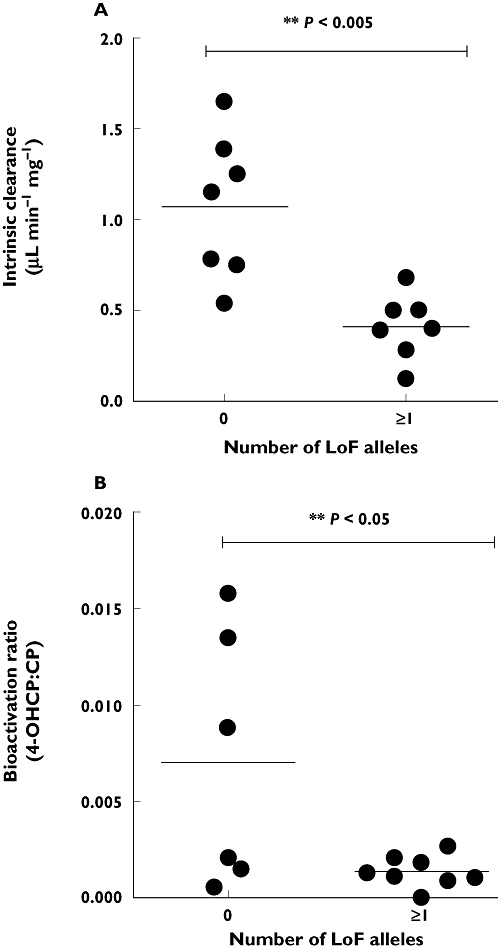
The relationship between decreased bioactivation of cyclophosphamide (A) in vitro and (B) in vivo and the presence of at least one (≥1) loss of function (LoF) allele at either CYP2C19 and CYP2B6. A) Presence of one or more LoF alleles at either CYP2C19 or CYP2B6 results in a significant (P = 0.0017, t = 4.03, d.f. 12) decrease in intrinsic clearance compared with livers that have zero LoF alleles at these genes. (B) Presence of one or more LoF alleles at either CYP2C19 or CYP2B6 results in a significant (P = 0.0316, t = 2.43, d.f. 12) decrease in bioactivation ratio compared with patients that have zero LoF alleles at these genes
Cyclophosphamide bioactivation in vivo
The mean cyclophosphamide dose in the sixteen patients with lupus nephritis was 0.63 g m−2, 95% CI 0.50, 0.76 (Table 2). There was a greater than 27-fold variability in the bioactivation to 4-OH CP across 16 patients (4-OH CP : cyclophosphamide range <0.0005–0.0164, mean 0.0048, 95% CI 0.002, 0.008, Table 3). One patient (#1) had no detectable 4-OH CP and was therefore classified as a null bioactivator with a ratio < 0.0005, however, this patient had only one LoF allele (CYP2C19*3). Furthermore, this patient was not on any co-medication known to influence CYP2C19 or CYP2B6 activity (Table 2). Therefore it remains unclear whether or not this patient was a phenotypic CYP2C19 poor metabolizer, similar to liver HL5.
Table 2.
Patient demographics
| Patient ID | Dose* (g m−2) | Cycle* | Concomitant treatment for lupus | Co-medications that may be inducers or inhibitors of CYP2C19 or CYP2B6 | Ethnicity | Gender | Age (years) |
|---|---|---|---|---|---|---|---|
| 1 | 0.5 | 2 | Prednisone, HCQ | – | Fijian European | F | 26 |
| 2 | 1.16 | 6 | Prednisone, HCQ | Cotrimoxazole | Samoan | F | 30 |
| 3 | 0.44 | 1 | Prednisone, HCQ | Omeprazole | Cook Island Maori | F | 20 |
| 4 | 0.75 | 2 | Prednisone, HCQ | Cotrimoxazole | Tongan | F | 37 |
| 5 | 0.424 | 1 | Prednisone, HCQ | Omeprazole, cotrimoxazole | Cambodian | F | 60 |
| 6 | 0.698 | 2 | Prednisone | Omeprazole, cotrimoxazole | Tongan | F | 33 |
| 7 | 0.52 | 7 | Prednisone, HCQ | – | Samoan | M | 66 |
| 8 | 0.45 | 2 | Prednisone, HCQ | Omeprazole, cotrimoxazole | Chinese | F | 58 |
| 9 | 0.546 | 1 | Prednisone, HCQ | Ondansetron | Cambodian | F | 45 |
| 10 | 0.291 | 1 | Prednisone, HCQ | Cotrimoxazole | Samoan | F | 27 |
| 11 | 0.5 | 2 | Prednisone, HCQ | Omeprazole, diclofenac | NZ European | F | 33 |
| 12 | 0.75 | 5 | Prednisone | Thyroxine | NZ European | M | 60 |
| 13 | 0.949 | 9 | Prednisone, HCQ | Omeprazole, fluoxetine, cotrimoxazole | Laotian | F | 30 |
| 14 | 0.38 | 10 | Prednisone, HCQ | Citalopram, omeprazole, cotrimoxazole | Tongan | F | 31 |
| 15 | 0.75 | 1 | Prednisone, HCQ | Omeprazole | Samoan | F | 42 |
| 16 | 1 | 5 | Prednisone | Carbamazepine | Chinese | F | 35 |
HCQ hydroxychloroquine.
Dose and cycle at which the bioactivation ratio was tested.
Table 3.
The relationship between CYP2C19 and CYP2B6 genotype and the bioactivation of cyclophosphamide in lupus nephritis patients
| Patient ID | CYP2C19 | CYP2B6 | 4-OH CP† (µg ml−1) | CP‡ (µg ml−1) | Bioactivation ratio (4-OHCP : CP) |
|---|---|---|---|---|---|
| 1 | *1/*3 | *1/*6 | <0.06 | 111.4 | <0.0005 |
| 2 | *1/*1 | *6/*6 | 0.09 | 153 | 0.0006 |
| 3 | *1/*2 | *1/*6 | 0.06 | 65.2 | 0.0009 |
| 4 | *1/*1 | *5/*6 | 0.11 | 98.6 | 0.0011 |
| 5 | *1/*2 | *1/*6 | 0.08 | 75.5 | 0.0011 |
| 6 | *1/*2 | *1/*6 | 0.16 | 121.4 | 0.0013 |
| 7 | *1/*1 | *1/*6 | 0.07 | 47.5 | 0.0015 |
| 8 | *1/*2 | *4/*4 | 0.15 | 78.8 | 0.0019 |
| 9 | *1/*1 | *6/*6 | 0.17 | 81.7 | 0.0021 |
| 10 | *1/*2 | *1/*1 | 0.11 | 52.4 | 0.0021 |
| 11 | *2/*2 | *1/*6 | 0.18 | 66.5 | 0.0027 |
| 12 | *1/*2 | *1/*5 | 0.66 | 95.9 | 0.0069§ |
| 13 | *1/*1 | *1/*6 | 0.54 | 61.7 | 0.0088 |
| 14 | *1/*1 | *1/*6 | 0.28 | 20.8 | 0.0135 |
| 15 | *1/*1 | *1/*6 | 0.54 | 34.1 | 0.0158 |
| 16 | *1/*3 | *1/*6 | 0.8 | 48.7 | 0.0164§ |
LOQ was 0.06 µg ml−1.
LOQ was 0.03 µg ml−1.
Patients 16 and 12 were treated as outliers due to prolonged treatment with carbamazepine and thyroxine, respectively.
Six patients were heterozygous for CYP2C19*2 and one patient was homozygous for this variant (Table 3). In addition, two patients were heterozygous for the CYP2C19*3 allele. Hence the overall frequency of CYP2C19 LoF alleles was 0.31. This relatively high frequency compared with the liver bank, which are from Caucasian donors, may be due to the mixed ethnicity of the patient population which included five Asian and nine Polynesian subjects (Table 2). No patients had CYP2C19*17 variant alleles. Two patients were heterozygous for CYP2B6*5 and one patient was homozygous for CYP2B6*4. The frequency of the CYP2B6*6 variant was 0.47, with two patients homozygous for this allele.
The CYP2C19*2*2 patient (#11) did not display null bioactivation (ratio = 0.0027). Two patients (#16 and #12) who were heterozygous variant for CYP2C19 had been on long-term treatment with carbamazepine or thyroxine (Table 2). Both these drugs are inducers of CYP and these patients were excluded from the analysis. However, the other six patients who were heterozygous for the CYP2C19 LoF alleles had lower, although not significant (P = 0.0758, t = 1.94.15, d.f. 12), bioactivation compared with those patients who were wild-type at this locus (mean ratio = 0.0012, 95% CI 0.0004, 0.002 vs. 0.0062, 95% CI 0.0002, 0.012).
There was a significantly lower bioactivation (P = 0.0316, t = 2.432, d.f. 12) in patients with at least one LoF variant allele at either CYP2B6 or CYP2C19 compared with patients who did not have a CYP2C19*2, CYP2C19*3, or CYP2B6*5 variant allele (mean ratio = 0.0014, 95% CI 0.0007, 0.002 vs. 0.0071, 95% CI 0.0001, 0.014, Figure 4B).
Discussion
In the present study the formation of 4-OH CP was determined over a wide range of substrate concentrations and intrinsic clearance via this route was calculated. 4-OH CP formation was highly variable (>13 fold), but there was a significant correlation between CYP2C19 protein expression and cyclophosphamide intrinsic clearance. This confirmed the preliminary in vitro evidence [7] of a possible role for CYP2C19 in the inter-individual bioactivation of cyclophosphamide. However, the one liver that was homozygous variant (CYP2C19*2/*2) did not have null 4-hydroxylase activity. To our knowledge there have been no previous reports of cyclophosphamide enzyme kinetics in a human liver that is homozygous variant for CYP2C19 LoF alleles.
It has been reported previously that CYP2B6 protein and activity correlates with the bioactivation of cyclophosphamide by human liver [3]. In contrast, the present study did not observe a significant correlation between CYP2B6 protein and cyclophosphamide CLint (i.e. catalytic efficiency). This difference may be due to the use of two low (0.25 and 1 mm) substrate concentrations in the previous study [3] compared with the extensive kinetic determination of CLint (Vmax/Km) over a range of substrate concentrations in this study. Further, although there was no impact of overall CYP2B6 expression on bioactivation, there was a significant decrease in CLint in livers that were carriers of one variant CYP2B6*5 allele compared with livers that were homozygous wild-type at this loci (CYP2B6*1/*1). Recent reports [24] indicate that the A785G variant associated with CYP2B6*6 results in low protein expression and low metabolism of bupropion. However, the functional effect of the CYP2B6*6 variant is often contradictory and substrate dependent [10]. CYP2B6*6 has been previously reported to result in increased turnover of cyclophosphamide [3] and increased bioactivation in vivo[13] and this was also observed in the present study as two livers that were heterozygous for CYP2B6*6 had high CLint (1.25 and 1.39 µl min−1 mg−1). This discrepancy between the impact of genotype vs. protein expression in relation to functional activity is most likely as a result of the fact that CYP2B6 variant alleles were not significantly correlated with the expression of CYP2B6 protein, which is in agreement with the previous data [3]. Although CYP2B6*5 has been previously reported to result in eight-fold lower protein expression compared with wild-type livers [23], it is possible that these variants also alter the functional activity of the CYP2B6 enzyme. Furthermore, no livers were homozygous for CYP2B6*5, which may also explain the lack of correlation in the present study.
We confirmed the in vitro ability of both CYP2C19 and CYP2B6 to hydroxylate cyclophosphamide using expressed recombinant CYP2C19 and CYP2B6 (supersomes®). The relatively high catalytic efficiency of CYP2B6 (CLint 56.92 µl min−1 nmol−1) is similar to that reported previously [5]. CYP2C19 had a lower catalytic efficiency (CLint 5.0 µl min−1 nmol−1) and moreover a low affinity (Km) of CYP2C19 for cyclophosphamide compared with CYP2B6 has been described previously [6–8]. Thus it appears that CYP2B6 is a high affinity, low capacity isozyme whereas CYP2C19 may have lower catalytic efficiency for cyclophosphamide hydroxylation in vitro. Consequently, the relative contribution of these isozymes to hepatic metabolism will be dependent on the concentration of cyclophosphamide in the liver as well as the relative expression of functional CYP2B6 and CYP2C19 protein.
Translation of this relationship to in vivo clinical pharmacogenetic studies is important since controversy exists as to the relative importance of CYP2C19 or CYP2B6 in the clinical disposition of this drug [14–18]. To determine whether one or more LoF alleles at either the CYP2B6 or the CYP2C19 loci results in decreased bioactivation clinically, we measured the plasma concentrations of cyclophosphamide and 4-OH CP in patients receiving treatment for lupus nephritis. We utilized a metabolic index approach (bioactivation ratio) with measurement at a single time point (at approximately tmax) as a significant linear correlation between cyclophosphamide and 4-OH CP AUC which has been reported previously [14, 25] and the bioactivation ratio 4-OH CP : cyclophosphamide at tmax gives the same ratio as 4-OH CP AUC : cyclophosphamide AUC.
Ten patients in the present study had at least one LoF variant allele at either CYP2C19 or CYP2B6. Although the sample size is small the relatively high frequency of these LoF alleles compared with the Caucasian liver biobank should be noted and may be due to the mixed ethnicity of the New Zealand patient population, since lupus nephritis is more prevalent in Asians [26] and Polynesians [27] than in Caucasians. This once again highlights the differences in CYP variant allele frequencies between ethnically different populations.
One patient in our study had been on long-term treatment with carbamazepine, a known inducer of CYP2C19 and CYP3A [28] and this patient had the highest bioactivation ratio (0.0164). The ability of carbamazepine to influence cyclophosphamide bioactivation has been noted before [14] with a 3.5-fold increased bioactivation reported. Another patient in the study had been on long-term treatment with thyroxine, a thyroid hormone that has recently been demonstrated to be important in the expression of constitutive androstane receptor (CAR) and controls the induction of CYP2B protein [29]. Hence this patient may also have elevated bioactivation of cyclophosphamide.
With regard to CYP2C19 genotype, a decreased elimination rate constant (ke) for clearance of cyclophosphamide has been associated with the CYP2C19*2 LoF genotype [15], with a trend for decreased clearance [16] and a significant increase in t1/2[13] for cyclophosphamide in patients with the LoF variant of CYP2C19. Other studies in cancer patients have not reported a relationship between CYP2C19 and clearance or bioactivation (AUC) ratio. In the current study the bioactivation ratio was somewhat lower, although not statistically significant, in the patients with CYP2C19 LoF variant alleles. As expected from the dual role of CYP2C19 and CYP2B6 in the bioactivation of cyclophosphamide, the one patient who was homozygous variant for CYP2C19*2 did not display null bioactivation.
Intriguingly, one patient had no detectable 4-OH CP and therefore was considered to be a null bioactivator. From our in vitro study we predicted that in order to have null activity a patient would have to be homozygous variant at both CYP2B6 and CYP2C19 genes. However, this person had only one LoF allele (CYP2C19*3). It is important to remember that additional gene–environment effects such as disease [30] and drug–drug interactions may obscure genotype–phenotype correlations. A genotype–phenotype discordance of 37% has been observed previously for CYP2C19 in patients with cancer [30]. Whether the three patients with no LoF alleles who had bioactivation ratios similar to the patients with ≥1LoF had a discordant CYP2C19 metabolizer status is not known. Studies to determine the correlation between measured activity of CYP2C19 and CYP2B6 and cyclophosphamide bioactivation in patients are underway. In addition, a number of drugs which are prescribed to lupus nephritis patients during cyclophosphamide therapy may influence bioactivation. For example, prednisone, although an inhibitor of CYP2C19 in vitro[31], is also an inducer of cyclophosphamide metabolism [32], and as such, may enhance the bioactivation ratio. Such gene–environment interactions, combined with the ability of cyclophosphamide to auto-induce its own metabolism [33], may preclude using a simple genotyping approach to identify patients at risk of therapeutic failure with this drug.
Nonetheless, irrespective of the cause, a low bioactivation ratio may predict therapeutic response and this will be the focus of our continuing observation of this cohort of patients.
In conclusion, our observations from the in vitro liver biobank kinetic study and this preliminary data set of clinical samples indicate that the presence of at least one LoF allele(s) at either the CYP2B6 or the CYP2C19 locus does appear to result in decreased bioactivation of cyclophosphamide. However, further studies are required to translate our in vitro prediction into the clinic. We suggest that a metabolic index approach (bioactivation ratio) at a single time point, combined with dual genotype analysis, may enable further clarification of the clinical pharmacogenetics of cyclophosphamide.
Acknowledgments
This study was supported by funding from the Auckland Medical Research Foundation and Arthritis NZ. J.K. Coller is a recipient of an FTT Fricker Fellowship (University of Adelaide Medical Endowment Funds). We wish to thank Dr Jason Ly and Dr Michael Lam for their help in the recruitment of patients.
Competing interests
There are no competing interests to declare.
REFERENCES
- 1.Cohen JL, Jao JY. Enzymatic basis of cyclophosphamide activation by hepatic microsomes of the rat. J Pharmacol Exp Ther. 1970;174:206–10. [PubMed] [Google Scholar]
- 2.Chang TKH, Weber GF, Crespi CL, Waxman DJ. Differential activation of cyclophosphamide and ifosfamide by cytochromes P450 2B and 3A in human liver microsomes. Cancer Res. 1993;53:5629–37. [PubMed] [Google Scholar]
- 3.Xie H-J, Yasar U, Lundgren S, Griskevicius L, Terelius Y, Hassan M, Rane A. Role of polymorphic human CYP2B6 in cyclophosphamide bioactivation. Pharmacogenomics J. 2003;3:53–61. doi: 10.1038/sj.tpj.6500157. [DOI] [PubMed] [Google Scholar]
- 4.Chen C-H, Lin JT, Goss K, He Y, Halpert JR, Waxman DJ. Activation of the anti-cancer prodrugs cyclophosphamide and ifosfamide: identification of cytochrome P450 2B enzymes and site-specific mutants with improved enzyme kinetics. Mol Pharmacol. 2004;65:1278–85. doi: 10.1124/mol.65.5.1278. [DOI] [PubMed] [Google Scholar]
- 5.Huang Z, Roy P, Waxman DJ. Role of human liver microsomal CYP3A4 and CYP2B6 in catalyzing N-dechloroethylation of cyclophosphamide and ifosfamide. Biochem Pharmacol. 2000;59:961–72. doi: 10.1016/s0006-2952(99)00410-4. [DOI] [PubMed] [Google Scholar]
- 6.Chang TKH, Yu L, Goldstein JA, Waxman DJ. Identification of the polymorphically expressed CYP2C19 and the wild type CYP2C9-ILE359 allele as low Km catalysts of cyclophosphamide and ifosfamide activation. Pharmacogenetics. 1997;7:211–21. doi: 10.1097/00008571-199706000-00006. [DOI] [PubMed] [Google Scholar]
- 7.Griskevicius L, Yasar U, Sandberg M, Hidestrand M, Eliasson E, Tybring G, Hassan M, Dahl M-L. Bioactivation of cyclophosphamide: the role of polymorphic CYP2C enzymes. Eur J Clin Pharmacol. 2003;59:103–9. doi: 10.1007/s00228-003-0590-6. [DOI] [PubMed] [Google Scholar]
- 8.Roy P, Yu LJ, Crespi C, Waxman DJ. Development of a substrate-activity based approach to identify the major human liver P-450 catalysts of cyclophosphamide and ifosfamide activation based in cDNA expressed activities and liver microsomal P450 profiles. Drug Metab Dispos. 1999;2:655–66. [PubMed] [Google Scholar]
- 9.Goldstein JA, Blaisdell J. Genetic tests which identify the principal defects in CYP2C19 responsible for the polymorphism in mephenytoin metabolism. Methods Enzymol. 1996;272:210–18. doi: 10.1016/s0076-6879(96)72025-6. [DOI] [PubMed] [Google Scholar]
- 10.Zanger UM, Klein K, Saussele T, Blievernicht J, Hofmann MH, Schwab M. Polymorphic CYP2B6: molecular mechanisms and emerging clinical significance. Pharmacogenomics. 2007;8:743–59. doi: 10.2217/14622416.8.7.743. [DOI] [PubMed] [Google Scholar]
- 11.Sim SC, Risinger C, Dahl M-J, Aklillu E, Christensen M, Bertilsson L, Ingelman-Sundberg M. A common novel CYP2C19 gene variant causes ultrarapid drug metabolism relevant for the drug response to proton pump inhibitors and antidepressants. Clin Pharmacol Ther. 2006;79:103–13. doi: 10.1016/j.clpt.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 12.Baldwin RM, Ohlsson S, Pedersen RS, Mwinyi J, Ingelman-Sunderg M, Eliasson E, Bertilsson L. Increased omeprazole metabolism in carriers of the CYP2C19*17 allele; a pharmacokinetic study in healthy volunteers. Br J Clin Pharmacol. 2008;65:767–74. doi: 10.1111/j.1365-2125.2008.03104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakajima M, Komagat S, Fujiki Y, Kanada Y, Ebi H, Itoh K, Mukai H, Yokoi T, Minami H. Genetic polymorphisms of CYP 2B6 affect the pharmacokinetics /pharmacodynamics of cyclophosphamide in Japanese cancer patients. Pharmacogenet Genomics. 2007;17:431–45. doi: 10.1097/FPC.0b013e328045c4fb. [DOI] [PubMed] [Google Scholar]
- 14.Xie H, Griskevicius L, Ståhle L, Hassan Z, Yasar U, Rane A, Broberg U, Kimby E, Hassan M. Pharmacogenetics of cyclophosphamide in patients with hematological malignancies. Eur J Pharm Sci. 2006;27:54–61. doi: 10.1016/j.ejps.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 15.Timm R, Kaiser R, Lotsch J, Heider U, Sezer O, Weisz K, Montemurro M, Roots I, Cascorbi I. Association of cyclophosphamide pharmacokinetics to polymorphic cytochrome P450 2C19. Pharmacogenomics J. 2005;5:365–73. doi: 10.1038/sj.tpj.6500330. [DOI] [PubMed] [Google Scholar]
- 16.Ekhart C, Doodeman VD, Rodenhuis S, Smits P, Beijnen JH, Huitema ADR. Influence of polymorphisms of drug metabolizing enzymes (CYP2B6, CYP2C9, CYP2C19, CYP3A4, CYP3A5, GSTA1, GSTP1, ALDH1A1 and ALDH3A1) on the pharmacokinetics of cyclophosphamide and 4-hydroxycyclophosphamide. Pharmacogenet Genomics. 2008;18:515–23. doi: 10.1097/FPC.0b013e3282fc9766. [DOI] [PubMed] [Google Scholar]
- 17.Takada K, Arefayene M, Desta Z, Yarboro CH, Boumpas DT, Balow JE, Flockhart DA, Illei GG. Cytochrome P450 pharmacogenetics as a predictor of toxicity and clinical response to pulse cyclophosphamide in lupus nephritis. Arthritis Rheum. 2004;50:2202–10. doi: 10.1002/art.20338. [DOI] [PubMed] [Google Scholar]
- 18.Singh G, Saxena N, Aggarwal A, Misra R. Cytochrome P450 polymorphism as a predictor of ovarian toxicity to pulse cyclophosphamide in systemic lupus erythematosus. J Rheumatol. 2007;34:731–3. [PubMed] [Google Scholar]
- 19.Zhou S, Paxton JW, Tingle MD, Kestell P. Identification of the human liver cytochrome P450 isoenzyme responsible for the 6-methylhydroxylation of the novel anticancer drug 5,6-dimethylxanthenone-4-acetic acid. Drug Metab Dispos. 2000;28:1449–56. [PubMed] [Google Scholar]
- 20.Griskevicius L, Meurling L, Hassan M. Simple method based on fluorescent detection for the determination of 4-hydroxycyclophosphamide in plasma. Ther Drug Monit. 2002;24:405–9. doi: 10.1097/00007691-200206000-00013. [DOI] [PubMed] [Google Scholar]
- 21.Takamizawa A, Matsumoto S, Iwata T, Tochino Y, Katagiri K, Yamaguchi K. Studies on cyclophosphamide metabolites and their related compounds. 2. Preparation of an active species of cyclophosphamide and related compounds. J Med Chem. 1975;18:376–83. doi: 10.1021/jm00238a011. [DOI] [PubMed] [Google Scholar]
- 22.Liu J, Kestell P, Findlay M, Riley G, Ackland S, Simpson A, Issacs R, McKeage M. Application of liquid chromatography-mass spectrometry to monitoring plasma cyclophosphamide in phase I trial cancer patients. Clin Exp Pharmacol Physiol. 2004;31:677–82. doi: 10.1111/j.1440-1681.2004.03065.x. [DOI] [PubMed] [Google Scholar]
- 23.Lang T, Klein K, Fischer J, Nussler AK, Neuhaus P, Hofman U, Eichelbaum M, Schwab M, Zanger UM. Extensive genetic polymorphism in the human CYP2B6 gene with impact on expression and function in human liver. Pharmacogenetics. 2001;11:399–415. doi: 10.1097/00008571-200107000-00004. [DOI] [PubMed] [Google Scholar]
- 24.Hofmann MH, Blievernicht JK, Klien K, Saussele T, Schaeffeler E, Schwab M, Zanger UM. Aberrant splicing caused by single nucleotide polymorphism c.516G>T [Q172H], a marker of CYP2B6*6, is responsible for decreased expression and activity of CY2B6 in liver. J Pharmacol Exp Ther. 2008;325:284–92. doi: 10.1124/jpet.107.133306. [DOI] [PubMed] [Google Scholar]
- 25.Chen T-L, Kennedy MJ, Anderson LW, Kiraly SB, Black KC, Colvin OM, Growchow LB. Non linear pharmacokinetics of cyclophosphamide and 4-hydroxycyclophosphamide/aldophosphamide in patients with metastatic breast cancer receiving high dose chemotherapy followed by autologous bone marrow transplantation. Drug Metab Dispos. 1997;25:544–51. [PubMed] [Google Scholar]
- 26.Patel M, Clarke AM, Bruce IN, Symmons DPM. The prevalence and incidence of biopsy-proven lupus nephritis in the UK-evidence of an ethnic gradient. Arthritis Rheum. 2006;54:2963–9. doi: 10.1002/art.22079. [DOI] [PubMed] [Google Scholar]
- 27.Hart HH, Grigor RR, Caughey DE. Ethnic difference in the prevalence of systemic lupus erythematosus. Ann Rheum Dis. 1983;42:529–32. doi: 10.1136/ard.42.5.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kato M, Chiba K, Horikawa M, Sugiyama Y. The quantitative prediction of in vivo enzyme-induction caused by drug exposure from in vitro information on human hepatocytes. Drug Metab Pharmacokinet. 2005;20:236–43. doi: 10.2133/dmpk.20.236. [DOI] [PubMed] [Google Scholar]
- 29.Ooe H, Kon J, Oshima H, Mikata T. Thyroid hormone is necessary for expression of constitutive androstane receptor in rat hepatocytes. Drug Metab Dispos. 2009;37:1963–9. doi: 10.1124/dmd.108.022905. [DOI] [PubMed] [Google Scholar]
- 30.Helsby NA, Lo W-Y, Sharples K, Riley G, Murray M, Spells K, Dzhelai M, Simpson A, Findlay M. CYP2C19 pharmacogenetics in advanced cancer: compromised function independent of genotype. Br J Cancer. 2008;99:1251–5. doi: 10.1038/sj.bjc.6604699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jurima M, Inaba T, Kalow W. Mephenytoin hydroxylase activity in human liver: inhibition by steroids. Drug Metab Dispos. 1985;13:746–9. [PubMed] [Google Scholar]
- 32.Faber OK, Mouridsen HT, Skovsted L. The biotransformation of cyclophosphamide in man: influence of prednisone. Acta Pharmacol Toxicol. 1974;35:195–200. doi: 10.1111/j.1600-0773.1974.tb00739.x. [DOI] [PubMed] [Google Scholar]
- 33.de Jonge ME, Huitema ADR, Rodenhuis S, Beijnen JH. Clinical pharmacokinetics of cyclophosphamide. Clin Pharmacokinet. 2005;44:1135–64. doi: 10.2165/00003088-200544110-00003. [DOI] [PubMed] [Google Scholar]