

Abstract
Dynamic and structural information has been obtained for an analogue of acetylcholine while bound to the agonist binding site on the nicotinic acetylcholine receptor (nAcChoR), using wide-line deuterium solid-state NMR. Analysis of the deuterium lineshape obtained at various temperatures from unoriented nAcChoR membranes labeled with deuterated bromoacetylcholine (BAC) showed that the quaternary ammonium group of the ligand is well constrained within the agonist binding site when compared with the dynamics observed in the crystalline solids. This motional restriction would suggest that a high degree of complementarity exists between the quaternary ammonium group of the ligand and the protein within the agonist binding site. nAcChoR membranes were uniaxially oriented by isopotential centrifugation as determined by phosphorous NMR of the membrane phospholipids. Analysis of the deuterium NMR lineshape of these oriented membranes enriched with the nAcChoR labeled with N+(CD3)3-BAC has enabled us to determine that the angle formed between the quaternary ammonium group of the BAC and the membrane normal is 42° in the desensitized form of the receptor. This measurement allows us to orient in part the bound ligand within the proposed receptor binding site.
Keywords: 2H NMR, structure, 31P NMR
The ligand gated ion channel, the nicotinic acetylcholine receptor (nAcChoR), is a member of the four-transmembrane superfamily of receptors that includes γ-aminobutyric acid, glycine, and the 5-hydroxytryptamine receptors and is involved in the transmission of information across the neuromuscular junction (1). Structurally the nAcChoR is composed of five glycosylated subunits (α2, β,γ, δ) with a total molecular mass of 280 kDa (1). Electron diffraction studies have been used to resolve the structure of the receptor to 4.6 Å and to define how it is conformationally altered upon the binding of acetylcholine (ACh) to the synaptic surface of the protein (2). However, the resolution of the current structures is insufficient to resolve the bound agonist, hindering a molecular understanding of the structural and dynamic events associated with the ACh binding to the agonist site on the nAcChoR.
Solid-state NMR has demonstrated its applicability to the study of various dynamic processes occurring within the protein backbone, side chains, and bound ligands (3–11). Furthermore, deuterium NMR has found widespread application to the study of protein dynamics both in soluble proteins in the solid state and membrane proteins in their native environment (3–11). In this study, we have exploited the selective reactivity of α-Cys-192/193 toward bromoacetylcholine (BAC) (12–14) after the reduction of the nAcChoR to covalently incorporate N+(CD3)3-BAC. The incorporation of this agonist analogue containing NMR-sensitive nuclei into the agonist binding site has allowed a detailed study of the structure and dynamics of the ligand within the agonist binding site of the nAcChoR. Using the well-characterized lineshapes of deuterated quaternary ammonium compounds (15), we have been able to obtain information relating to the dynamics of the quaternary ammonium group of the covalently attached N+(CD3)3-BAC while in the agonist binding site of the nAcChoR. Additionally, by macroscopically orienting nAcChoR-enriched membranes labeled with N+(CD3)3-BAC, we have been able to exploit the anisotropy present in the deuterium spectrum to determine the angle formed between the C—N bond and the membrane normal.
The deuterium NMR spectra of deuterated quaternary ammonium groups in crystalline solids are dominated by the presence of rotational motion of the individual methyl groups (rotation about the C3 axis) and the entire quaternary ammonium group (rotation about the C3′ axis; Fig. 1) (15–17). These two classes of motions also have been used to describe the amplitude and frequency of motion of a quaternary ammonium group for a ligand bound to a membrane-bound anion transporter (11). In both cases rotation about these axes leads to an averaging of the observed quadrupolar splitting by an additional factor of 1/2(3cos2θ − 1), where θ represents the angle between the axis of averaging and the bond vector. Thus when rapid exchange occurs between the three equally populated sites then the observed quadrupolar splitting can be determined (15):
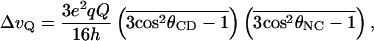 |
where θCD and θNC represent the angles formed between the bond vector and the C3 and C3′ axis of rotation, respectively. Additionally, it has been demonstrated that in such cases the lineshapes observed might reflect the motion propagated along the backbone of the ligand, leading to additional motional averaging, which manifests itself in the deuterium spectrum (17). Here, we have exploited the differing degrees of motional averaging of the quaternary ammonium group of BAC to describe qualitatively the motional processes of the ligand while resident in the agonist binding site of the nAcChoR.
Figure 1.

Diagram showing the linkage of the BAC to Cys-192/193 of the α subunit of the nAcChoR. Additionally the axes of rotation are shown that lead to the motional averaging of the deuterium lineshape (C3 and C3′). The orientation of the N+(CD3)3 group is determined between the C3′ axis of rotation and the magnetic field, with the membrane normal at both 0° and 90° with respect to the applied field.
In addition, this dynamic analysis of the quaternary ammonium
group of the BAC bound in the agonist binding site of the nAcChoR is
extended to provide angular constraints for the bound ligand.
Previously, solid-state NMR of macroscopically aligned membranes
together with isotopic labeling has exploited the uniaxial distribution
of both lipids and proteins about the membrane normal to extract
orientational constraints from the system (18–20). In this study we
aim to obtain the angle (θ) formed between the
C3′ axis of rotation and the membrane
normal. This angle can be obtained directly from the quadrupolar
splitting ΔvQ observed with a sample aligned
uniaxially with the magnetic field, using the equation
ΔvQ =
Δv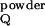 (3cos2θ
− 1). Using computer simulations (18–20) we have been able
to analyze the lineshapes obtained to calculate an angle for the
C3′ axis of rotation with respect to
the membrane normal.
(3cos2θ
− 1). Using computer simulations (18–20) we have been able
to analyze the lineshapes obtained to calculate an angle for the
C3′ axis of rotation with respect to
the membrane normal.
Materials and Methods
Synthesis of Crystalline Ligands.
N+(CD3)3-BAC bromide was synthesized according to the method of Rama Sastry and Chiou (21), through the addition of N+(CD3)3 choline bromide to bromoacetyl bromide. N+(CD3)3-BAC bromide subsequently was purified by recrystallization from hot isopropanol. N+(CD3)3-ACh perchlorate was synthesized by the methylation of ethanolamine with a slight excess of methyliodide in ethanol (22). After recrystallization from hot ethanol the choline iodide was acetylated by using acetic anhydride in the presence of the catalyst dimethylaminopyridine (22). N+(CD3)3-ACh perchlorate was produced through the recrystallization of ACh iodide in an ethanol solution saturated with ammonium perchlorate (22). N+(CD3)3-ACh chloride (MSD Isotopes) was recrystallized from a hot ethanol solution. The nature, purity, and isotopic enrichment of each compound was determined by 1H-NMR and electrospray mass spectrometry. The nature of the counterion was determined by 13C-cross polarization magic angle spinning NMR and paper chromatography (23).
Purification and Labeling of Enriched nAcChoR Membranes.
Membranes were prepared according to Sobel's method (24) from Torpedo nobilana and resuspended in 20 mM Tris buffer (pH 7.0) to a protein concentration of 1 mg⋅ml-1. The samples then were treated with 10 mM diisopropyl fluorophosphate to inhibit acetylcholine esterase action and to prevent nonspecific binding of the BAC to the esterase (24). The activity of the sample was 0.8 nmol being site per milligram of protein as determined by using a dansylcholine displacement assay (25). The receptor membranes were labeled with BAC as described (26). Briefly, membranes were treated with N-ethyl maleimide (3 mM, 24 h, 4°C) to mask free cysteines within the membrane suspension. The receptor membranes then were washed and subsequently reduced by incubation with DTT (300 μM) for 1 h to reduce the vicinal disulphide bonds. After extensive washing, the membranes were incubated with 100 μM BAC (1 h room temperature). Afterward, the membranes were washed thoroughly with Tris buffer to remove free N+(CD3)3-BAC before orientation. It has been shown previously that under these conditions more than 80% of the BAC incorporated is associated with the α-chain of the nAcChoR (26, 27). Furthermore it has been shown that the binding of α-neurotoxin is completely abolished (27) and the binding of α-bungarotoxin reduced by 85% (28), indicating that the BAC is present in the agonist binding sites. Labeling of the reduced nAcChoR with the alkylating reagent [4-(N-maleimido)benzyl]trimethylammonium iodidie (MBTA) has been shown to inhibit the alkylation of the nAcChoR by BAC, suggesting that these two molecules alkylate a common site within the agonist binding pocket (27). For MBTA this site has been demonstrated, by sequencing of peptide cleavage products of the labeled α-chain of the nAcChoR, to be located at Cys-192 (12). Binding studies of a variety of ligands to membranes prepared according to such protocol have suggested that under these conditions the nAcChoR has adopted its desensitized conformation (28, 29).
Orientation of Enriched nAcChR Membranes.
nAcChoR membranes were oriented by using an isopotential spinning technique (30). Alignment was achieved through pelletting 1 ml of nAcChoR membranes (1 mg⋅ml-1) onto a Mellanex sheet (Agar Scientific, Stansted, U.K.) by centrifigation (90,000 g for 15 h at 4°C) without drying. The samples were removed from the apparatus and the bulk solvent were removed under a stream of nitrogen; the samples then were left to equilibrate at 85% humidity (3 h, 20°C). Similar conditions have been used to perform attenuated total reflectance–Fourier transform IR and fluorescence experiments on oriented nAcChoR membranes. Under these conditions the reversible binding of ACh has been detected and demonstrated to be pharmacologically specific through the use of the specific inhibitor α-bungarotoxin (31, 32). For NMR measurements the sheets were stacked in either an 8- or 10-mm NMR tube and sealed to prevent dehydration.
NMR Measurements.
31P NMR measurements were performed on a Bruker MSL 400 spectrometer at 161.98 MHz with a 8-mm double-resonance cross polarization probe. Data were acquired by using a standard Hahn-echo pulse sequence with broadband proton decoupling during acquisition; 90° pulse lengths were typically 5 μs with an interpulse delay of 30 μs with a recycle delay of 3.5 s. Powder deuterium spectra of crystalline solids were acquired on a Bruker MSL 400 at 61 MHz by using a 10-mm single-resonance static probe. Temperature was regulated to within ±1 K with a Bruker VT1000 temperature control unit. Both powder and oriented samples of nAcChoR membranes labeled with N+(CD3)3-BAC bromide were acquired on a CMX 500 Infinity spectrometer at 75 MHz by using a 10-mm single-resonance static probe. Deuterium spectra were acquired by using a standard quadrupolar echo pulse sequence with a 5-μs 90° pulse, 30-μs interpulse delay, and 0.5-s recycle time.
Lineshape Simulations.
To obtain values for both the mosaic spread and the angular dependence of the aligned membranes computer simulations were performed. For both the oriented phosphorous and deuterium spectra the lineshape simulation algorithm based on that of Ulrich and Watts (33) and Seelig (34, 35) and subsequently modified by Glaubitz and colleagues (18, 19) was used. The lineshape simulations were written in C++ using gamma (36) and typically took less than 30 s on a SGI Octane workstation (Reading, U.K.). These simulations allowed the mosaic spread to be modeled as a three-dimensional distribution about the membrane director, as well as the angular dependence of the N+(CD3)3 group of the BAC bound to the receptor to be extracted from the experimental spectra.
Results and Discussion
Dynamics of Crystalline N+(CD3)3-Labeled Ligands.
To determine the motional characteristics of the quaternary ammonium groups in N+(CD3)3-BAC bromide, N+(CD3)3-ACh chloride, and N+(CD3)3-ACh perchlorate, the deuterium lineshape obtained for each salt was studied as a function of temperature and the spectra are shown in Fig. 2 A–C, respectively. At 293 K the deuterium spectra of N+(CD3)3-BAC bromide is characterized by a sharp central intensity superimposed on a powder pattern with an inner splitting of 13 kHz, which is consistent with rotation about both the C3 and C3′ axis of rotation. However, the appearance of a sharp central feature and the rather rounded nature of the Pake doublet suggests that either the C3 or the C3′ axis of rotation is entering the intermediate motional regime (k = 103-106 s-1) (15–17, 37). As the temperature is lowered through 273 K to 250 K, a coalescence of the inner wings and a broadening of the outer wings is observed, such that at 250 K the lineshape is characterized by a sharp central intensity superimposed on top of a broad low intensity component. These spectra are indicative of motions about either the C3 or C3′ axis occurring on an intermediate time scale (k = 103-106 s-1). The same motional characteristics are observed for N+(CD3)3-ACh chloride (Fig. 2B), although at the highest temperature measured (293 K) the lineshapes are still dominated by the intermediate motions about either the C3 or C3′ axis. As the temperature at which the spectra of N+(CD3)3-BAC bromide are recorded is lowered from 230 K to 190 K, the sharp central intensity disappears and an increase in intensity of a powder lineshape occurs, the inner splitting of which is 40 kHz. This feature shows that the intermediate motions that previously dominated the lineshape are largely suppressed. This results in a deuterium powder pattern whose lineshape is now governed by the single rapid rotation about either the C3 or the C3′ axis, giving rise to a scaling of the quadrupolar coupling constant by a factor 1/2(3cos2θ) − 1 and the observed quadrupolar coupling of 40 kHz. Such lineshapes also are observed for N+(CD3)3-ACh chloride (Fig. 2B) between 210 and 190 K, suggesting that the lineshapes are again governed by a single rapid axial rotation about either the C3 or C3′ axis of rotation. At 190 K the lineshapes for the N+(CD3)3-BAC bromide (Fig. 2A) and N+(CD3)3-ACh chloride (Fig. 2B) both show the appearance of a broad central component between the inner wings. As the temperature is lowered to 170 K this central broad component grows in intensity at the expense of the inner wings at 40 kHz, such that at 170 K the lineshapes are dominated by a broad central feature 40 kHz wide superimposed on a broader (80 kHz) wide component. This lineshape is consistent with the remaining axis of averaging entering an intermediate motional regime (15–17, 37).
Figure 2.

Deuterium variable temperature NMR spectra (1,024 acquisitions) of crystalline N+(CD3)3-BAC bromide (A), N+(CD3)3 ACh chloride (B), and N+(CD3)3-ACh perchlorate (C). Data were acquired at the temperatures indicated.
In contrast to the strong temperature dependence of the deuterium lineshapes of both the N+(CD3)3-BAC bromide and N+(CD3)3-ACh chloride salts the lineshape of the N+(CD3)3-ACh perchlorate (Fig. 2C) shows relatively little variation until the temperature reaches 210 K. Above this temperature range the lineshapes are governed by the rapid axial rotation about both the C3 and C3′ axis, which leads to the averaging of the quadrupolar coupling constant to the observed 13 kHz. Below 210 K, the appearance of a broad central intensity indicates the slowing of motions about either the C3 or C3′ axis of rotation, such that it begins to enter the intermediate motional regime.
The motional characteristics of the N+(CD3)3-ACh chloride and N+(CD3)3-ACh perchlorate can be readily rationalized in terms of their crystal structures (38, 39), where the rotation of the quaternary ammonium group in the ACh chloride salt would be sterically hindered by contacts between the quaternary ammonium group and the ester oxygen (3.17 Å, rvdw = 3.40 Å) and with the OCH2 group (3.20 Å, rvdw = 4.00 Å). In contrast, in the perchlorate salt there are fewer intramolecular contacts within the backbone of the ACh molecule and only limited contacts between the crystal planes. The observed dynamics for the ACh chloride and perchlorate salts also have been observed in 13C-cross polarization magic angle spinning studies (F. Riddell, personal communication).
Dynamics of N+(CD3)3-BAC Bound to the nAcChoR.
Deuterium NMR spectra of nAcChoR membranes labeled with N+(CD3)3-BAC were acquired between 278 K and 153 K and are shown in Fig. 3. At 278 K the lineshape is dominated by a narrow resonance whose width approaches the homogeneity of the magnet. On lowering the temperature to 253 K the resonance broadens, adopting a Lorentzian lineshape with a line width of 500 Hz, reflecting a more motionally restricted quaternary ammonium group. At 233 K the lineshape continues to broaden with wings appearing with a splitting of 36 kHz. The quadrupolar splitting of 36 kHz is similar to that observed for the crystalline N+(CD3)3-BAC bromide and N+(CD3)3-ACh chloride when motion about either the C3 or C3′ axis of rotation has been suppressed at low temperatures (Fig. 2 A and B) and is similar to that observed for deuterated methyl groups within the retinals of both bacteriorhodopsin and rhodopsin (8, 18–19). This finding suggests that motion of the quaternary ammonium group about either the C3 or C3′ axis in the agonist binding site is slowly being suppressed at these temperatures. As the temperature is lowered to 153 K, a broad spectral envelope appears underneath the narrower spectral feature that is 36 kHz wide. Although the spectra do not allow an accurate analysis of this broad component due to the inefficient excitation of the broader spectral components (as a result of the limited power available during these experiments), there is evidence of at least two spectral components. The first has an apparent splitting of 76 kHz whereas the second and more pronounced component has an apparent splitting of 160 kHz. These lineshapes are broader than previously observed for the crystalline ligands, indicating that there is a greater degree of immobilization of the quaternary ammonium group in the agonist binding site compared with those previously observed in all three crystalline solids studied (Fig. 2). The lineshapes observed for the N+(CD3)3-BAC bromide bound to the nAcChoR membranes between 213 K and 153 K mirror those observed previously for the inorganic salt trimethyl ammonium iodide (TMAI) (15). For TMAI these lineshapes have been shown to arise due to hindered rotation about both the C3 and C3′ axes of rotation, suggesting that motion is similarly hindered in these experiments for the quaternary ammonium group when bound in the agonist binding site in the nAcChoR. We have demonstrated here that for the crystalline salts studied, the observed lineshapes for the quaternary ammonium group highly depend on the motional averaging about the C3 and C3′ axes of rotation, and that the dynamics observed can be readily rationalized through both intermolecular and intramolecular steric contacts between the quaternary ammonium group and neighboring atoms. Extrapolating these observations to the receptor, it is possible to conclude that the quaternary ammonium group within the agonist binding site is well constrained sterically. In the absence of any structure for the bound BAC, it is not possible to determine whether the restricted motions observed occur through either interactions with the choline backbone or residues within the binding site. The restricted motions observed appear to be consistent with previous power saturation ESR studies that also show that a variety of spin labels attached to Cys-192/193 in the ligand binding site experience restricted motions on the ESR time scale (40). This observation suggests that the motional restriction observed in both the ESR and NMR measurements arises through interactions of the ligands with the binding site, as the restricted motions observed are not restricted to one class of ligand. The observation of restricted motions within the ligand binding site appears consistent with the current electron diffraction data (2), which suggests that a high degree of complementarity probably exists between the ACh and the putative agonist binding sites. These conclusions drawn from both structural and dynamic data would appear to agree with structure-activity relationship studies (41). These studies also suggest that a high degree of complementarity exists between the quaternary ammonium group of the ACh and the agonist binding site, as even minor modifications such as the substitution of a single ethyl for a methyl group, leads to dramatic drops in the binding affinity of the ligand (41).
Figure 3.

Deuterium NMR spectra of 50 mg of nAcChoR membranes labeled with N+(CD3)3-BAC as described. Spectra were acquired as described with 60,000 acquisitions at the temperatures indicated and processed with 500-Hz line broadening.
Macroscopic Alignment of Enriched nAcChoR Membranes.
To determine the degree of orientation present in the macroscopically aligned receptor membrane phosphorous-31 NMR spectra were acquired with the membrane normal at 0° and 90° with respect to the applied magnetic field (Fig. 4). At 0° a broad resonance covering 40 ppm to 10 ppm is observed with a main peak at 25 ppm (Fig. 4A). Changing the tilt angle of the membrane to 90° results in a shift in the resonance to −15 ppm (Fig. 4B) as expected for macroscopically aligned lamellar membranes (30). The observed chemical shift anisotropy is typical for a mixed phosphatidylcholine/phosphatidylethanolamine lipid system in its Lα phase (30). The broad nature of the 0° and the 90° spectrum can be attributed to the distribution of different lipid types (phosphatidylcholine, phosphatidylethanolamine, and phosphatidylserine) within the sample, each of which exhibits a slightly different chemical shift anisotropy (42). Although this effect may partially account for the broad distribution, there is also a significant contribution from the sample mosaic spread around the membrane normal. Simulations of the lineshapes observed at both 0° and 90° indicated that the mosaic spread of 20–25° is present in the system studied (30).
Figure 4.

Static 31P NMR spectra of oriented nAcChoR-rich membranes labeled with deuterated BAC. Spectra were recorded by using two plates containing 2 mg of protein each. Spectra were the result of 4,000 acquisitions and acquired under the conditions described in Materials and Methods. Membrane normal orientated at 0° to the magnetic field (A). Membrane normal orientated at 90° to the magnetic field (B).
Deuterium NMR of N+(CD3)3-BAC in Orientated Enriched nAcChoR Membranes.
The deuterium spectra of oriented nAcChoR membranes labeled with N+(CD3)3-BAC bromide were obtained at 0° and 90° to allow the orientational dependence of the ligand to be determined with respect to the membrane normal. The spectra obtained at 0° and 90° are shown in Fig. 5A (black spectra throughout). The 0° orientation is characterized by a central intensity with a splitting of 30 kHz, beneath a 100-kHz wide broad component (Fig. 5A). In contrast, the 90° spectra shows a rather featureless lineshape, whose base is 50 kHz wide (Fig. 5D). Notably though, edges appear on this spectrum with an apparent splitting of 30 kHz. Even though the powder spectra at this temperature does not show a static lineshape, it is already apparent that the N+(CD3)3 group of the BAC shows an angular dependence with respect to the membrane norm, indicating that the ligands located in the agonist binding sites are well constrained in agreement with the dynamic data presented above.
Figure 5.

Deuterium spectra of 40 mg of orientated nAcChoR membranes labeled with N+(CD3)3-BAC oriented with the membrane normal parallel (A) and perpendicular (D) to the magnetic field (data shown in black). Spectra were acquired with 80,000 acquisitions at 153 K. Optimal simulated lineshapes are shown in red (A–F). Effect on simulated spectra of small changes in the angle (γ) formed between the C3′ axis of rotation and the membrane normal for both the parallel and perpendicular cases are shown in B and E, respectively (black, experimental; red, best fit as shown in A and D; green, +5° from best fit; blue, −5° degrees from best fit). Effect on simulated spectra of small changes in the mosaic spread for the parallel and perpendicular cases are shown in C and F, respectively (black, experimental; red, best fit as shown in A and D; green, +5° from best fit; blue, −5° degrees from best fit).
Using the methods outlined above, the lineshapes for the 0° and the 90° orientations have been simulated (Fig. 5 A and D, respectively; red spectra). For the sample with the membrane normal oriented at 0° the best fit with the experimental data were obtained by using an angle of 42° with a mosaic spread of 40° and a line broadening of 50 Hz (Fig. 5A; red spectrum). The major discrepancies between the experimental and simulated data arise at the wings of the spectrum at ±50 kHz and have been attributed to the finite power levels used during these experiments, which leads to inefficient excitation of the wings of wider spectra. An optimal fit for the sample with the membrane normal oriented at 90° with respect to the magnetic field was obtained by using an angle of 42° with a mosaic spread of 7.5° and a line broadening of 50 Hz (Fig. 5D; red spectrum). Variations of the tilt angle (γ) by + 5° (green spectra) and −5° (blue spectra) for the both the 0° and 90° orientations (Fig. 5 B and E, respectively) indicate that very little deviation from this angle can be tolerated without the quality of the fit being reduced.
Although the angle formed between the magnetic field and the cone axis remains constant, the mosaic spreads used to simulate the lineshapes obtained at 0° and 90° do not correlate with each other or with that determined by phosphrous-31 NMR (≈20°). Such discrepancies between mosaic spread of the lipid environment and the embedded protein have been observed previously (18). However, significant changes in mosaic spread are not expected as the angle of the membrane normal is altered with respect to the magnetic field.
In making these simulations several assumptions have been made that may contribute to these observations. First, we assume that the BAC molecules located at each agonist binding site are similarly orientated with respect to the bilayer normal, and second, that the deuterium lineshapes are not dominated by intermediate motions.
In principle this technique should allow us to distinguish the orientation of the quaternary ammonium group located within each of the agonist binding pockets as the resulting spectral intensity will arise from the superimposition of two uniaxially symmetric distributions. However, our ability to resolve this situation is compromised because of the poor signal to noise obtained in these spectra, thus hindering any attempt at spectral deconvolution. Biochemical data (43) and medium-resolution electron diffraction data (2) suggests that the binding sites share significant structural homology, with any perturbations arising through the interaction of the α subunit with its neighbors. This similarity of binding would suggest that the ligand might adopt a similar orientation with respect to the membrane normal with any discrepancies being small compared with the resolution of the experiment.
The deuterium spectra of the same sample before orientation acquired at a similar temperature indicated that significant motions were still present on the intermediate time scale even at these low temperatures (−120°C). Alternative simulations are available to calculate the lineshapes of uniaxially oriented samples (44, 45); however, these also assume that intermediate motions do not dominate the lineshape. Lineshape simulations that incorporate such dynamic processes have been developed (46). However, these are highly model-dependent and have not been used to date to study such complex systems as either powder or uniaxially oriented samples. In an attempt to resolve whether this angular dependence of mosaic spread significantly affects the interpretation of the data, we have systematically varied the mosaic spread for both the 0° and 90° spectra by both +5° (Fig. 5C, green) and −5° (Fig. 5F, blue). These simulations suggest that although the overall lineshape shows a significant dependence the inner and outer wings of the 0° lineshape and the overall width of the 90° spectrum show very little dependence. Such weak correlation between the inner and outer wings of the deuterium spectra of uniaxially aligned samples and the mosaic spread has previously been reported on similar studies of the bacterial proton pump bacteriorhodopsin (44, 45). Both of these observations would indicate that the estimate of the angle formed between the quaternary ammonium group and the membrane normal is not significantly compromised by the simulations used.
The cavities in the electron diffraction structure, which have been proposed to represent the agonist binding site on the basis of their structural responses upon the addition of agonist (2, 47, 48), and other fluorescence energy transfer measurements (49, 50), indicate that the binding sites would best accommodate the bound agonist with the choline backbone lying parallel to the membrane normal, with alternative orientations being sterically unfavorable (2, 47, 48). Such an orientation of the bound ligand would position the quaternary ammonium group within the binding site at an angle consistent with the 42° obtained by using the above approach. The position and orientation of the ligand based on electron diffraction data, fluorescence energy transfer measurements, and our own NMR data are summarized in Fig. 6. Due to the relatively constrained nature of the quaternary ammonium group within the agonist binding site, we suggest that little deviation from this geometry could be tolerated, and thus a similar geometry would be shared by all ligands that bind the nAcChoR through the quaternary ammonium pharmacophore.
Figure 6.

Diagram showing the relative position and orientation of the agonist ACh while bound to the nAcChoR based on electron diffraction data, fluorescence energy transfer measurements, and the data presented here. Note, our data would be equally consistent with the ligand inverted with respect to the orientation shown.
Conclusions
Deuterium NMR studies of the nAcChoR labeled with N+(CD3)3-BAC bromide have shown that once the overall reorientation of the complex is suppressed, the deuterium lineshapes obtained for the N+(CD3)3 group are qualitatively similar to those obtain from other quaternary ammonium salts studied both here and elsewhere. A comparison of the deuterium lineshapes obtained for the N+(CD3)3-BAC bound to the receptor with that of the crystalline solid suggests that within the agonist binding site the quaternary ammonium group exhibits a reduction in the degree of motion about both the C3 and C3′ axis of rotation.
By orienting nAcChoR-enriched bilayers we have been able to obtain oriented deuterium spectra from N+(CD3)3-BAC within the receptor binding site. Through the simulation of these lineshapes we are able to determine the average angle between the C3′ axis of rotation of the quaternary ammonium group and the membrane normal to be 42°.
Acknowledgments
Gerhard Gröbner, University of Umeå, is gratefully acknowledged for his valuable comments in the preparation of this manuscript. Clemens Glaubitz is acknowledged for his assistance with the simulation of the deuterium lineshapes. Frank Ridell is acknowledged for his useful comments on the dynamics in quaternary ammonium groups. This work was supported by grants from the Higher Education Funding Council, Biotechnology and Biological Sciences Research Council, and European Community (to A.W.), a Biotechnology and Biological Sciences Research Council-CASE (Glaxo Wellcome) studentship (to P.T.F.W.), and National Institution on Alcohol Abuse and Alcoholism Grant NIAAA07040 (to K.W.M.).
Abbreviations
- nAcChoR
nicotinic acetylcholine receptor
- ACh
acetylcholine
- BAC
bromoacetylcholine
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Stroud R M, McCarthy M P, Schuster M. Biochemistry. 1990;29:11009–11023. doi: 10.1021/bi00502a001. [DOI] [PubMed] [Google Scholar]
- 2.Miyazawa A, Fujiyoshi Y, Stowell M, Unwin N. J Mol Biol. 1999;288:765–786. doi: 10.1006/jmbi.1999.2721. [DOI] [PubMed] [Google Scholar]
- 3.Watts A, Burnett I J, Glaubitz C, Gröbner G, Middleton D A, Spooner P J R, Watts J A, Williamson P T F. Nat Prod Rep. 1999;16:419–423. doi: 10.1039/a801189c. [DOI] [PubMed] [Google Scholar]
- 4.Watts A. Curr Opin Biotechnol. 1999;10:48–53. doi: 10.1016/s0958-1669(99)80009-3. [DOI] [PubMed] [Google Scholar]
- 5.Watts A. Pharm Pharmacol Commun. 1999;5:7–13. [Google Scholar]
- 6.Watts A, Burnett I J, Glaubitz C, Gröbner G, Middleton D A, Spooner P J R, Williamson P T F. Eur Biophys J. 1998;28:84–90. [Google Scholar]
- 7.Zhang H, Bryant R G. Biophys J. 1997;72:363–372. doi: 10.1016/S0006-3495(97)78675-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Copie V, McDermott A E, Bashah K, Williams J C, Spijker-Assink M, Gebhard R, Lugtenburg J, Herzfeld J, Griffin R G. Biochemistry. 1994;33:3280–3286. doi: 10.1021/bi00177a019. [DOI] [PubMed] [Google Scholar]
- 9.Oldfield E, Kinsey R A, Kintanar A. Methods Enzymol. 1982;88:310–325. [Google Scholar]
- 10.Kinery M A, Kintanar A, Smith R L, Gutowsky H S, Oldfield E. Biochemistry. 1984;23:288–298. doi: 10.1021/bi00297a018. [DOI] [PubMed] [Google Scholar]
- 11.Taylor A M, Grobner G, Williamson P T F, Watts A. Biochemistry. 1999;38:11172–11179. doi: 10.1021/bi990618p. [DOI] [PubMed] [Google Scholar]
- 12.Kao P N, Dwork A J, Kaldany R R J, Silver M L, Wideman J, Stein S, Karlin A. J Biol Chem. 1984;259:11662–11665. [PubMed] [Google Scholar]
- 13.Walker J W, Richardson C A, McNamee M G. Biochemistry. 1984;23:2329–2338. doi: 10.1021/bi00306a002. [DOI] [PubMed] [Google Scholar]
- 14.Silman I, Karlin A. Science. 1969;164:1420–1421. doi: 10.1126/science.164.3886.1420. [DOI] [PubMed] [Google Scholar]
- 15.Penner G H, Polson J M, Daleman S I, Reid K. Can J Chem. 1992;71:417–426. [Google Scholar]
- 16.Penner G H, Zhao B, Jeffrey K R. Z Naturforsch. 1994;50:81–89. [Google Scholar]
- 17.Ratcliffe C I, Ripmeester J A. Can J Chem. 1986;64:1348–1354. [Google Scholar]
- 18.Gröbner G, Choi G, Burnett I J, Glaubitz C, Verdegem P J E, Lugtenburg J, Watts A. FEBS Lett. 1998;422:201–204. doi: 10.1016/s0014-5793(97)01591-3. [DOI] [PubMed] [Google Scholar]
- 19.Gröbner G, Burnett I J, Glaubitz C, Choi G, Mason A J, Watts A. Nature (London) 2000;405:810–813. doi: 10.1038/35015604. [DOI] [PubMed] [Google Scholar]
- 20.Ulrich A S, Watts A. Nat Struct Biol. 1995;2:190–192. doi: 10.1038/nsb0395-190. [DOI] [PubMed] [Google Scholar]
- 21.Rama Sastry B V, Chiou C. Biochem Pharmacol. 1968;17:805–815. doi: 10.1016/0006-2952(68)90017-8. [DOI] [PubMed] [Google Scholar]
- 22.Williamson P T F, Gröbner G, Miller K W, Watts A. Biochemistry. 1998;37:10854–10859. doi: 10.1021/bi980390q. [DOI] [PubMed] [Google Scholar]
- 23.Whittaker V P, Wijesundera S. J Biol Chem. 1952;51:348–351. [PMC free article] [PubMed] [Google Scholar]
- 24.Brasswell L M, Miller K W, Sauter J F. Br J Pharmacol. 1984;83:305–311. doi: 10.1111/j.1476-5381.1984.tb10147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neubig R R, Cohen J B. Biochemistry. 1979;18:5464–5475. doi: 10.1021/bi00591a032. [DOI] [PubMed] [Google Scholar]
- 26.Wolosin J M, Lyddiatt A, Dolly J O, Bernard E A. Eur J Biochem. 1980;109:495–506. doi: 10.1111/j.1432-1033.1980.tb04821.x. [DOI] [PubMed] [Google Scholar]
- 27.Damle V N, McLaughlin M, Karlin A. Biochem Biophys Res Commun. 1978;84:845–851. doi: 10.1016/0006-291x(78)91661-3. [DOI] [PubMed] [Google Scholar]
- 28.Ratman M, Gullick W, Spiess J, Wan K, Criado M, Lindstrom J. Biochemistry. 1986;25:4268–4275. doi: 10.1021/bi00363a014. [DOI] [PubMed] [Google Scholar]
- 29.Delegeane A M, McNamee M G. Biochemistry. 1980;19:890–895. doi: 10.1021/bi00546a010. [DOI] [PubMed] [Google Scholar]
- 30.Grobner G, Taylor A, Williamson P T F, Choi G, Glaubitz C, Watts J A, deGrip W J, Watts A. Anal Biochem. 1997;254:132–138. doi: 10.1006/abio.1997.2415. [DOI] [PubMed] [Google Scholar]
- 31.Baezinger J E, Miller K W, Rothschild K J. Biophys J. 1992;61:983–992. doi: 10.1016/S0006-3495(92)81905-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baezinger J E, Miller K W, Rothschild K J. Biochemistry. 1993;32:5448–5454. doi: 10.1021/bi00071a022. [DOI] [PubMed] [Google Scholar]
- 33.Ulrich A, Watts A. Solid State Nucl Magn. 1993;2:21–36. doi: 10.1016/0926-2040(93)90060-z. [DOI] [PubMed] [Google Scholar]
- 34.Seelig J. Q Rev Biophys. 1977;10:353–410. doi: 10.1017/s0033583500002948. [DOI] [PubMed] [Google Scholar]
- 35.Seelig J. Biochim Biophys Acta. 1978;515:105–140. doi: 10.1016/0304-4157(78)90001-1. [DOI] [PubMed] [Google Scholar]
- 36.Smith S A, Levante T O, Meier B H, Ernst R R. J Magn Reson. 1994;100:75–105. [Google Scholar]
- 37.Torchia D. Annu Rev Biophys Bioeng. 1984;13:125–144. doi: 10.1146/annurev.bb.13.060184.001013. [DOI] [PubMed] [Google Scholar]
- 38.Herdklotz J K, Sass R L. Biochem Biophys Res Commun. 1970;40:583–588. doi: 10.1016/0006-291x(70)90942-3. [DOI] [PubMed] [Google Scholar]
- 39.Mahajan V, Sass R L. J Crystallogr Mol Struct. 1974;4:15–21. [Google Scholar]
- 40.Addona G H, Andrews S H, Cafiso D S. Biochim Biophys Acta. 1997;1329:74–84. doi: 10.1016/s0005-2736(97)00089-8. [DOI] [PubMed] [Google Scholar]
- 41.Michelson M J, Zeimal E V. Acetylcholine: An Approach to Molecular Mechanism and Action. Oxford: Permagon; 1973. p. 82. [Google Scholar]
- 42.Barrantes F J. Crit Rev Biochem Mol Biol. 1989;24:437–478. doi: 10.3109/10409238909086961. [DOI] [PubMed] [Google Scholar]
- 43.Galzi J L, Revah F, Bessis A, Changeux J P. Annu Rev Pharmacol. 1991;31:37–72. doi: 10.1146/annurev.pa.31.040191.000345. [DOI] [PubMed] [Google Scholar]
- 44.Moltke S, Nevzorov A A, Sakai N, Wallat I, Job C, Nakanishi K, Heyn M P, Brown M F. Biochemistry. 1998;37:11821–11835. doi: 10.1021/bi980676v. [DOI] [PubMed] [Google Scholar]
- 45.Moltke S, Wallat I, Sakai N, Nakanishi K, Brown M F, Heyn M P. Biochemistry. 1999;38:11762–11772. doi: 10.1021/bi990593u. [DOI] [PubMed] [Google Scholar]
- 46.Greenfield M S, Ronemus A D, Vold R L, Vold P D, Ellis P D, Raidy T E. J Magn Reson. 1987;2:89–107. [Google Scholar]
- 47.Unwin N. Nature (London) 1995;373:37–43. doi: 10.1038/373037a0. [DOI] [PubMed] [Google Scholar]
- 48.Unwin N. Cell. 1988;107:1123–1138. doi: 10.1083/jcb.107.3.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herz J M, Johnson D A, Taylor P M. J Biol Chem. 1989;264:12439–12448. [PubMed] [Google Scholar]
- 50.Fernando Valenzuela C, Weign P, Yguerabide J, Johnson D A. Biophys J. 1994;66:674–682. doi: 10.1016/s0006-3495(94)80841-0. [DOI] [PMC free article] [PubMed] [Google Scholar]