

Abstract
Chikungunya virus (CHIKV) is an arthritogenic mosquito-transmitted alphavirus that is undergoing reemergence in areas around the Indian Ocean. Despite the current and potential danger posed by this virus, we know surprisingly little about the induction and evasion of CHIKV-associated antiviral immune responses. With this in mind we investigated innate immune reactions to CHIKV in human fibroblasts, a demonstrable in vivo target of virus replication and spread. We show that CHIKV infection leads to activation of the transcription factor interferon regulatory factor 3 (IRF3) and subsequent transcription of IRF3-dependent antiviral genes, including beta interferon (IFN-β). IRF3 activation occurs by way of a virus-induced innate immune signaling pathway that includes the adaptor molecule interferon promoter stimulator 1 (IPS-1). Despite strong transcriptional upregulation of these genes, however, translation of the corresponding proteins is not observed. We further demonstrate that translation of cellular (but not viral) genes is blocked during infection and that although CHIKV is found to trigger inactivation of the translational molecule eukaryotic initiation factor subunit 2α by way of the double-stranded RNA sensor protein kinase R, this response is not required for the block to protein synthesis. Furthermore, overall diminution of cellular RNA synthesis is also observed in the presence of CHIKV and transcription of IRF3-dependent antiviral genes appears specifically blocked late in infection. We hypothesize that the observed absence of IFN-β and antiviral proteins during infection results from an evasion mechanism exhibited by CHIKV that is dependent on widespread shutoff of cellular protein synthesis and a targeted block to late synthesis of antiviral mRNA transcripts.
Chikungunya virus (CHIKV) was first isolated in Tanzania in 1953 (52, 69). The virus causes an acute febrile illness associated with severe joint pain that can persist long after viral clearance (9, 26, 42, 47, 49, 62, 68, 75). In 2005 and 2006 CHIKV reemerged on a number of Indian Ocean islands and subsequently in India in 2006 and 2007 (78). Since these locations are popular tourist destinations, such outbreaks represent significant threats to European countries from traveler-associated infections. Because the virus is transmitted most commonly via mosquitoes (23, 67), changing patterns of vector distribution and abundance in response to climate change (36) and increased vector-human contact following human encroachment into undeveloped areas renders CHIKV an emerging pathogen of high potential danger for future generations. Unfortunately, information regarding many basic aspects of CHIKV molecular biology, immunology, and pathology are lacking.
CHIKV is a member of the family Togaviridae and genus Alphavirus. The enveloped virion contains an icosahedral nucleocapsid and an ∼12-kb plus-strand single-stranded RNA genome that includes a 5′ cap and 3′ polyadenylation. The genome includes two open reading frames (ORFs) separately encoding polyproteins that are subsequently processed into four nonstructural proteins (nsP1 to nsP4) and three structural proteins (capsid [C], envelope glycoproteins E1 and E2). Sindbis virus (SINV) represents the most thoroughly investigated Alphavirus species yet most of our knowledge regarding pathogenesis, replication, and immunobiology is derived from murine animal or cellular models (see reference 70). As such, the immune reactions to and immunomodulatory counteractions exhibited by potentially destructive alphaviruses such as CHIKV in the context of human infection are incompletely examined.
It is becoming clear, however, that innate immunity, in particular the type I interferon (IFN) system, represents one of the most important antiviral responses to CHIKV due to its immediate onset upon infection and susceptibility of the virus to IFN′s antiviral effects. Type I IFNs include IFN-β and IFN-α subtypes 1 to 14. Release of IFN-α/β from infected cells results in autocrine and paracrine stimulation of the IFN-α/β receptor (IFNAR), which leads through associated tyrosine (TYK) and Janus (JAK) kinases to the phosphorylation of STAT (signal transducers and activators of transcription) 1 and 2. STAT1/2 heterodimers associate with IFN regulatory factor 9 (IRF9) and bind to IFN-stimulated response elements (ISREs) upstream of so-called IFN-stimulated genes (ISGs). ISG-encoded proteins represent the antiviral effector molecules that directly inhibit molecular and biochemical activities required for virus replication. Indeed, when added to cells prior to infection, IFN-α/β is extremely suppressive to in vitro growth of all examined alphaviruses (3, 20, 21, 59, 71), including CHIKV (83). Moreover, whereas adult wild-type (WT) mice do not typically die following high-titer CHIKV infection (15, 31, 76, 92), infection of mice lacking either IFNAR (15, 76) or STAT1 (76) is quickly and invariably fatal. Interestingly, CHIKV replication can be detected in joint and muscle tissues of adult IFNAR+/− but not WT mice, indicating a possible IFN “dose effect” of permissiveness (15).
Induction of IFN-β expression is an increasingly well-characterized process that occurs after exposure to pathogen-associated molecular patterns (PAMPs). PAMPs initiate signaling cascades that lead to formation of a complex containing IFN regulatory factor 3 (IRF3) and nuclear factor κB (NF-κB) on the IFN-β promoter (50, 54, 87, 91). IRF3 can itself initiate expression of a subset of ISGs independently of IFN (6, 10, 13, 19, 37, 58, 61) and, unlike NF-κB (86), is necessary for virus-induced IFN synthesis in fibroblasts (19, 74). IRF3 is constitutively expressed and normally shuttles between the cytoplasm and the nucleus. Phosphorylation of C-terminal serine and threonine residues by the kinases IKKɛ or TBK1 results in IRF3 homodimerization, coactivator association, and nuclear accumulation (25, 50, 79, 81, 91). These phosphorylation signals originate from pattern recognition receptors (PRRs) that react with specific PAMPs. Viral PRRs include Toll-like receptors (TLRs) 3 reacting with double-stranded RNA (dsRNA), as well as cytoplasmic helicases that contain caspase recruitment domains (CARDs). These include retinoic acid inducible gene I (RIG-I), recognizing RNA species containing 5′ triphosphates and shorter dsRNA fragments (44, 90) and melanoma differentiation-associated gene 5 (MDA5) (89) reacting with poly(I:C) and longer dsRNA (16, 43, 44, 63, 72, 73). Although TLRs are predominantly expressed on immune cells (dendritic cells, monocytes, and macrophages), the cytoplasmic helicases are expressed in nearly all cells, including fibroblasts and epithelial cells. Importantly, PRR signaling occurs via essential adaptor molecules that act as integration points linking PAMP detection with activation of IRF3-directed kinases. RIG-I and MDA5 require the mitochondrion-associated adaptor protein IFN promoter stimulator 1 (IPS-1; also called MAVS, Cardif, or VISA) (46, 55, 80, 88, 90). IPS-1 is emerging as an extremely important antiviral signaling molecule involved in the type I IFN response to both DNA and RNA viruses (1, 14, 41). Currently, the transcription factors, PRR(s), and CHIKV-specific PAMP(s) required for IFN-α/β induction during infection are unknown. However, IPS-1 was shown to be important to CHIKV-triggered IFN-α/β induction in mouse cells (76), thus implying a role for MDA5 or RIG-I in Alphavirus-triggered IFN-α/β synthesis. However, despite investigation into this question using MDA5 and RIG-I knockout mice, a definitive answer has not been reached for any Alphavirus species (11, 64, 76).
In addition to questions surrounding the induction of innate immune responses by CHIKV, mechanisms used by CHIKV to evade these responses remain largely uninvestigated. We therefore decided to more closely examine events comprising the innate immune response to infection with a recently emerged epidemic strain of the virus, as well as potential strategies used by CHIKV to evade this response. Our detailed investigation of the mechanism of CHIKV-mediated IRF3 activation, IFN-β and ISG expression revealed that CHIKV strongly induces the transcription of IFN and ISGs via the adaptor molecule IPS-1. However, we observed that these mRNA messages are not translated into protein and that this accompanied both a widespread block of cellular translation, as well as a late block of RNA synthesis. Interestingly, the translation block may represent a novel mechanism since it occurs independently of virus-induced phosphorylation of eukaryotic initiation factor subunit 2α (eIF2α) via the protein kinase activated by dsRNA (PKR).
MATERIALS AND METHODS
Reagents and antibodies.
The dsRNA mimic polyinosine-polycytosine [poly(I:C)] was obtained from Amersham Biosciences and resuspended in millipure water at 1 μg ml−1. Hygromycin B was obtained from InvivoGen and used at 300 μg ml−1 cell culture medium. Puromycin was obtained from Clontech and used at 2 μg ml−1 cell culture medium. Lipofectamine-LTX transfection reagent was obtained from Invitrogen and used according to the manufacturer's instructions. HiPerfect transfection reagent was obtained from Qiagen. Human recombinant IFN-β was obtained from PBL. ONE-Glo cell lysis/luciferin reagent was obtained from Promega. 4-Thiouridine and actinomycin D were obtained from Sigma. The antibodies used against the following antigens are indicated in parentheses: GAPDH (SC-51906; Santa Cruz) and IRF3 for immunofluorescence analysis (IFA; 550428; BD Biosciences); IRF3 for immunoblotting (SC-9082; Santa Cruz); and Ser398 phospho-IRF3 (07-581; Millipore), eIF2α (SC-11386; Santa Cruz), Ser51 phospho-eIF2α (9721; Cell Signaling), PKR (SC-707; Santa Cruz), and Thr446 phospho-PKR (1120-1; Epitomics). Puromycin was as described previously (77), dsRNA (clone K1) was obtained from English and Scientific Consulting, ISG56 was kindly provided by Ganes Sen, Viperin was kindly provided by Peter Cresswell, and Alphavirus capsid was kindly provided by Irene Greiser-Wilke (38).
Virus and cell culture.
Primary human foreskin fibroblasts (HFs) were obtained from the American Type Culture Collection. HFs stably transfected with the catalytic subunit of the human telomerase gene to extend passage life were kindly provided by Wade Bresnahan (8). Cells were propagated in Dulbecco minimal essential medium (DMEM) containing 10% fetal calf serum (FCS) and antibiotics at 37°C in 5% CO2. Sendai virus (SeV) was obtained from Charles River Laboratories and exposed to cells in duplicate at 160 hemagglutination (HA) units cell culture medium ml−1. BHK-21 and C6/36 cells were obtained from Jay Nelson. CHIKV strain LR2006 OPY1 was obtained from Stephen Higgs (84). SINV strain Ar 339 was obtained from the American Type Culture Collection, and stocks were grown by infecting BHK-21 cells at a multiplicity of infection (MOI) of 0.001. CHIKV viral stocks were prepared by infecting either BHK-21 or C6/36 mosquito cells at an MOI of 0.001 with passage 1 virus derived from an infectious clone as described previously (84). At 72 h postinfection the supernatant was harvested, cleared, and pelleted through a 20% (wt/vol) sucrose cushion in Hanks balanced salt solution by ultracentrifugation at 23,000 rpm in a Beckman SW28 rotor. Virus pellets were then resuspended in phosphate-buffered saline (PBS), and titers were determined by using an endpoint dilution assay (66). Transfection of poly(I:C) at 1 μg ml−1 of culture medium was performed in 6-, 12-, or 24-well dishes by adding 2 μl of Lipofectamine-LTX per 1 μg of poly(I:C).
Transient RNA interference.
Cells were plated at 30 to 40% confluence in 35-mm dishes the day before transfection with small interfering RNA (siRNA). Five microliters of siRNA (20 μM stock) was mixed with 10 μl of HiPerfect in 95 μl of Opti-MEM (Gibco) and added to cells containing 2.3 ml of Opti-MEM. The cells were transfected twice, 8 h apart, and incubated for 16 h, and the Opti-MEM was replaced with DMEM with 10% FCS. The cells were allowed to expand for 3 to 4 days to near confluence and transfected once more at 16 h before treatment. The siRNA sequences were as follows: nonspecific (NS), 5′-GGACGUAGAAGAGGGUGUAGAG-3′; and IPS-1, 5′-GGGUUCUUCUGAGAUUGAA-3′. PKR and IRF3 were targeted by using a SmartPool of four different sequences (Thermo Scientific).
Generation of stable cell lines.
Stable cell lines were constructed using lentiviral expression vectors. IRF3 was stably degraded in telomerized human fibroblasts by stably expressing the NPro open reading frame from bovine viral diarrhea virus as described previously (24). Replication-defective recombinant retrovirus was produced by transfecting the retroviral vector into Phoenix A or 293T cells (for expression of NPro from the pdl-NPro lentiviral vector [kindly provided by Roger Everett]) using Lipofectamine-LTX and harvesting supernatant after 48 h (48). The supernatant was centrifuged (3,000 × g for 10 min) and filtered through two 0.45-μm-pore-size filters to remove cell debris. Subconfluent target cells were exposed to retrovirus for 16 h in the presence of 5 μg of Polybrene ml−1. After the cells reached confluence they were split into DMEM plus 10% FCS containing 1 μg of puromycin ml−1. Transduced cells were passaged in the presence of increasing puromycin (to 3 μg ml−1) until the cultures were fully resistant. Short hairpin RNA (shRNA) directed against PKR was stably transfected into telomerized human fibroblasts using the pGIPZ lentivirus vector obtained from Open Biosystems (catalog no. RHS4430-98844125) according to the manufacturer's protocol.
Type I IFN quantification assays.
Secretion of type I IFN by target cells was quantified as described earlier (18). Briefly, confluent HFs were treated as indicated in 24-well dishes. At 6 h posttreatment, the cells were washed three times with 1× PBS, and then 500 μl of DMEM plus 10% FCS was added for 16 h. The medium was harvested and added to telomerized HF cells stably transfected with an expression cassette containing firefly luciferase under the control of the IFN-dependent response element (termed THF-ISRE) grown to confluence in a 96-well dish (50 μl of medium was added to each of four wells). At 6 h after medium transfer, 50 μl of ONE-Glo lysis/luciferin reagent was added to each well, and the luminescence was measured on a Veritas luminometer (Turner Biosystems).
RNA isolation and semiquantitative reverse transcription-PCR (RT-PCR).
Total RNA was isolated from cells by using a Mini-RNA Isolation II kit according to the manufacturer's protocol (Zymo Research, product R1030) and quantified by UV spectrometry. RNA samples were treated with DNase using the DNA-Free RNA kit according to the manufacturer's protocol (Zymo Research, product 1013). Single-stranded cDNA for use as a PCR template was made from total RNA using random hexamers to prime first-strand synthesis by Superscript III reverse transcriptase (Invitrogen, product 11754) as described in the manufacturer's protocol. Comparison of mRNA expression between samples (e.g., infected versus untreated) was performed using SYBR green-based semiquantitative real-time RT-PCR (qPCR) with the Applied Biosystems sequence detection system according to the ΔΔCT method of Livak and Schmittgen (51). GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was used as a housekeeping gene to establish a baseline against which target genes were compared between samples (described in reference 18). Other primer sequences were as follows: Mx1-For, 5′-ATGATTGTCAAGTGCCG-3′; Mx1-Rev 5′-GCCTTTCCTTCCTCCA-3′; β-Actin-For, 5′-TCACCCACACTGTGCCCATCTACGA-3′; and β-Actin-Rev, 5′-CAGCGGAACCGCTCATTGCCAATGG-3′.
Indirect immunofluorescence assays.
HFs were grown on coverslips in 24-well plates and treated as described above. The cells were washed twice with PBS, fixed for 30 min in 3.7% formaldehyde, washed, and quenched for 10 min using 50 mM NH4Cl. Cells were permeabilized with 0.1% Triton X-100 for 7 min and washed three times with PBS containing 2% bovine serum albumin (BSA). The cells were incubated with primary antibody in PBS containing 2% BSA at 37°C for 1 h, washed three times in PBS containing 2% BSA (10 min each wash), and incubated with fluorescently labeled secondary antibody diluted 1:1,000 in PBS containing 2% BSA for 1 h. The cells were washed twice in PBS containing 2% BSA (10 min each) and once in PBS. Secondary antibodies incuded goat anti-mouse 488 (A-11001; Invitrogen) and goat anti-mouse 594 (A-11020; Invitrogen). Coverslips were mounted on a microscope slide with Vectashield mounting medium (Vector Laboratories, Burlingame, CA) containing DAPI (4′,6′-diamidino-2-phenylindole).
Immunoblotting.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) immunoblots were performed as follows. After trypsinization and cell pelleting at 2,000 × g for 10 min, whole-cell lysates were harvested in 2% SDS lysis buffer (50 mM Tris-HCl, 20% glycerol). Lysates were electrophoresed in 10% polyacrylamide gels and transferred onto polyvinylidene difluoride membranes (Millipore) using semidry transfer at 400 mA for 1.5 h. The blots were blocked at room temperature for 2 h using 10% nonfat milk in 1× PBS containing 0.1% Tween 20. The blots were exposed to primary antibody in 5% nonfat milk in 1× PBS containing 0.1% Tween 20 for 16 h at 4°C. The blots were then washed in 1× PBS containing 0.1% Tween 20 for 20, 15, and 5 min, followed by deionized water for 5 min. A 1-h exposure to horseradish peroxidase-conjugated secondary antibodies and subsequent washes were performed as described for the primary antibodies. The antibody was visualized using enhanced chemiluminescence (Pierce).
Puromycin pulse-chase.
To examine de novo protein synthesis, we used a recently described technique that involves measuring the incorporation of puromycin into growing polypeptide chains of live cells through the use of a puromycin-specific antibody (77). This involves exposing cells to 10 μg of puromycin ml−1 in DMEM-FCS for 15 min (pulse), removing the puromycin-containing medium, washing the cells three times with PBS, and adding puromycin-free DMEM-FCS for 1 h (chase). The cells were then harvested in SDS lysis buffer, and protein-incorporated puromycin was examined by using SDS-PAGE with the puromycin-specific antibody as described above.
RNA metabolic labeling and separation.
To quantify newly synthesized RNA, we used a technique described earlier (22). Briefly, 4-thiouridine in culture medium (200 μM, final concentration) was added to confluent HFs in T-75 culture flasks for 1 h. The cells were then treated with trypsin and collected by centrifugation, and the total RNA was isolated by using a Qiagen RNeasy kit according to the manufacturer's protocol. Biotinylation of the RNA-incorporated 4-thiouridine was then performed using EZ-Link biotin-HPDP (Pierce) in dimethylformamide at 1 mg ml−1. Biotinylation took place in 10 mM Tris (pH 7.4), 1 mM EDTA, and 0.2 mg of biotin-HPDP ml−1 using RNA at 100 ng μl−1 for 1.5 h at room temperature. Approximately 70 μg of total RNA was used per reaction. Unbound biotin-HPDP was removed by using chloroform-isoamyl alcohol (24:1) extraction and heavy phase-lock gels (5 Prime), followed by precipitation. Samples were then denatured at 65°C for 10 min and rapidly cooled on ice for 5 min. A μMACS streptavidin bead/column system (Miltenyi) was used to collect biotinylated (newly synthesized) RNA. RNA was separately pooled from the column (biotinylated) and wash buffer flowthrough (unbiotinylated).
RESULTS
Infection with CHIKV induces the accumulation of mRNA from IFN-β and ISGs.
Fibroblasts are known to be a major target of CHIKV replication in humans (15, 76). However, information regarding fundamental aspects of the innate immune response to CHIKV infection of these cells is lacking. We therefore decided to examine the transcriptional induction of innate antiviral genes in primary human fibroblasts following exposure to CHIKV at various MOIs. As shown in Fig. 1, at 24 h postinfection CHIKV induces the expression of mRNA for the IFN-β gene, as well as for the so-called ISGs Viperin and ISG56. The degree of this transcription closely correlates with the MOI used and induction is evident at an MOI as low as 0.01. Human fibroblasts thus appear to be capable of responding to CHIKV infection through an innate immune response that involves expression of IFN-β and antiviral effector genes.
FIG. 1.

CHIKV infection triggers MOI-dependent transcription of IFN-β and ISGs. Primary HFs were infected with CHIKV at the indicated MOIs for 24 h. Values presented are fold changes in the expression of IFN-β, Viperin, and ISG56 mRNA relative to untreated cells as determined by qPCR. The data are representative of results from duplicate experiments.
CHIKV infection triggers phosphorylation and nuclear accumulation of IRF3.
We next decided to investigate whether the strong, MOI-dependent induction of antiviral mRNA by CHIKV was accompanied by and correlated with activation of IRF3. Whether CHIKV infection activates IRF3 and the dynamics of that activation have thus far remained unexplored. We therefore sought to determine whether infection leads to the phosphorylation of IRF3 and its accumulation in the cell nucleus. To do this, we infected HFs at three different MOIs (0.1, 1, or 10) for 16 h and probed whole-cell lysates following SDS-PAGE with antibody specific to IRF3 phosphorylated on Ser398. As shown in Fig. 2A infection with CHIKV resulted in levels of IRF3 phosphorylation that increase with MOI. CHIKV-dependent IRF3 phosphorylation occurs between 8 and 16 h postinfection (Fig. 2B). We next examined whether CHIKV-induced IFN-β mRNA accumulation correlates temporally with IRF3 phosphorylation. As shown in Fig. 2C, accumulation of IFN-β mRNA is evident by 8 h and is increased at 16 and 24 h postinfection. Early IFN-β transcription could either occur independently of IRF3 or perhaps in response to phosphorylated IRF3 that is undetectable on immunoblots. The essential role of IRF3 in CHIKV-induced gene expression is addressed in greater detail below. Since phosphorylation of IRF3 results in the nuclear accumulation of the protein, we therefore monitored the protein's subcellular localization in HFs following CHIKV infection. As shown in Fig. 2D, while the protein is distributed nearly homogeneously throughout untreated cells, it exhibits almost exclusively nuclear localization after infection with CHIKV. These data indicate that activation of IRF3 in human fibroblasts occurs in response to infection with CHIKV.
FIG. 2.

CHIKV infection triggers Ser398 phosphorylation and nuclear accumulation of IRF3. (A) IRF3 Ser398 phosphorylation in primary HFs at 16 h postinfection with CHIKV at MOIs of 0.1, 1, and 10. (B) IRF3 Ser398 phosphorylation in primary HFs infected with CHIKV (MOI = 10) between 2 and 24 h postinfection. (C) Fold change relative to untreated cells as determined by qPCR of IFN-β mRNA in HFs infected with CHIKV (MOI = 1) for 4, 8, 16, and 24 h. (D) Nuclear accumulation of IRF3 in primary HFs infected with CHIKV (MOI = 10) at 16 h postinfection.
Accumulation of IFN-β and ISG mRNA in response to CHIKV infection is directly dependent on IRF3.
As shown above (Fig. 1), strong transcriptional induction of IFN-β and ISGs, along with IRF3 activation (Fig. 2), occurs following infection of HFs with CHIKV. Virus- and PAMP-triggered induction of type I IFN expression is conventionally known to require activation of IRF3 (see references 45 and 60 for reviews). IFN-independent expression of a subset of ISGs, including ISG56 and Viperin, has also been shown to be induced directly by IRF3 (6, 10, 13, 19, 37, 58, 61). Alternatively, other ISGs, such as Mx1, are only transcribed in response to IFN-dependent signaling and are not directly activated by IRF3 itself (37). Hidmark et al. have demonstrated the necessity of IRF3 for type I IFN induction by Semliki Forest virus (SFV) in murine fibroblasts and myeloid dendritic cells (39). Intriguingly, however, exceptions to this rule have been described for Hantavirus (65) and West Nile virus (17).
We used two approaches to determine whether IFN-β and ISG mRNA accumulation following CHIKV infection requires IRF3. First, we constructed human fibroblasts that stably express the NPro protein from bovine viral diarrhea virus which efficiently targets IRF3 for proteasomal degradation (12, 24, 40). Next, we transiently transfected siRNA directed against IRF3 into HFs as described previously (19). As shown in Fig. 3A, IRF3 protein is depleted upon stable expression of NPro or transfection of IRF3-directed siRNA. As shown in Fig. 3B, when HF-NPro or parental HF cells were treated with IFN-β, normal transcriptional induction of ISGs was observed. However, when HF-NPro cells were exposed to a stimulus such as SeV that is known to activate IRF3-dependent transcription of IFN-β and ISGs, no such induction was seen (Fig. 3B). These results thus indicate that while the IFN-dependent (JAK/STAT) signaling pathway is functional in these cells, IRF3-dependent gene expression is not. We next examined whether CHIKV infection was capable of inducing transcription of IFN-β and ISGs during IRF3 depletion. As shown in Fig. 3B, treatment of HF-NPro cells or HF transfected with IRF3-directed siRNA with CHIKV at an MOI of 10 failed to induce IFN-β or ISG mRNA expression as seen in parental HFs and HFs transfected with NS siRNA. To verify that the IFN-dependent pathway is not being stimulated during CHIKV infection and that ISGs are being activated directly by IRF3, we examined accumulation of Mx1 mRNA. Transcription of this gene occurs in response to IFN-dependent signaling but not direct IRF3 activation (37). As shown in Fig. 3C, CHIKV infection did not stimulate accumulation of Mx1 as did treatment with SeV or IFN-β. Based on these results, we conclude that CHIKV infection triggers IRF3-dependent transcription of IFN-β and ISGs.
FIG. 3.

IRF3 is essential for CHIKV-induced IFN-β/ISG transcription. (A) Immunoblot showing GAPDH and total IRF3 protein from HFs in the presence or absence of stably expressed NPro or transiently transfected nonspecific (NS) or IRF3-directed siRNA as described in text. (B) Accumulation of IFN-β, ISG56, or Viperin mRNA in the indicated cell type following treatment with IFN-β (1,000 U/ml), infection with SeV (160 HA units/ml), or infection with CHIKV (MOI = 10) for 16 h. CHIKV-induced accumulation of IFN-β, ISG56, or Viperin mRNA is also shown in HFs after transfection of NS or IRF3-directed siRNA. The values presented are the mRNA fold changes relative to untreated cells as determined by qPCR and are representative of experiments performed at least in duplicate. (C) Accumulation of Mx1 mRNA in HF following treatment with SeV (160 HA units/ml), IFN-β (1,000 U/ml), or CHIKV (MOI = 10) for 16 h. The values presented are mRNA fold changes relative to untreated cells as determined by qPCR and are representative of experiments performed in duplicate.
IPS-1 is required for CHIKV-mediated activation of IRF3-dependent transcription.
Since CHIKV is a positive-sense single-stranded RNA virus, we presumed that (like other alphaviruses [see references 27 and 82]) its replication involves the synthesis of dsRNA, a powerful inducer of IRF3 activation and synthesis of type I IFN. We therefore examined whether CHIKV-infected HFs accumulate dsRNA and the kinetics of this accumulation using IFA with a dsRNA-specific antibody. As shown in Fig. 4, dsRNA is evident at 2 h postinfection and is maximal between 6 and 8 h postinfection. Cytoplasmic dsRNA is known to be capable of stimulating IRF3-terminal signaling after interacting with RIG-I or MDA5. Signaling pathways activated by these PRR molecules require the adaptor molecule IPS-1. As such, we next sought to determine whether IPS-1 was also essential to IRF3 phosphorylation triggered by CHIKV infection. To address this, we used transfected siRNA targeting IPS-1. In contrast to nonspecific (NS) siRNA, transfection of IPS-1-directed siRNA greatly reduced levels of IPS-1 protein (Fig. 4B). siRNA-mediated knockdown of IPS1 protein subsequently inactivated CHIKV-stimulated IRF3 phosphorylation, which occurred in control cells transfected with NS siRNA (Fig. 4B). Furthermore, as shown in Fig. 4C, CHIKV-induced transcription of IFN-β, Viperin, and ISG56 was nearly eliminated following treatment of cells with IPS-1-directed siRNA. Based on these observations, we conclude that the infection of HFs with CHIKV leads to IRF3 activation and IRF3-dependent gene expression via an IPS-1-dependent pathway.
FIG. 4.

CHIKV infection of HF leads to accumulation of dsRNA and IRF3-dependent transcription via IPS1. (A) Indirect immunofluorescence showing dsRNA in CHIKV-infected HFs (MOI = 10) at 0, 2, 4, 6, and 8 h postinfection. (B) Immunoblot showing levels of IPS1, Ser398-phosphorylated IRF3, total IRF3, and GAPDH after treatment of HFs with NS or IPS1-directed siRNA in the presence or absence of CHIKV infection (MOI = 10) at 24 h postinfection. (C) Transcriptional induction of IFN-β, Viperin, and ISG56 as measured by qPCR after treatment of HFs with NS or IPS1-directed siRNA. The cells were either left uninfected or were infected with SeV (░⃞) or CHIKV (▪; MOI = 10), and total RNA was harvested at 24 h postinfection. Values presented are normalized to transcriptional induction in the presence of NS siRNA (set to 1) and are representative of experiments performed in duplicate.
CHIKV does not induce IFN-α/β secretion or ISG protein translation.
Taken together, our data indicate that infection of HFs with CHIKV strongly activates the innate immune response via the IPS-1/IRF3-dependent pathway resulting in induction of IRF3-dependent genes such as IFN-β, ISG56, and Viperin. To verify that CHIKV also induced levels of the corresponding proteins under these conditions, we monitored IFN-α/β secretion and the protein expression of selected ISGs. To determine whether IFN-β was secreted from CHIKV-infected HFs, we used IFN-responsive reporter cells as previously described (18). Briefly, HFs were either left untreated or exposed to SeV (160 HA units/ml), SINV, or CHIKV at three different MOIs (1, 10, and 100). Media from these cells were subsequently transferred to confluent reporter cells expressing firefly luciferase (LUC) under the control of a type I IFN-dependent promoter (ISRE). As shown in Fig. 5A, treatment of reporter cells with IFN-β induced an ∼8-fold increase in LUC expression relative to untreated cells. Likewise, the use of media from cells infected with SeV also led to strong IFN-dependent LUC expression. Infection of cells with SINV, an alphavirus related to CHIKV, triggered secretion of IFN-α/β that was clearly proportional to the MOI used. In sharp contrast, cells infected with CHIKV secreted little to no IFN-α/β regardless of the MOI. We next examined whether the synthesis of other genes transcriptionally upregulated during CHIKV infection occurred. This was done by using immunoblotting to measure Viperin and ISG56 protein in CHIKV-infected cells. Figure 5B shows the levels of Viperin and ISG56 proteins from whole HF lysates collected after exposure to IFN-β, SeV, or CHIKV. The expression of CHIKV capsid protein is also shown for CHIKV-infected cells. Although treatment with IFN-β and SeV induced strong expression of Viperin and ISG56 protein, exposure of HFs to CHIKV did not result in ISG protein expression even at high MOI. IFN-β transcriptional induction but no IFN-α/β secretion was also observed after infection with CHIKV grown on C6/36 insect cells (data not shown). We thus conclude that, surprisingly, CHIKV infection of HFs triggers the transcription but not the translation of IRF3-dependent genes.
FIG. 5.

CHIKV infection of HFs does not induce secretion of IFN-α/β or synthesis of ISG proteins. (A) Expression of IFN-α/β-dependent luciferase from reporter cells after exposure to media collected from HF that were left untreated, treated with media containing 1,000 U of IFN-β/ml, infected with SeV (160 HA units/ml), or infected with SINV or CHIKV at indicated MOIs for 24 h. Treatments were performed in quadruplicate, and media were transferred to confluent THF-ISRE in six wells of a 96-well plate per treatment. Values presented are average luminescence readings from four separate treatments ± the standard error of the mean (SEM). (B) Immunoblot for Viperin, ISG56, GAPDH, and CHIKV capsid proteins after a 24-h treatment with IFN-β, SeV, or CHIKV at the indicated MOIs.
CHIKV triggers the widespread shutoff of host cell protein translation.
As shown above, CHIKV triggers strong accumulation of IRF3-dependent mRNA without translation of induced transcripts. This led us to ask whether the lack of protein synthesis during infection was accompanied by a widespread block in translation or, rather, is the result of virus-targeted inhibition of IRF3-dependent antiviral genes. To address this, we used a nonradioactive technique for examining protein synthesis (77). This method involves the incorporation of puromycin into growing polypeptide chains and the subsequent detection of puromycin-containing proteins in whole-cell lysates using a specific antibody. We thus infected HFs with CHIKV at an MOI of 10 as described above. At 4, 8, 16, and 24 h postinfection, the cell culture media was replaced (“pulsed”) with medium containing puromycin for 15 min and “chased” for 60 min with puromycin-free medium, after which whole-cell lysates were harvested and subjected to SDS-PAGE. As shown in Fig. 6, protein synthesis, as measured by abundance of polypeptide-incorporated puromycin, was diminished by 8 h postinfection and absent by 16 h postinfection. Synthesis of viral capsid, however, is visible at 8 h and maximal at 16 h postinfection. Translational inhibition was also observed at MOIs of 0.1 and 1 (data not shown). It is worth noting here that while the puromycin pulse-chase method is effective for demonstrating gross differences in de novo protein synthesis between samples, labeling of individual proteins (e.g., CHIKV capsid) may not generate enough signal to be detected following SDS-PAGE. Nevertheless, based on these data, we conclude that CHIKV infection results in a widespread shutoff of host protein, but not viral capsid protein, synthesis which likely contributes to the absence of IFN-β secretion and ISG protein expression from infected cells.
FIG. 6.

CHIKV infection of HFs induces cellular translational shutoff. An immunoblot shows the puromycin incorporated into newly synthesized protein in untreated HF or after exposure to 200 μg of cycloheximide (CHX) ml−1 for 6 h or CHIKV (MOI = 10) (top), CHIKV capsid protein (middle), and GAPDH (bottom) for the indicated durations.
CHIKV infection and infection-associated RNA induce PKR phosphorylation.
Protein kinase activated by dsRNA (PKR) is a PRR that is autophosphorylated following interaction with dsRNA, a process that enables the protein's downstream kinase activity. Since replication of CHIKV involves synthesis of dsRNA (Fig. 4), we decided to examine whether PKR is phosphorylated during infection. This was done by using immunoblotting with an antibody specific to PKR protein phosphorylated on Thr446. As shown in Fig. 7A, PKR phosphorylation is clearly evident by 4 h after CHIKV infection and increases through time to become maximal at 24 h postinfection. We next verified that RNA species generated during virus infection are capable of inducing PKR phosphorylation. To do this, we isolated total RNA from uninfected HFs or HFs infected with CHIKV (MOI = 10) at 2, 4, 6, 8, 12, 16, and 24 h postinfection. The total RNA samples were DNase treated as described above. We next individually transfected 0.5 μg of RNA from each of these time points into subconfluent HFs grown in 12-well dishes and harvested whole-cell lysates at 6 h posttransfection. As shown in Fig. 7B, PKR phosphorylation is evident in cells transfected with RNA harvested at 8 h postinfection, and the RNA appears to be maximally stimulatory at 16 h postinfection. The expression of CHIKV capsid protein in transfected cells was not observed (data not shown). Based on these data, we conclude that both CHIKV infection and cell-associated RNA synthesized during infection are capable of triggering PKR autophosphorylation.
FIG. 7.

CHIKV infection and infection-associated RNA induce phosphorylation of PKR. (A) Immunoblot showing PKR phosphorylated on Thr446, total PKR, CHIKV capsid, and GAPDH after infection of HFs with CHIKV for the indicated duration (left) and densitometry ratio of phosphorylated PKR to total PKR with ratio in untreated cells set to 1 (right). (B) Immunoblot showing phosphorylated PKR, total PKR, and GAPDH from HFs treated only with transfection reagent (TF) or transfected for 6 h with total RNA harvested from HFs infected with CHIKV (MOI = 10) for the indicated duration.
Phosphorylation of eIF2α during CHIKV infection is dependent on PKR.
Cellular stress such as virus infection can trigger a shutoff of protein translation through the inactivation via phosphorylation of eukaryotic initiation factor 2 subunit α (eIF2α). This can occur as a result of dsRNA-mediated activation of PKR, as well as via kinases activated by other types of cellular stress. Since CHIKV induces autophosphorylation of PKR (Fig. 7), it next became of interest to examine whether eIF2α is phosphorylated during infection and, if so, to determine whether PKR is the responsible kinase. As shown in Fig. 8, phosphorylation of eIF2α Ser51 occurs after CHIKV infection in an MOI-dependent manner. To investigate a specific role for PKR in eIF2α phosphorylation triggered by CHIKV, we created an HF cell line that stably expresses shRNA directed against PKR. As shown in Fig. 8, shRNA expression nearly eliminates the PKR protein in these cells. Furthermore, relative to parental HFs, the phosphorylation of eIF2α is severely reduced in the absence of PKR. Based on these results, we conclude that CHIKV triggers PKR-dependent phosphorylation of eIF2α, most likely through the synthesis of dsRNA during virus replication.
FIG. 8.

CHIKV induces PKR-dependent phosphorylation of eIF2α. Immunoblots show PKR, Ser51-phosphorylated eIF2α, total eIF2α, CHIKV capsid, and GAPDH in HFs and HFs stably expressing shRNA directed against PKR following no infection or 24 h postinfection with CHIKV at the indicated MOIs (top), the corresponding densitometry ratio of phosphorylated eIF2α to total eIF2α in parental HFs (░⃞), and HFs expressing shRNA directed against PKR (▪) in response to CHIKV at an MOI of 0 (ratio set to 1), an MOI of 1, an MOI of 10, and an MOI of 100 (right).
PKR is not required for CHIKV-associated cellular translational shutoff.
Our results demonstrate that infection with CHIKV leads to a widespread shutoff of cellular protein synthesis, along with PKR-dependent phosphorylation of eIF2α, a process known to block translation. We therefore next sought to determine whether PKR is required for the virus-associated block to protein translation. To address this, we examined total protein synthesis following CHIKV infection of HFs that were transiently transfected with siRNA directed against PKR (since HF-shPKR is puromycin resistant, the compound will not be incorporated into polypeptides in these cells). As shown in Fig. 9A, transfection of siRNA directed against PKR leads to diminishment of the protein to a nearly undetectable level. However, protein synthesis as detected by incorporated puromycin following CHIKV infection was absent in cells pretreated with either nonspecific or PKR-directed siRNA (Fig. 9A). Since puromycin incorporation is not sensitive enough to detect individual proteins, as evidenced by the fact that bands corresponding to capsid protein are not detectable in anti-puromycin immunoblots (Fig. 6 and 9), we decided to specifically examine whether IFN-β or ISG proteins are synthesized in the absence of PKR. As shown in Fig. 9B, CHIKV infection of HF-shPKR did not result in synthesis of ISG56 or Viperin proteins. Moreover, CHIKV-induced secretion of IFN-α/β was also not observed in these cells (Fig. 9C). We therefore conclude that while CHIKV infection triggers PKR-dependent phosphorylation of eIF2α, the observed virus-associated block to cellular protein synthesis is unrelated to this process.
FIG. 9.

CHIKV-associated cellular translational shutoff occurs in the absence of PKR. (A) Immunoblot showing PKR, CHIKV capsid, GAPDH, and puromycin incorporated into newly synthesized protein in HFs either left untreated or infected with CHIKV for 4, 8, 12, or 16 h at an MOI of 10 following the transfection of nonspecific (NS) or PKR-directed siRNA. (B) Immunoblot showing PKR, ISG56, Viperin, CHIKV capsid, and GAPDH in HF or HF-shPKR following no treatment, transfection with poly(I:C), or infection with CHIKV for 24 h. (C) Expression of IFN-α/β-dependent luciferase from reporter cells after exposure to media collected from HFs or HF-shPKRs that were left untreated or infected with SeV (160 HA units/ml) or CHIKV (MOI = 10) for 24 h. Treatments were performed in quadruplicate, and media were transferred to confluent THF-ISRE in six wells of a 96-well plate per treatment. Values presented are average luminescence readings from four separate treatments ± the SEM.
CHIKV induces shutoff of host cell transcription of IFN-β and ISGs.
Old World Alphavirus species such as SINV are known to inhibit both cellular translation (28) and transcription (32) by way of distinct mechanisms (34, 35). Since neither phenomenon has previously been examined during CHIKV infection, we decided to next determine whether the virus also inhibits host cell transcription, which could contribute to the diminution of cellular protein synthesis observed (Fig. 6). To address this, we used a previously described technique (22) involving the addition of 4-thiouridine to culture medium that is incorporated into newly synthesized RNA. Biotin is then enzymatically added to the thiol groups following the isolation of total RNA, which subsequently enables separation into newly synthesized (biotinylated) and preexisting (unbiotinylated) RNA using streptavidin bead-based positive selection.
As shown in Fig. 10A, treatment of HFs with the RNA transcription inhibitor actinomycin D (ActD) leads to a ratio of newly synthesized (biotinylated) to total RNA that is greatly reduced relative to the ratio observed in untreated cells. When HFs were infected with CHIKV or SINV at an MOI of 10 a time-dependent decrease in the ratio of newly synthesized to total RNA was also seen (Fig. 10A). However, while the ratio levels off by 16 h after SINV infection, the ratio is maximal at 24 h after CHIKV infection. Moreover, the ratio of new to total RNA at 24 h after CHIKV infection is actually slightly below that seen after treatment with ActD, indicating that infection with the virus leads to potent inhibition of RNA synthesis. However, our data indicate that IFN-β and ISG mRNAs are abundant at 24 h after CHIKV infection (Fig. 1). We therefore hypothesize that this is due to transcription that occurred prior to this time point. To address this, we performed RT-PCR to examine the presence of these transcripts in newly synthesized (biotinylated) and preexisting (unbiotinylated) mRNA pools following infection with CHIKV or SINV. As shown in Fig. 10B at 6 h postinfection mRNAs for IFN-β, ISG56, and Viperin, as well as β-actin and GAPDH housekeeping genes, are present in both the newly synthesized and preexisting RNA fractions from virus-infected cells. Yet while cells infected with CHIKV or SINV synthesize housekeeping genes at 24 h postinfection, only SINV-infected cells synthesize IFN-β/ISGs (Fig. 10B). These results indicate that while both viruses lead to diminution of overall RNA synthesis in infected cells, transcription of IFN-β/ISGs, but not housekeeping genes, appears specifically blocked only in CHIKV-infected cells. Whether this observation represents a CHIKV-specific inhibitory phenotype directed at IRF3-dependent genes that contributes to the observed translational block will require further exploration.
FIG. 10.

Infection with CHIKV or SINV reduces levels of cellular transcription. (A) Ratio of biotinylated (newly synthesized) to total RNA in HF following (i) no treatment, (ii) exposure to 5 μg of actinomycin D (ActD) ml−1, or (iii) infection with CHIKV or SINV (MOI = 10) for the indicated durations. The ratio obtained from untreated cells was set to 1. (B) Agarose gel showing amplification products of RT-PCR generated using biotinylated or unbiotinylated RNA fractions as templates that were harvested from HFs either (i) left untreated, (ii) exposed to 5 μg of ActD ml−1, or (iii) infected with CHIKV or SINV (MOI = 10) for 6 or 24 h.
DISCUSSION
The nature of human immune responses to CHIKV and the strategies used by the virus to withstand these are poorly characterized phenomena. In light of this, we undertook an investigation of the innate antiviral reactions to CHIKV by human fibroblasts, a known in vivo target of the virus. Our goal was to acquire information on some of the basic responses, processes, and molecules involved in immune activation by CHIKV in host cells that are both nontransformed (see reference 4) and relevant to the virus's replication and pathogenesis. We focused on one of the most rapid and consequential innate immune responses to alphaviral infection, namely, the induction of type I IFN and the expression of ISGs through the activation of IRF3. Since CHIKV is highly susceptible to the actions of IFN-α/β, information regarding the molecular and biochemical bases of IFN induction by the virus and its ability to replicate in the face of this induction is likely to be of great utility to the design of antiviral therapies.
Our data show MOI-dependent upregulation of IFN-β, ISG56, and Viperin mRNA during CHIKV infection. We further observed Ser398 phosphorylation and nuclear accumulation of IRF3 during infection that occurs after the appearance of dsRNA. While shown for other alphaviruses in nonhuman cells (11), activation of IRF3 during CHIKV infection of human cells has until this point not been described. Importantly, we also show that CHIKV-triggered IFN-β/ISG mRNA accumulation is directly dependent on IRF3 and does not require JAK/STAT activity since (i) Transcription of these genes does not occur following siRNA-directed depletion or NPro-mediated degradation of IRF3, (ii) these genes are induced despite the fact that CHIKV does not stimulate IFN-α/β secretion in these cells (Fig. 5A), and (iii) infection does not induce mRNA accumulation of the IFN-dependent ISG Mx1. Interestingly, however, while IFN-β/ISG expression is evident at an MOI of 0.1 (Fig. 1) and as early as 6 h postinfection (Fig. 10), IRF3 Ser398 phosphorylation is only weakly detected at this MOI and is not substantial until after 8 h postinfection (Fig. 2). It is possible that the amount of Ser398-phosphorylated IRF3 protein is below the detection limit of this assay yet is still functionally active at this MOI and time point. It is also possible that innate responses to CHIKV at early times postinfection or following low MOI exposure result in the phosphorylation of other serine or threonine residues that result in activation of the protein (e.g., see reference 57). We are currently attempting to distinguish between these alternatives. To our knowledge, this represents the first demonstration of the direct requirement of IRF3 for alphavirus-mediated induction of IFN-β and ISGs. It is worth noting that IRF3 is not required for such transcriptional induction by all viruses, however. Prescott et al. showed that ISG56 and Mx1 were transcriptionally induced in HUH-7 cells infected with Sin Nombre virus (a Hantavirus) following siRNA-mediated knockdown of IRF3 (65). Daffis et al. recently showed type I IFN secretion in mice lacking both IRF3 and IRF7 (DKO) after infection with West Nile virus (WNV) (17). Interestingly, these authors also examined virus-triggered IFN-β transcription in macrophages harvested from these mice and saw no difference between WT and DKO macrophages infected with WNV, encephalomyocarditis virus, or CHIKV strain 142 (17). While this result may appear to contrast with data presented here, the disparity could be related to differences in cell type or viral strain.
We also show that CHIKV-mediated phosphorylation of IRF3 and subsequent activation of IRF3-dependent transcription requires the adaptor protein IPS-1. As shown in Fig. 4, CHIKV infection of HFs involves cytoplasmic accumulation of dsRNA, a strong stimulator of IRF3-dependent gene expression. Cytoplasmic dsRNA is detected by two known IRF3-terminal PRRs, MDA5 and RIG-I, that both signal via IPS-1. Our results are thus consistent with activation of IRF3 through detection of dsRNA by MDA5 and/or RIG-I. However, while we are currently examining the potential roles of these proteins for CHIKV-triggered IRF3 activation, we have thus far been unable to determine the essentiality of either molecule. Our results in human cells agree with recent data from Schilte et al. in which an essential role was found for IPS-1 in IFN-β induction by murine cells following CHIKV infection (76). Consistent with this, IFN-α/β secretion was diminished relative to WT cells in MDA5−/− MEFs infected with SINV (11). However, Schilte et al. observed a reduction in CHIKV-triggered IFN-β transcription in RIG-I−/− murine embryonic fibroblasts (MEFs) relative to WT cells exceeding that seen in MDA5−/− MEFs (76). In another recent study, IFN-stimulatory total RNA was harvested from cells infected with the alphavirus SFV and transfected into MEFs, but no difference in IFN-α/β secretion was observed between WT, RIG-I−/−, or MDA5−/− cells (64). Thus, great observational disparities exist regarding Alphavirus-triggered IFN responses that may be due to biological differences between viral species or strains or perhaps is related to the species or genetic background of host cells (see reference 76). However, the role of an IPS-1-dependent signaling pathway in CHIKV-induced IFN-β synthesis appears clear even across host species.
In contrast to these previous reports, however, transcriptional induction of IFN-β and ISGs by CHIKV was not reflected in the synthesis of corresponding proteins. Thus, unlike the RNA virus SeV and the Alphavirus SINV, infection of our target cells with CHIKV (even at high MOI) led to no appreciable IFN-β, ISG56, or Viperin protein, and yet viral protein synthesis was obvious. While these findings represent, to our knowledge, the first examination of ISG protein induction by CHIKV, the results we obtained for IFN-β secretion seemingly contrast with those published in a previous study (76). The authors of that study show that CHIKV-triggered IFN-β secretion from human lung (MRC-5) and foreskin fibroblasts that is MOI dependent. Although barely detectable IFN secretion from CHIKV-infected relative to untreated HFs was observed in our case, this was in no way MOI dependent. Our data are in agreement, however, with a study by Burke et al. in which the authors detect no type I IFN secretion from MEFs infected with CHIKV for 8, 12, 24, or 32 h (11). It is possible that the disparity between the two studies is related to the strains of CHIKV used. While Burke et al. (11) and the present study used the CHIKV-LR strain (2, 3, 84), Schilte et al. (76) used CHIKV-21 strain. Whether this is responsible for the differences observed between these studies will require further exploration.
We next examined whether the absence of IFN-β secretion and ISG protein synthesis in response to CHIKV infection could be due to a virus-associated, widespread block in cellular translation. To address this, we examined polypeptide-incorporated puromycin using SDS-PAGE in CHIKV-infected cells. Infection of HFs led to the diminishment of puromycin in cellular protein by 8 h postinfection and puromycin was undetectable by 16 h postinfection. Based on this, we conclude that a global shutoff of cellular gene translation may contribute to the lack of IFN-β and ISG protein synthesis during CHIKV infection. Determining whether cellular translational shutoff is essential to the lack of these proteins will require experimental approaches involving inhibition of this response. We nevertheless hypothesize that this represents a strategy of immune evasion in which virus infection leads to prevention of the synthesis of cellular proteins, especially those that are actually or potentially detrimental to virus replication (e.g., ISGs). Although translational shutoff has not been previously investigated for CHIKV, it has been described for other alphaviruses such as SINV (28) and SFV (53). In SFV this is at least partially due to virus-triggered phosphorylation of eIF2α (53). Moreover, the nsP2 proteins of SINV (32-34) and SFV (7, 33) have been implicated in both the suppression of host transcription and IFN evasion, although the molecular bases for these have not been characterized. Interestingly, we also observed translational shutoff in SINV-infected HFs (data not shown) and, as such, this phenomenon is unlikely to provide the sole explanation for the absence of CHIKV-induced IFN/ISG protein. The causal relationship between antiviral gene translation and the Alphavirus-associated shutoff of protein synthesis clearly requires more thorough examination. Intriguing work by Frolov and coworkers has shown that little to no IFN is secreted from NIH 3T3 mouse cells infected with wild-type SINV (strain TE12) yet when a mutant is used that fails to induce translational shutoff strong IFN secretion is induced (28, 32). This result is consistent with our observations for and inferences about CHIKV replication in human cells. However, in contrast to those findings we detected high levels of secreted IFN-α/β from SINV-infected cells (Fig. 5A). It is also important to note that we observed translational shutoff in both MEFs and human fibroblasts after infection with SINV and in MEFs after CHIKV infection (data not shown). As such, differences in host cell species are not likely to account completely for the observed SINV and CHIKV differences in IFNα/β secretion. This may, however, be related to differences in the virus strain (TE12 versus Ar339), and our results do agree with other studies examining SINV-triggered IFN secretion (29, 71). The bases of these disparities will require more careful examination.
Phosphorylation of eIF2α on Ser51 leads to shutoff of mRNA translation by reducing levels of ternary complex eIF2-GTP-Met-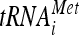 . While never examined for CHIKV, phosphorylation of eIF2α Ser51 has been demonstrated for SINV (85) and SFV (53). eIF2α phosphorylation can occur via four upstream kinases that react to cytoplasmic dsRNA (PKR), heme deficiency, heat shock and oxidative stress (heme-regulated inhibitor [HRI]), amino acid or serum deprivation or UV irradiation (general control nonderepressible-2 [GCN2]), or unfolded proteins (PKR-like endoplasmic reticulum kinase [PERK]). Since replication of alphaviruses (including CHIKV; Fig. 4) involves the synthesis of dsRNA, a role for PKR in eIF2α phosphorylation has been hypothesized. Indeed, Gorchakov et al. have shown SINV-induced eIF2α phosphorylation in PKR-positive NIH 3T3 MEFs but not PKR−/− MEFs (35). These authors also describe increased eIF2α phosphorylation in NIH 3T3 cells infected with SINV replicons that express WT PKR and a lack of phosphorylation in cells infected with replicons expressing a PKR dominant-negative mutant (35). Ventoso et al. have also described dependence of eIF2α phosphorylation on PKR during SINV infection (85). However, in contrast to these data, Berlanga et al. have shown diminished SINV-associated eIF2α phosphorylation in GCN2−/− MEFs relative to WT cells (5). Curiously, however, these authors observed no change in eIF2α phosphorylation in PKR−/− MEFs relative to WT cells following SINV infection (5). Our finding that CHIKV-induced eIF2α phosphorylation is greatly diminished following shRNA-mediated knockdown of PKR is in agreement with the data of Gorchakov et al. and Ventoso et al., and yet a potential role for GCN2 remains to be examined.
. While never examined for CHIKV, phosphorylation of eIF2α Ser51 has been demonstrated for SINV (85) and SFV (53). eIF2α phosphorylation can occur via four upstream kinases that react to cytoplasmic dsRNA (PKR), heme deficiency, heat shock and oxidative stress (heme-regulated inhibitor [HRI]), amino acid or serum deprivation or UV irradiation (general control nonderepressible-2 [GCN2]), or unfolded proteins (PKR-like endoplasmic reticulum kinase [PERK]). Since replication of alphaviruses (including CHIKV; Fig. 4) involves the synthesis of dsRNA, a role for PKR in eIF2α phosphorylation has been hypothesized. Indeed, Gorchakov et al. have shown SINV-induced eIF2α phosphorylation in PKR-positive NIH 3T3 MEFs but not PKR−/− MEFs (35). These authors also describe increased eIF2α phosphorylation in NIH 3T3 cells infected with SINV replicons that express WT PKR and a lack of phosphorylation in cells infected with replicons expressing a PKR dominant-negative mutant (35). Ventoso et al. have also described dependence of eIF2α phosphorylation on PKR during SINV infection (85). However, in contrast to these data, Berlanga et al. have shown diminished SINV-associated eIF2α phosphorylation in GCN2−/− MEFs relative to WT cells (5). Curiously, however, these authors observed no change in eIF2α phosphorylation in PKR−/− MEFs relative to WT cells following SINV infection (5). Our finding that CHIKV-induced eIF2α phosphorylation is greatly diminished following shRNA-mediated knockdown of PKR is in agreement with the data of Gorchakov et al. and Ventoso et al., and yet a potential role for GCN2 remains to be examined.
Since CHIKV infection leads to both PKR-mediated eIF2α phosphorylation and shutoff of cellular protein synthesis, we next sought to determine whether the two events were causally linked. However, a CHIKV-associated block to cellular protein translation was still seen following siRNA-mediated knockdown of PKR expression (Fig. 8). In addition, knockdown of PKR failed to reestablish synthesis of ISG56, Viperin, or IFN-β protein following CHIKV infection (Fig. 8). Based on these results, we conclude that the translational shutoff observed does not result from CHIKV-induced, PKR-dependent phosphorylation of eIF2α. Although a specific role for PKR in CHIKV infection has not previously been examined, the molecule has been investigated with respect to SINV replication. In this case translational inhibition was shown to occur following SINV infection as efficiently in PKR−/− MEFs, as in wild-type NIH 3T3 cells (35). In addition, viable SINV mutants have been constructed that fail to induce a block in cellular translation, suggesting that the virus achieves this effect via an active genomically encoded factor (34, 35). Much additional work thus remains regarding the molecular basis of Alphavirus-mediated shutoff of cellular translation. Recently, a related phenomenon was described during in vitro infection with hepatitis C virus (HCV) (30). Infection was found to lead to PKR-dependent phosphorylation of eIF2α. Furthermore, this process inhibited cellular (including ISG) protein synthesis, which was restored during PKR knock down. Thus, HCV appears to effectively exploit an innate antiviral response for a proviral objective. It is also possible that this represents a strategy used by many RNA viruses to evade the effects of antiviral gene expression.
We next examined whether CHIKV infection leads to a decrease in RNA transcription, a response described for other alphaviruses and that could contribute to the differences in IFN/ISG translation observed here between SINV and CHIKV. To address this, we used a technique that allows the separation of newly synthesized from preexisting cellular RNA using biotinylation of incorporated 4-thiouridine (22). Using this approach we found that while infection of HFs with either SINV or CHIKV leads to reduced host cell transcription, CHIKV leads to an overall greater transcriptional reduction at 24 h postinfection (Fig. 10A). Moreover, while cells infected with SINV appear to be actively synthesizing mRNA as late as 24 h postinfection, cells infected with CHIKV are synthesizing mRNA for housekeeping genes but not IFN-β/ISGs (Fig. 10B). Based on these observations we conclude that CHIKV and SINV differ in their abilities to inhibit both overall cellular RNA transcription and transcription of IRF3-dependent mRNAs. A role for this more potent and potentially target-specific CHIKV-induced transcriptional block in the lack of IFN-β/ISG protein synthesis observed during infection will require further exploration and is currently being investigated.
Our work aims to describe and characterize basic aspects of innate immune induction and evasion by CHIKV in a clinically relevant cell model. We have shown that the virus strongly induces, via IPS-1, accumulation of IRF3-dependent mRNAs but that it also efficiently prevents synthesis of corresponding proteins, perhaps through blocking global cellular protein synthesis. The extent to which this translational block represents an immune evasion strategy whose purpose is to avoid the antiviral effects of IFN and ISG proteins is unknown but is a current research focus in our laboratory. In addition, we have shown that CHIKV leads to PKR-dependent phosphorylation of eIF2α but that this process is not essential to translational shutoff. It is possible that the action of PKR during CHIKV infection leads to changes in the kinetics of viral or cellular protein synthesis. In addition, CHIKV also induces shutoff of RNA transcription that may specifically target IRF3-dependent genes and the relationship between this effect and the absence of ISG proteins requires further elucidation. The molecular basis of Alphavirus-induced shutoff of cellular transcription and translation remain important areas of inquiry and detailed characterization of these phenomena are likely to have profound implications for the development of anti-Alphavirus therapies.
Acknowledgments
We thank Steven Higgs, Ganes Sen, Irene Greiser-Wilke, Peter Cresswell, Wade Bresnahan, Jay Nelson, Klaus Früh, and Roger Everett for kindly providing reagents.
This study was supported by NIH career development grant CD001 from the Pacific Northwest Regional Center of Excellence (V.R.D.) and by HSFP, ANR DC-TRANS, and LNCC (P.P.).
Footnotes
Published ahead of print on 20 October 2010.
REFERENCES
- 1.Ablasser, A., F. Bauernfeind, G. Hartmann, E. Latz, K. A. Fitzgerald, and V. Hornung. 2009. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 10:1065-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aguilar, P. V., S. C. Weaver, and C. F. Basler. 2007. Capsid protein of eastern equine encephalitis virus inhibits host cell gene expression. J. Virol. 81:3866-3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anishchenko, M., S. Paessler, I. P. Greene, P. V. Aguilar, A. S. Carrara, and S. C. Weaver. 2004. Generation and characterization of closely related epizootic and enzootic infectious cDNA clones for studying interferon sensitivity and emergence mechanisms of Venezuelan equine encephalitis virus. J. Virol. 78:1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartee, E., and G. McFadden. 2009. Human cancer cells have specifically lost the ability to induce the synergistic state caused by tumor necrosis factor plus interferon-beta. Cytokine 47:199-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berlanga, J. J., I. Ventoso, H. P. Harding, J. Deng, D. Ron, N. Sonenberg, L. Carrasco, and C. de Haro. 2006. Antiviral effect of the mammalian translation initiation factor 2α kinase GCN2 against RNA viruses. EMBO J. 25:1730-1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boudinot, P., S. Riffault, S. Salhi, C. Carrat, C. Sedlik, N. Mahmoudi, B. Charley, and A. Benmansour. 2000. Vesicular stomatitis virus and pseudorabies virus induce a vig1/cig5 homologue in mouse dendritic cells via different pathways. J. Gen. Virol. 81:2675-2682. [DOI] [PubMed] [Google Scholar]
- 7.Breakwell, L., P. Dosenovic, G. B. Karlsson-Hedestam, M. D'Amato, P. Liljestrom, J. Fazakerley, and G. M. McInerney. 2007. Semliki Forest virus nonstructural protein 2 is involved in suppression of the type I interferon response. J. Virol. 81:8677-8684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bresnahan, W. A., G. E. Hultman, and T. Shenk. 2000. Replication of wild-type and mutant human cytomegalovirus in life-extended human diploid fibroblasts. J. Virol. 74:10816-10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brighton, S. W., O. W. Prozesky, and A. L. de la Harpe. 1983. Chikungunya virus infection. A retrospective study of 107 cases. S Afr. Med. J. 63:313-315. [PubMed] [Google Scholar]
- 10.Browne, E. P., B. Wing, D. Coleman, and T. Shenk. 2001. Altered cellular mRNA levels in human cytomegalovirus-infected fibroblasts: viral block to the accumulation of antiviral mRNAs. J. Virol. 75:12319-12330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burke, C. W., C. L. Gardner, J. J. Steffan, K. D. Ryman, and W. B. Klimstra. 2009. Characteristics of alpha/beta interferon induction after infection of murine fibroblasts with wild-type and mutant alphaviruses. Virology 395:121-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen, Z., R. Rijnbrand, R. K. Jangra, S. G. Devaraj, L. Qu, Y. Ma, S. M. Lemon, and K. Li. 2007. Ubiquitination and proteasomal degradation of interferon regulatory factor-3 induced by Npro from a cytopathic bovine viral diarrhea virus. Virology 366:277-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chew, T., R. Noyce, S. E. Collins, M. H. Hancock, and K. L. Mossman. 2009. Characterization of the interferon regulatory factor 3-mediated antiviral response in a cell line deficient for IFN production. Mol. Immunol. 46:393-399. [DOI] [PubMed] [Google Scholar]
- 14.Chiu, Y. H., J. B. Macmillan, and Z. J. Chen. 2009. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138:576-591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Couderc, T., F. Chretien, C. Schilte, O. Disson, M. Brigitte, F. Guivel-Benhassine, Y. Touret, G. Barau, N. Cayet, I. Schuffenecker, P. Despres, F. Arenzana-Seisdedos, A. Michault, M. L. Albert, and M. Lecuit. 2008. A mouse model for Chikungunya: young age and inefficient type-I interferon signaling are risk factors for severe disease. PLoS Pathog. 4:e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cui, S., K. Eisenacher, A. Kirchhofer, K. Brzozka, A. Lammens, K. Lammens, T. Fujita, K. K. Conzelmann, A. Krug, and K. P. Hopfner. 2008. The C-terminal regulatory domain is the RNA 5′-triphosphate sensor of RIG-I. Mol. Cell 29:169-179. [DOI] [PubMed] [Google Scholar]
- 17.Daffis, S., M. S. Suthar, K. J. Szretter, M. Gale, Jr., and M. S. Diamond. 2009. Induction of IFN-beta and the innate antiviral response in myeloid cells occurs through an IPS-1-dependent signal that does not require IRF-3 and IRF-7. PLoS Pathog. 5:e1000607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DeFilippis, V. R., D. Alvarado, T. Sali, S. Rothenburg, and K. Früh. 2010. Human cytomegalovirus induces the interferon response via the DNA sensor ZBP1. J. Virol. 84:585-598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DeFilippis, V. R., B. Robinson, T. M. Keck, S. G. Hansen, J. A. Nelson, and K. Früh. 2006. Interferon regulatory factor 3 is necessary for induction of antiviral genes during human cytomegalovirus infection. J. Virol. 80:1032-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Despres, P., J. W. Griffin, and D. E. Griffin. 1995. Antiviral activity of alpha interferon in Sindbis virus-infected cells is restored by anti-E2 monoclonal antibody treatment. J. Virol. 69:7345-7348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deuber, S. A., and J. Pavlovic. 2007. Virulence of a mouse-adapted Semliki Forest virus strain is associated with reduced susceptibility to interferon. J. Gen. Virol. 88:1952-1959. [DOI] [PubMed] [Google Scholar]
- 22.Dolken, L., Z. Ruzsics, B. Radle, C. C. Friedel, R. Zimmer, J. Mages, R. Hoffmann, P. Dickinson, T. Forster, P. Ghazal, and U. H. Koszinowski. 2008. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA 14:1959-1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Enserink, M. 2006. Infectious diseases. Massive outbreak draws fresh attention to little-known virus. Science 311:1085. [DOI] [PubMed] [Google Scholar]
- 24.Everett, R. D., D. F. Young, R. E. Randall, and A. Orr. 2008. STAT-1- and IRF-3-dependent pathways are not essential for repression of ICP0-null mutant herpes simplex virus type 1 in human fibroblasts. J. Virol. 82:8871-8881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fitzgerald, K. A., S. M. McWhirter, K. L. Faia, D. C. Rowe, E. Latz, D. T. Golenbock, A. J. Coyle, S. M. Liao, and T. Maniatis. 2003. IKKɛand TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4:491-496. [DOI] [PubMed] [Google Scholar]
- 26.Fourie, E. D., and J. G. Morrison. 1979. Rheumatoid arthritic syndrome after Chikungunya fever. S. Afr. Med. J. 56:130-132. [PubMed] [Google Scholar]
- 27.Friedman, R. M., and J. A. Sonnabend. 1965. Inhibition by interferon of production of double-stranded Semliki Forest virus ribonucleic acid. Nature 206:532. [DOI] [PubMed] [Google Scholar]
- 28.Frolov, I., and S. Schlesinger. 1994. Comparison of the effects of Sindbis virus and Sindbis virus replicons on host cell protein synthesis and cytopathogenicity in BHK cells. J. Virol. 68:1721-1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frolova, E. I., R. Z. Fayzulin, S. H. Cook, D. E. Griffin, C. M. Rice, and I. Frolov. 2002. Roles of nonstructural protein nsP2 and alpha/beta interferons in determining the outcome of Sindbis virus infection. J. Virol. 76:11254-11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garaigorta, U., and F. V. Chisari. 2009. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host Microbe 6:513-522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gardner, J., I. Anraku, T. T. Le, T. Larcher, L. Major, P. Roques, W. A. Schroder, S. Higgs, and A. Suhrbier. 2010. Chikungunya virus arthritis in adult wild-type mice. J. Virol. 84:8021-8032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garmashova, N., R. Gorchakov, E. Frolova, and I. Frolov. 2006. Sindbis virus nonstructural protein nsP2 is cytotoxic and inhibits cellular transcription. J. Virol. 80:5686-5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garmashova, N., R. Gorchakov, E. Volkova, S. Paessler, E. Frolova, and I. Frolov. 2007. The Old World and New World alphaviruses use different virus-specific proteins for induction of transcriptional shutoff. J. Virol. 81:2472-2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gorchakov, R., E. Frolova, and I. Frolov. 2005. Inhibition of transcription and translation in Sindbis virus-infected cells. J. Virol. 79:9397-9409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorchakov, R., E. Frolova, B. R. Williams, C. M. Rice, and I. Frolov. 2004. PKR-dependent and -independent mechanisms are involved in translational shutoff during Sindbis virus infection. J. Virol. 78:8455-8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gould, E. A., and S. Higgs. 2009. Impact of climate change and other factors on emerging arbovirus diseases. Trans. R. Soc. Trop. Med. Hyg. 103:109-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grandvaux, N., M. J. Servant, B. tenOever, G. C. Sen, S. Balachandran, G. N. Barber, R. Lin, and J. Hiscott. 2002. Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J. Virol. 76:5532-5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Greiser-Wilke, I., V. Moenning, O. R. Kaaden, and L. T. Figueiredo. 1989. Most alphaviruses share a conserved epitopic region on their nucleocapsid protein. J. Gen. Virol. 70(Pt. 3):743-748. [DOI] [PubMed] [Google Scholar]
- 39.Hidmark, A. S., G. M. McInerney, E. K. Nordstrom, I. Douagi, K. M. Werner, P. Liljestrom, and G. B. Karlsson Hedestam. 2005. Early alpha/beta interferon production by myeloid dendritic cells in response to UV-inactivated virus requires viral entry and interferon regulatory factor 3 but not MyD88. J. Virol. 79:10376-10385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hilton, L., K. Moganeradj, G. Zhang, Y. H. Chen, R. E. Randall, J. W. McCauley, and S. Goodbourn. 2006. The NPro product of bovine viral diarrhea virus inhibits DNA binding by interferon regulatory factor 3 and targets it for proteasomal degradation. J. Virol. 80:11723-11732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hiscott, J., R. Lin, P. Nakhaei, and S. Paz. 2006. MasterCARD: a priceless link to innate immunity. Trends Mol. Med. 12:53-56. [DOI] [PubMed] [Google Scholar]
- 42.Hochedez, P., S. Jaureguiberry, M. Debruyne, P. Bossi, P. Hausfater, G. Brucker, F. Bricaire, and E. Caumes. 2006. Chikungunya infection in travelers. Emerg. Infect. Dis. 12:1565-1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hornung, V., J. Ellegast, S. Kim, K. Brzozka, A. Jung, H. Kato, H. Poeck, S. Akira, K. K. Conzelmann, M. Schlee, S. Endres, and G. Hartmann. 2006. 5′-Triphosphate RNA is the ligand for RIG-I. Science 314:994-997. [DOI] [PubMed] [Google Scholar]
- 44.Kato, H., O. Takeuchi, E. Mikamo-Satoh, R. Hirai, T. Kawai, K. Matsushita, A. Hiiragi, T. S. Dermody, T. Fujita, and S. Akira. 2008. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 205:1601-1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kawai, T., and S. Akira. 2006. Innate immune recognition of viral infection. Nat. Immunol. 7:131-137. [DOI] [PubMed] [Google Scholar]
- 46.Kawai, T., K. Takahashi, S. Sato, C. Coban, H. Kumar, H. Kato, K. J. Ishii, O. Takeuchi, and S. Akira. 2005. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 6:981-988. [DOI] [PubMed] [Google Scholar]
- 47.Kennedy, A. C., J. Fleming, and L. Solomon. 1980. Chikungunya viral arthropathy: a clinical description. J. Rheumatol. 7:231-236. [PubMed] [Google Scholar]
- 48.Kinsella, T. M., and G. P. Nolan. 1996. Episomal vectors rapidly and stably produce high-titer recombinant retrovirus. Hum. Gene Ther. 7:1405-1413. [DOI] [PubMed] [Google Scholar]
- 49.Lam, S. K., K. B. Chua, P. S. Hooi, M. A. Rahimah, S. Kumari, M. Tharmaratnam, S. K. Chuah, D. W. Smith, and I. A. Sampson. 2001. Chikungunya infection: an emerging disease in Malaysia. Southeast Asian J. Trop. Med. Public Health 32:447-451. [PubMed] [Google Scholar]
- 50.Lin, R., C. Heylbroeck, P. M. Pitha, and J. Hiscott. 1998. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 18:2986-2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar]
- 52.Mason, P. J., and A. J. Haddow. 1957. An epidemic of virus disease in Southern Province, Tanganyika Territory, in 1952-53: an additional note on Chikungunya virus isolations and serum antibodies. Trans. R. Soc. Trop. Med. Hyg. 51:238-240. [DOI] [PubMed] [Google Scholar]
- 53.McInerney, G. M., N. L. Kedersha, R. J. Kaufman, P. Anderson, and P. Liljestrom. 2005. Importance of eIF2α phosphorylation and stress granule assembly in alphavirus translation regulation. Mol. Biol. Cell 16:3753-3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Merika, M., A. J. Williams, G. Chen, T. Collins, and D. Thanos. 1998. Recruitment of CBP/p300 by the IFNβ enhanceosome is required for synergistic activation of transcription. Mol. Cell 1:277-287. [DOI] [PubMed] [Google Scholar]
- 55.Meylan, E., J. Curran, K. Hofmann, D. Moradpour, M. Binder, R. Bartenschlager, and J. Tschopp. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167-1172. [DOI] [PubMed] [Google Scholar]
- 56.Reference deleted.
- 57.Noyce, R. S., S. E. Collins, and K. L. Mossman. 2009. Differential modification of interferon regulatory factor 3 following virus particle entry. J. Virol. 83:4013-4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Paladino, P., D. T. Cummings, R. S. Noyce, and K. L. Mossman. 2006. The IFN-independent response to virus particle entry provides a first line of antiviral defense that is independent of TLRs and retinoic acid-inducible gene I. J. Immunol. 177:8008-8016. [DOI] [PubMed] [Google Scholar]
- 59.Perri, S., C. E. Greer, K. Thudium, B. Doe, H. Legg, H. Liu, R. E. Romero, Z. Tang, Q. Bin, T. W. Dubensky, Jr., M. Vajdy, G. R. Otten, and J. M. Polo. 2003. An alphavirus replicon particle chimera derived from Venezuelan equine encephalitis and Sindbis viruses is a potent gene-based vaccine delivery vector. J. Virol. 77:10394-10403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Perry, A. K., G. Chen, D. Zheng, H. Tang, and G. Cheng. 2005. The host type I interferon response to viral and bacterial infections. Cell Res. 15:407-422. [DOI] [PubMed] [Google Scholar]
- 61.Peters, K. L., H. L. Smith, G. R. Stark, and G. C. Sen. 2002. IRF-3-dependent, NFκB- and JNK-independent activation of the 561 and IFN-beta genes in response to double-stranded RNA. Proc. Natl. Acad. Sci. U. S. A. 99:6322-6327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pialoux, G., B. A. Gauzere, S. Jaureguiberry, and M. Strobel. 2007. Chikungunya, an epidemic arbovirosis. Lancet Infect. Dis. 7:319-327. [DOI] [PubMed] [Google Scholar]
- 63.Pichlmair, A., O. Schulz, C. P. Tan, T. I. Naslund, P. Liljestrom, F. Weber, and C. Reis e Sousa. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314:997-1001. [DOI] [PubMed] [Google Scholar]
- 64.Pichlmair, A., O. Schulz, C. P. Tan, J. Rehwinkel, H. Kato, O. Takeuchi, S. Akira, M. Way, G. Schiavo, and C. Reis e Sousa. 2009. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J. Virol. 83:10761-10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Prescott, J. B., P. R. Hall, V. S. Bondu-Hawkins, C. Ye, and B. Hjelle. 2007. Early innate immune responses to Sin Nombre hantavirus occur independently of IFN regulatory factor 3, characterized pattern recognition receptors, and viral entry. J. Immunol. 179:1796-1802. [DOI] [PubMed] [Google Scholar]
- 66.Reed, L., and H. Muench. 1938. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 27:493-497. [Google Scholar]
- 67.Reiter, P., D. Fontenille, and C. Paupy. 2006. Aedes albopictus as an epidemic vector of Chikungunya virus: another emerging problem? Lancet Infect. Dis. 6:463-464. [DOI] [PubMed] [Google Scholar]
- 68.Robinson, M. C. 1955. An epidemic of virus disease in Southern Province, Tanganyika Territory, in 1952-53. I. Clinical features. Trans. R. Soc. Trop. Med. Hyg. 49:28-32. [DOI] [PubMed] [Google Scholar]
- 69.Ross, R. W. 1956. The Newala epidemic. III. The virus: isolation, pathogenic properties and relationship to the epidemic. J. Hyg. (Lond.) 54:177-191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ryman, K. D., and W. B. Klimstra. 2008. Host responses to alphavirus infection. Immunol. Rev. 225:27-45. [DOI] [PubMed] [Google Scholar]
- 71.Ryman, K. D., L. J. White, R. E. Johnston, and W. B. Klimstra. 2002. Effects of PKR/RNase L-dependent and alternative antiviral pathways on alphavirus replication and pathogenesis. Viral Immunol. 15:53-76. [DOI] [PubMed] [Google Scholar]
- 72.Saito, T., and M. Gale, Jr. 2008. Differential recognition of double-stranded RNA by RIG-I-like receptors in antiviral immunity. J. Exp. Med. 205:1523-1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Saito, T., D. M. Owen, F. Jiang, J. Marcotrigiano, and M. Gale, Jr. 2008. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454:523-527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sato, M., H. Suemori, N. Hata, M. Asagiri, K. Ogasawara, K. Nakao, T. Nakaya, M. Katsuki, S. Noguchi, N. Tanaka, and T. Taniguchi. 2000. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 13:539-548. [DOI] [PubMed] [Google Scholar]
- 75.Saxena, S. K., M. Singh, N. Mishra, and V. Lakshmi. 2006. Resurgence of Chikungunya virus in India: an emerging threat. Euro Surveill. 11:E0608102. [DOI] [PubMed] [Google Scholar]
- 76.Schilte, C., T. Couderc, F. Chretien, M. Sourisseau, N. Gangneux, F. Guivel-Benhassine, A. Kraxner, J. Tschopp, S. Higgs, A. Michault, F. Arenzana-Seisdedos, M. Colonna, L. Peduto, O. Schwartz, M. Lecuit, and M. L. Albert. 2010. Type I IFN controls Chikungunya virus via its action on nonhematopoietic cells. J. Exp. Med. 207:429-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schmidt, E. K., G. Clavarino, M. Ceppi, and P. Pierre. 2009. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 6:275-277. [DOI] [PubMed] [Google Scholar]
- 78.Schuffenecker, I., I. Iteman, A. Michault, S. Murri, L. Frangeul, M. C. Vaney, R. Lavenir, N. Pardigon, J. M. Reynes, F. Pettinelli, L. Biscornet, L. Diancourt, S. Michel, S. Duquerroy, G. Guigon, M. P. Frenkiel, A. C. Brehin, N. Cubito, P. Despres, F. Kunst, F. A. Rey, H. Zeller, and S. Brisse. 2006. Genome microevolution of Chikungunya viruses causing the Indian Ocean outbreak. PLoS Med. 3:e263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Servant, M. J., B. ten Oever, C. LePage, L. Conti, S. Gessani, I. Julkunen, R. Lin, and J. Hiscott. 2001. Identification of distinct signaling pathways leading to the phosphorylation of interferon regulatory factor 3. J. Biol. Chem. 276:355-363. [DOI] [PubMed] [Google Scholar]
- 80.Seth, R. B., L. Sun, C. K. Ea, and Z. J. Chen. 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 122:669-682. [DOI] [PubMed] [Google Scholar]
- 81.Sharma, S., B. R. tenOever, N. Grandvaux, G. P. Zhou, R. Lin, and J. Hiscott. 2003. Triggering the interferon antiviral response through an IKK-related pathway. Science 300:1148-1151. [DOI] [PubMed] [Google Scholar]
- 82.Simmons, D. T., and J. H. Strauss. 1972. Replication of Sindbis virus. II. Multiple forms of double-stranded RNA isolated from infected cells. J. Mol. Biol. 71:615-631. [DOI] [PubMed] [Google Scholar]
- 83.Sourisseau, M., C. Schilte, N. Casartelli, C. Trouillet, F. Guivel-Benhassine, D. Rudnicka, N. Sol-Foulon, K. Le Roux, M. C. Prevost, H. Fsihi, M. P. Frenkiel, F. Blanchet, P. V. Afonso, P. E. Ceccaldi, S. Ozden, A. Gessain, I. Schuffenecker, B. Verhasselt, A. Zamborlini, A. Saib, F. A. Rey, F. Arenzana-Seisdedos, P. Despres, A. Michault, M. L. Albert, and O. Schwartz. 2007. Characterization of reemerging Chikungunya virus. PLoS Pathog. 3:e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tsetsarkin, K., S. Higgs, C. E. McGee, X. De Lamballerie, R. N. Charrel, and D. L. Vanlandingham. 2006. Infectious clones of Chikungunya virus (La Reunion isolate) for vector competence studies. Vector Borne Zoonotic Dis. 6:325-337. [DOI] [PubMed] [Google Scholar]
- 85.Ventoso, I., M. A. Sanz, S. Molina, J. J. Berlanga, L. Carrasco, and M. Esteban. 2006. Translational resistance of late alphavirus mRNA to eIF2α phosphorylation: a strategy to overcome the antiviral effect of protein kinase PKR. Genes Dev. 20:87-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang, X., S. Hussain, E. J. Wang, X. Wang, M. O. Li, A. Garcia-Sastre, and A. A. Beg. 2007. Lack of essential role of NF-κB p50, RelA, and cRel subunits in virus-induced type 1 IFN expression. J. Immunol. 178:6770-6776. [DOI] [PubMed] [Google Scholar]
- 87.Wathelet, M. G., C. H. Lin, B. S. Parekh, L. V. Ronco, P. M. Howley, and T. Maniatis. 1998. Virus infection induces the assembly of coordinately activated transcription factors on the IFN-β enhancer in vivo. Mol. Cell 1:507-518. [DOI] [PubMed] [Google Scholar]
- 88.Xu, L. G., Y. Y. Wang, K. J. Han, L. Y. Li, Z. Zhai, and H. B. Shu. 2005. VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol. Cell 19:727-740. [DOI] [PubMed] [Google Scholar]
- 89.Yoneyama, M., M. Kikuchi, K. Matsumoto, T. Imaizumi, M. Miyagishi, K. Taira, E. Foy, Y. M. Loo, M. Gale, Jr., S. Akira, S. Yonehara, A. Kato, and T. Fujita. 2005. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 175:2851-2858. [DOI] [PubMed] [Google Scholar]
- 90.Yoneyama, M., M. Kikuchi, T. Natsukawa, N. Shinobu, T. Imaizumi, M. Miyagishi, K. Taira, S. Akira, and T. Fujita. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5:730-737. [DOI] [PubMed] [Google Scholar]
- 91.Yoneyama, M., W. Suhara, Y. Fukuhara, M. Fukuda, E. Nishida, and T. Fujita. 1998. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO J. 17:1087-1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ziegler, S. A., L. Lu, A. P. da Rosa, S. Y. Xiao, and R. B. Tesh. 2008. An animal model for studying the pathogenesis of Chikungunya virus infection. Am. J. Trop. Med. Hyg. 79:133-139. [PubMed] [Google Scholar]