

Abstract
Metazoan cells form cytoplasmic mRNA granules such as stress granules (SG) and processing bodies (P bodies) that are proposed to be sites of aggregated, translationally silenced mRNAs and mRNA degradation. Poliovirus (PV) is a plus-strand RNA virus containing a genome that is a functional mRNA; thus, we investigated if PV antagonizes the processes that lead to formation of these structures. We have previously shown that PV infection inhibits the ability of cells to form stress granules by cleaving RasGAP-SH3-binding protein (G3BP). Here, we show that P bodies are also disrupted during PV infection in cells by 4 h postinfection. The disruption of P bodies is more rapid and more complete than disruption of stress granules. The kinetics of P body disruption correlated with production of viral proteinases and required substantial viral gene product expression. The organizing mechanism that forms P body foci in cells is unknown; however, potential scaffolding, aggregating, or other regulatory proteins found in P bodies were investigated for degradation. Two factors involved in 5′-end mRNA decapping and degradation, Xrn1 and Dcp1a, and the 3′ deadenylase complex component Pan3 underwent accelerated degradation during infection, and Dcp1a may be a direct substrate of PV 3C proteinase. Several other key factors proposed to be essential for P body formation, GW182, Edc3, and Edc4, were unaffected by poliovirus infection. Since deadenylation has been reported to be required for P body formation, viral inhibition of deadenylation, through Pan3 degradation, is a potential mechanism of P body disruption.
Poliovirus (PV), the causative agent of poliomyelitis, is a naked icosahedral, positive-sense RNA virus, which has a cytolytic replication cycle. Infection of susceptible cells with PV leads to significant disruption of cellular gene expression at several levels including transcription, nucleocytoplasmic transport, and cap-dependent translation (7, 14, 29, 49). Poliovirus 2A protease (2Apro) and cellular proteases mediate the cleavage of host cell eIF4GI and eIF4GII. These events, coupled with poliovirus 3C protease (3Cpro)-mediated cleavage of poly(A)-binding protein (PABP) and eIF5b, result in the abrogation of 7-methylguanosine cap-dependent translation (8, 42-44, 49, 51, 52). Poliovirus translation is driven by an internal ribosome entry site (IRES) rather than the cap-mediated ribosome recruitment employed by most cellular mRNAs (60, 61). The cleavage of eIF4G occurs shortly after infection, and the creation of the eIF4GI C-terminal cleavage product enhances the IRES-mediated translation of virus mRNA (30).
Translation control mechanisms now extend into mRNA silencing and RNA decay, which are all closely linked via cross talk among proteins that regulate translation initiation, silencing, and RNA decay. eIF4E, eIF4G, PABP, and certain mRNP proteins such as HuR all counteract silencing functions when bound to mRNPs and also regulate access to mRNA by decapping complexes and deadenylases (59). Deadenylase complexes must interact with PABP to be recruited to mRNA (91).
Rapid accumulation of translationally silenced mRNAs from eIF2α phosphorylation or eIF4G cleavage results in formation of cytoplasmic stress granules (SG) (3, 38, 53, 70). Stress granules are cytoplasmic foci consisting of concentrations of silenced mRNPs and are readily visualized through immunofluorescence tagging of certain RNA binding proteins such as TIA-1, TIAR, or RasGAP-SH3-binding protein (G3BP). SG assembly is proposed to be driven by the self-aggregation of certain RNA binding proteins, especially TIA-1, TIAR, and G3BP (3, 38, 70). Recently, we demonstrated that PV infection disrupts the ability of cells to form SG containing G3BP or TIAR in response to oxidative stress (85) via cleavage of G3BP by viral 3Cpro (85).
Processing bodies (P bodies) are another type of RNA granule containing translationally silenced, mostly deadenylated mRNPs, which are enriched for many proteins involved in mRNA decapping and decay (10, 17, 18, 24, 59). P bodies have been suggested to function in many pathways of mRNA decay and translation repression, ranging from nonsense-mediated decay and miRNA-mediated decay to mRNA storage and miRNA-mediated repression (10, 13, 32, 47, 48, 59, 64, 71, 72). The core constituents of P bodies are conserved throughout eukaryotic cells, many of which are involved in mRNA degradation. Most notably, P bodies contain the proteins involved in formation of active mRNA decapping complexes, Dcp1a/Dcp2 and Edc3, as well as the major 5′ exonuclease Xrn1 (15, 33, 50, 71, 82, 83). P bodies are also enriched for proteins involved in mRNA deadenylation, such as Ccr4, Caf1, Pan2, and Pan3 (17, 59, 91). Several of these proteins are proposed to be important for formation and maintenance of microscopically visible mRNA aggregates (15, 38, 82, 91). P bodies have been proposed to form through a mechanism similar to SG, involving self-aggregation of mRNA binding proteins bound to silenced mRNA molecules (24, 38). In Saccharomyces cerevisiae, Edc3 and LSM4 are required for formation of P bodies (15, 88). Many proteins thought to contribute to P body formation contain glutamine- and asparagine-rich regions proposed to interact and aggregate with other glutamine/asparagine-rich protein domains (15, 69). Aside from protein-mediated aggregation, the process of active deadenylation has been suggested as essential for efficient P body formation and recruitment of deadenylase proteins to P bodies (91). While several protein-protein interactions have been identified, the mechanisms surrounding the dynamic formation and dissolution of P bodies remain unclear.
Both stress granules and P bodies vary in size and number rapidly as a result of the translation status of the cell. The equilibrium between actively translating mRNA molecules associated with polysomes and those associated with stress granules or P bodies is a result of competitive binding between factors that promote translation and those that favor mRNA silencing (3, 9, 10, 17, 18, 24, 25, 59, 75, 79). Stress granules often form in close proximity to constitutively expressed P bodies, which increase in size upon addition of a stressor (3, 54, 58). This suggests a flow of proteins and mRNA between the granules, and fluorescence recovery after photobleaching (FRAP) studies have demonstrated rapid transit of granule components with the cytosol (1, 38, 45, 54). Stress granules have been proposed to exchange mRNP cargo with P bodies in a dynamic mRNA triage system (3, 38).
Plus-strand RNA viruses may be particularly vulnerable to cellular mRNA decay and storage pathways since their genomes are functional mRNAs. We hypothesized that PV has evolved to control such responses. Since PV infection, through the action of 3Cpro, disrupts the ability of susceptible cells to form stress granules (85), we hypothesized that viral infection would also interfere with P body formation. Here, we show that PV infection disrupts P bodies in the mid-phase of the infectious cycle. We have further identified several P body component proteins that undergo degradation during infection and determined their susceptibility to viral proteases. We also show that PV causes cleavage or enhanced degradation of proteins involved in both 5′- and 3′-mediated degradation of mRNA. This is the first description of an RNA virus attacking or destabilizing key components of the cellular mRNA decay machinery, and this may provide mechanisms to sustain high levels of viral RNA in the cell.
MATERIALS AND METHODS
Cell culture and virus infections.
HeLa and 293T cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and maintained at 37°C with 5% CO2. Sixteen hours prior to infection, HeLa or 293T cells were seeded at a density of 1 × 106 cells/35-mm well. The next day, HeLa or 293T cells were infected with DMEM containing 2% FBS and poliovirus type 1 (Mahoney strain) at a multiplicity of infection (MOI) of 20, or as indicated in figure legends, for 0 to 6 h. Mock-treated cells received 2% FBS-DMEM only. Poliovirus was grown in HeLa S3 cells and purified on CsCl gradients as described previously (35). PV was titrated by plaque assay on HeLa cell monolayers overlaid with 2% FBS-DMEM containing 1% methylcellulose.
Immunofluorescence.
For immunofluorescence, HeLa cells were plated on clean glass slides at a concentration of 3 × 105 cells per 35-mm well. Following infection on the subsequent day, the cells were fixed for 30 min in 4% paraformaldehyde in PEM [80 mM piperazine-N,N′-bis(2-ethanesulfonic acid (PIPES), 5 mM EGTA, 2 mM MgCl2]. Cells were treated with 1 mg/ml sodium borohydride (Sigma) for 5 min to quench autofluorescence. After several washes with PEM, the cells were permeabilized in 0.5% Triton X-100 for 30 min. Cells were then blocked with 5% skim milk in Tris-buffered saline-1% Tween 20 (TBS-T) for 30 min. Coverslips were then treated with primary antibodies against Dcp1a, Rck/p54, EDC4, and PV 2C at a dilution of 1:1,000 or anti-Dcp2 or anti-GW182 at 1:500 in blocking buffer for 1 h at room temperature. After several washes with blocking buffer and TBS-T, the cells were incubated with fluorescein isothiocyanate (FITC)-labeled goat anti-rabbit or rabbit anti-goat (Zymogen) and Texas Red-conjugated goat anti-mouse (Invitrogen) secondary antibodies (1:1,000) in blocking buffer at room temperature for 30 min. The cells were then fixed for 30 min in 4% paraformaldehyde in PEM and quenched with 1 mg/ml sodium borohydride for 5 min. Following quenching and three washes in TBS-T, cells were stained with 4′,6′-diamidino-2-phenylindole (DAPI) to visualize the nuclei before being mounted on microscope slides and stored at 4°C. Following mounting, the coverslips were analyzed by microscopy on a Zeiss Axioplan microscope. For P body quantification, 30 cells were selected from multiple fields of view and analyzed for P body size and number using NIH imaging software according to established protocols (56).
Immunoblot analysis.
HeLa cells were plated at a density of 1 × 106 cells/35-mm well and infected 16 h later with PV at an MOI of 20 for 0 to 6 h. Following infection, the cells were collected and lysed in 1% NP-40 (10 mM Tris-Cl, pH 7.4, 100 mM NaCl, 1 mM EDTA) to release cytoplasmic contents. Lysates were spun for 10 min at 8,100 × g to remove nuclei, and the cytoplasmic fraction was added to 2× SDS-PAGE buffer. Samples were then subjected to gel electrophoresis in 10% SDS-polyacrylamide gels. Proteins were transferred from the gel to nitrocellulose membranes for 100 min at 350 mA. The nitrocellulose membranes were then blocked with 5% skim milk in TBS-T for 30 min prior to the addition of primary antibodies and incubation overnight. The following antibodies were used in immunoblotting: anti-Dcp1a polyclonal (gift from A. B. Shyu), anti-Dcp1a monoclonal (Novus), anti-Dcp2 (Sigma), anti-Xrn1 (Novus), anti-GW182/TNCR6A (Novus), anti-EDC3/Lsm16 (Novus), anti-EDC4 (Novus), anti-V5 tag (Invitrogen), anti-G3BP (85), anti-Rck/p54(DDX6) (gift from C. E. Cameron), and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Millipore). After several washes with TBS-T, the membranes were incubated with secondary peroxidase-coupled anti-mouse (Pierce) or anti-rabbit (Bio-Rad) secondary antibodies diluted in 5% blocking buffer for 1 h. The membranes were washed three more times in TBS-T and then exposed to film following incubation with chemiluminescence Pico Reagent (Thermo) according to the manufacturer's directions.
Transfections and in vitro protease assays.
HeLa cells were plated at a density of 3 × 105 cells/35-mm well and transfected according to the Lipofectamine 2000 (Invitrogen) protocol. Cells were allowed to express the transgene for 24 to 36 h, at which time the cells were infected with PV or mechanically lysed for in vitro protease assays. Cells were collected for lysis by mechanical cell scraping and centrifugation for 5 min at 700 × g. Mechanical lysis was conducted through three freeze-thaw cycles or by incubation of cells for 15 min in ice-cold hypotonic buffer (20 mM HEPES, pH 7.2, 10 mM KCl, 1.5 mM MgCl2) followed by Dounce homogenization. Lysates were centrifuged for 10 min at 8,100 × g. Supernatants were collected and incubated with concentrations of purified PV 2A or 3C protease at 37°C for lengths of time and concentrations of protease indicated in the figure legends.
Pulse labeling and autoradiography of infected cells.
HeLa cells were plated at a density of 1 × 106 cells/35-mm well and infected with PV at an MOI of 20. For the final 30 min of each infection time point, the 2% FBS-DMEM was removed and replaced with methionine/cysteine-deficient medium supplemented with 0.3 μCi of 35S per 100 μl of labeling medium (10.5 mCi/ml; MP Biomedicals). The cells were allowed to translate for the remaining 30 min of the infection, allowing radiolabeling of actively translating proteins. At the end of the infection, cells were harvested and lysed as previously described. Following SDS-polyacrylamide gel electrophoresis, the gel was dried for 75 min at 80°C under a vacuum. The dried gel was exposed to single-emulsion film (Denville) for 16 to 24 h and then developed.
RESULTS
Poliovirus infection disrupts human P bodies.
Infection of human epithelial cells by PV has been shown to inhibit the ability of the cells to form cytoplasmic stress granules in response to oxidative stress through the cleavage of G3BP, a component crucial for stress granule formation, by PV 3C proteinase (85). To monitor effects of infection on P bodies, PV-infected cells were costained with antibody against commonly used P body marker proteins RCK/p54 and Dcp1a. Both markers colocalized to numerous cytoplasmic P body foci in mock-infected cells that averaged 7 to 10 per cell (Fig. 1A). The average number of P body foci present in infected cells diminished in number as the infection progressed (Fig. 1), and by 4 h postinfection (hpi) punctate foci stained with these marker proteins were nearly absent in infected cells. To explore if other enteroviruses could generate this response, cells were also infected with coxsackievirus B3 (CVB3), which resulted in a similar loss of P bodies in nearly all cells by 4 hpi (Fig. 1A). To further substantiate that P bodies were disrupted, PV-infected cells were stained with antibodies versus additional key marker proteins for P bodies. Dcp2, Edc4 (also known as HEDLS or Ge-1), and GW182 all localized to P bodies in uninfected cells; however, foci costaining these marker proteins were dispersed by 4 hpi (Fig. 1B). To quantify the observed loss of P bodies in PV-infected cells, P body size and number were analyzed by NIH image particle counting software (Fig. 2 C and D). These data revealed a trend toward a reduction in P body size and number even at 2 h, which progressed to a more drastic reduction at 4 hpi.
FIG. 1.

Disruption of cytoplasmic P bodies in HeLa cells during PV infection. (A) HeLa cells were infected with PV (left) or CVB3 (right) at an MOI of 20 for 4 h before fixation and processing for immunofluorescence by staining with antibodies against Rck/p54 and Dcp1a as markers for P bodies. (B) HeLa cells infected with PV for various times were processed as described for panel A before dual staining with antibodies to P body marker proteins Dcp2 and Edc4 or GW182 and Rck/p54 as indicated.
FIG. 2.

P bodies are disrupted in cells with substantial viral gene expression. (A) HeLa cells were infected for 0, 2, 4, and 6 h with PV at an MOI of 20 or 0.1 for 4 h (right panels only) and fixed and processed for immunofluorescence with antibodies against Dcp1a and PV 2C protein. (B) Autoradiograph of proteins labeled with [35S]methionine showing kinetics of translation shutoff and viral protein translation. (C) Immunofluorescence micrographs of 30 randomly selected cells stained for Rck/p54 were quantitated for the P body size and number per cell using NIH Image, and box plots showing medians, upper and lower quartiles and outliers, were graphed (**, P < 0.005). (D) Results were also plotted as a scatter plot showing average P body (PB) size /cell versus number of foci/cell. Black squares, 0 hpi; orange triangles, 2 hpi; red diamonds, 4 hpi.
Poliovirus gene product expression is required for P body disruption.
To determine the degree of viral gene expression required for disruption of P bodies, we performed immunofluorescence analysis of PV gene products in infected cells in concert with P body components. Figure 2 again shows that punctate staining of the P body marker Dcp1a was observed in uninfected cells, which shifted to a diffuse cytoplasmic staining by 4 hpi. Viral protein 2C was not detected above background in cells infected for 2 h; however, it was readily detectable in cells by 4 hpi and was sustained through 6 hpi (Fig. 2A). We observed an inverse correlation between levels of PV 2C expression and the number of P bodies observed in cells. Kinetically, the disappearance of P bodies between 2 and 4 hpi correlated with the onset of peak viral protein synthesis (Fig. 2B). A few rare cells containing P bodies late in infection were invariably found to display very low levels of viral protein production and also displayed no cytopathic effects (Fig. 2A, 6h). Similar results were also obtained using antibody to virus 3C protease (data not shown). We did not detect any cells with demonstrably high levels of viral protein expression that retained P bodies. To determine if infected cells transduce signals that repress P bodies in neighboring cells, low-MOI infections were performed using a one-step growth curve. In these infections, only cells demonstrably expressing viral proteins were deficient in P bodies, and other uninfected cells displayed largely unchanged P body content (Fig. 2A, right panels).
To determine if virus replication was required to disrupt P bodies, we infected HeLa cells in the presence of 2 mM guanidine-HCl, a potent inhibitor of PV RNA replication (39, 65, 66). Prevention of viral RNA replication greatly reduces but does not eliminate viral protein expression since viral proteins are still translated from the input viral genomic RNA. Figure 3 B shows that eIF4G cleavage still occurs due to activation of cellular proteases by PV 2Apro; however, 3Cpro cleavage of G3BP is blocked under these conditions, as expected (85, 89). Guanidine treatment of infected cells preserved P body foci throughout the 6-h infection (Fig. 3); however, guanidine had an intermediate effect with regard to P body size and number, with cells at 6 hpi expressing fewer P bodies than mock-infected cells (Fig. 3C). Interestingly, even though the total number of P bodies declined, the average size of the remaining P bodies increased (Fig. 3D).
FIG. 3.

Viral RNA replication is required for P body disruption. (A) HeLa cells were infected with PV for 2, 4, and 6 h in the presence of 2 mM guanidine-HCl. The cells were fixed and stained with two markers for P bodies, Dcp1a and Rck/p54. (B) Immunoblotting controls showing rapid cleavage of eIF4GI and lack of G3BP cleavage. P body (PB) number per cell (C) and average size (D) were quantitated for 30 randomly selected cells at each time point using NIH Image (**, P < 0.0005; *, P < 0.005).
Poliovirus infection causes degradation of P body protein components.
Poliovirus infection disrupts cellular metabolism principally through cleavage of key cellular proteins such as TBP, eIF4G, and G3BP. Since governing mechanisms of P body focus formation remain largely unknown, we screened a panel of key proteins that concentrate in P bodies that could be cleavage targets of viral proteinases and possibly contribute to P body dispersal. We included proteins proposed to be required for P body formation or mRNP aggregation and factors involved in 5′- and 3′-end-mediated mRNA decay. Immunoblot analysis of P body components revealed that PV infection did not affect the protein levels of the RNA helicase Rck/p54 throughout the course of infection, nor did it have an impact on the levels or structure of GW182, EDC4, and EDC3 (Fig. 4), proteins proposed to be required for the formation and structure of P bodies (15, 18, 37, 80, 82). These proteins were unchanged despite virus-induced translation shutoff; GW182 and GAPDH have reported half-lives longer than 16 h (23, 34). In contrast, Xrn1 and Dcp1a, which are two critical P body components in 5′-end-mediated mRNA decay mechanisms, displayed significantly diminished protein levels over the 6-h infection (Fig. 4). In typical infections, both proteins were observed to decline starting at about 3 to 4 hpi and were often undetectable at 6 hpi. Dcp2, which functions with Dcp1a in decapping, was not substantially altered though its concentration showed a modest decline over the course of infections. The reduction in Dcp1a or Xrn1 protein levels during PV infection could contribute mechanistically to the loss of P bodies in infected cells or could constitute an unrelated finding of destabilization of the 5′ mRNA decay machinery.
FIG. 4.
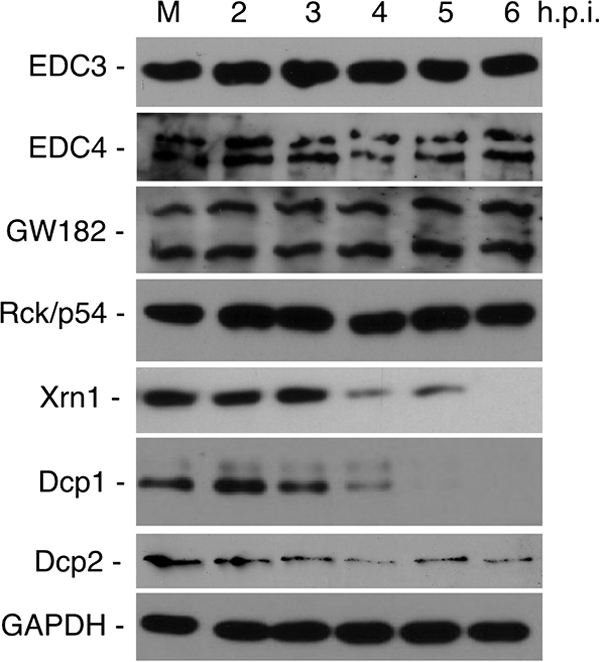
Effect of PV infection on P body protein components. HeLa cells were infected with PV, and then cytoplasmic lysates taken at various time points were subjected to immunoblot analysis with antibodies versus P body protein components. GAPDH immunoblot provides a loading control.
To determine the mechanism of Xrn1 degradation, we incubated uninfected HeLa cell lysates in vitro with purified PV 2A or 3C proteinases. Xrn1 showed no detectable changes in apparent molecular weight after a 60-min incubation with 2A or 3C proteinase and was thus unlikely to be a substrate for either proteinase (Fig. 5A). Both proteinases were active in these assays, resulting in rapid cleavage of eIF4GI and G3BP substrates (data not shown). PV infection blocks cellular cap-dependent translation; thus, degradation of Xrn1 could result from normal protein turnover combined with a lack of nascent Xrn1 protein synthesis. First, we determined that the normal half-life of Xrn-1 in HeLa cells was approximately 5.8 h by blocking translation in uninfected cells with cycloheximide (Fig. 5C). Parallel experiments show that PV significantly shortened the Xrn-1 half-life, which abruptly decreased to only 1.4 h after the 3-h time point, when viral proteins began pronounced expression (Fig. 2B). We also performed virus infections in the presence of MG132, a proteasome inhibitor, to determine the role of the proteasome in Xrn1 degradation. Treatment with MG132 retarded PV replication and the peak of maximal virus protein synthesis by about 3 h (Fig. 5B), so the time course of infections was extended 6 h to ensure completion of the viral replication cycle and development of equivalent levels of viral protein expression. The results show that in the context of proteasome inhibition, Xrn1 was stabilized throughout the course of the infection (Fig. 5B, bottom panel), suggesting that the observed reduction in Xrn1 protein levels is mostly a combined result of translational shutoff and proteasome degradation. However, the normal turnover rate of Xrn1 was significantly slower, suggesting that PV infection accelerated Xrn1 proteasomal decay.
FIG. 5.

Xrn1 undergoes proteasomal degradation. (A) HeLa cells were mechanically lysed, and postnuclear lysates were incubated with either purified PV 2A or 3C proteinase (50 ng/μl) for 10, 30, and 60 min before examination of Xrn1 by immunoblot analysis. (B) HeLa cells were infected for 3, 6, 9, and 12 h in the presence of the proteasome inhibitor MG132 (50 μM). The upper panel shows an autoradiograph of 35S-radiolabeled proteins. The lower panels are immunoblots. The GAPDH immunoblot provides a loading control. DMSO, dimethyl sulfoxide. (C) Xrn1 half-life in uninfected cells was determined by cycloheximide (CHX) treatment for various time points, followed by determination of protein levels in cytoplasmic extracts by immunoblot analysis and densitometry. Parallel quantitation of Xrn1 levels in infected cells is shown. The results in all panels are representative of three repeat experiments.
In contrast to Xrn1, the incubation of HeLa lysate with 3Cpro resulted in apparent cleavage of Dcp1a (Fig. 6A); however, incubation with 2Apro had no effect. 3Cpro caused an increase in relative migration of the Dcp1a doublet by about 6 kDa, suggesting that cleavage occurs near either the N or C terminus of the protein. HeLa cell lysates were incubated with decreasing concentrations of 3Cpro to determine the sensitivity of proteins to cleavage (Fig. 6B). Cleavage of Dcp1a was observed at 100 to 33 ng/μl of 3Cpro but not at lower concentrations in this assay (Fig. 6B). Thus, Dcp1a was less sensitive to cleavage by 3Cpro in static protease assays than the control G3BP substrate, as determined by efficiency of cleavage. Mixing experiments with uninfected and infected lysates revealed that an activity was present in infected cells that degraded the upper band of the Dcp1a doublet (Fig. 6D). Interestingly, the putative 3Cpro cleavage products of Dcp1a observed from direct incubation with protease are not readily detected by immunoblotting of lysates from infected cells or in the mixing experiments incubated in vitro (compare Fig. 4 and 6A and D). The upper band is a phosphorylated variant of Dcp1a (5), which may be a preferred substrate, as suggested by phosphatase treatment prior to incubation with 3Cpro (data not shown). We also examined Dcp1a turnover rates in uninfected versus infected cells. Dcp1a had a long half-life in cycloheximide-treated cells, over 10 h, which decreased to just 1 h after elevated expression of PV proteins began at 3 hpi (Fig. 6C). We also tested the effect of proteasome inhibition on the rate of Dcp1a degradation throughout PV infection. Interestingly, MG132 treatment resulted in significant restoration of Dcp1a at later time points during the infectious cycle (Fig. 6E) though Dcp1a levels were still significantly reduced at later points during the infection. Partial restoration of Dcp1a levels in the presence of MG132 suggests that both viral protease cleavage and proteasomal turnover contribute to loss of Dcp1a during poliovirus infection. Similar to Xrn1, proteasome decay of Dcp1a may be accelerated by infection, and, correspondingly, cleavage products of Dcp1a produced by 3Cpro may also be rapidly degraded in vivo.
FIG. 6.

Dcp1a is cleaved by 3C proteinase. (A) HeLa lysates were incubated with either purified poliovirus 2A or 3C proteinase for 10, 30, and 60 min with buffer (as described in Materials and Methods) and analyzed by immunoblotting. (B) HeLa cell lysates were incubated with various concentrations of 3Cpro to test the sensitivity of Dcp1a to cleavage by 3Cpro. G3BP served as a positive control for 3Cpro activity before immunoblot analysis. Both panels show immunoblots. (C) Dcp1a protein turnover in uninfected cells treated with cycloheximide (CHX) or infected with PV. Immunoblot analysis of cell lysates incubated for the time periods indicated were quantitated by densitometry. Results shown are representative of three repeat experiments. (D) Equal amounts of lysate from uninfected cells (lane C) and infected cells (lane PV) were mixed and incubated for 60 min before immunoblot analysis for Dcp1a. (E) Cells were treated with MG132 (50 μM) or dimethyl sulfoxide (DMSO) diluent during poliovirus infection. Cell extracts taken at the indicated times were evaluated for Dcp1a by immunoblotting.
Effect of poliovirus infection on deadenylase factors.
Deadenylation of mRNA has been demonstrated to be a prerequisite for P body formation (91); thus, we examined the effect of PV infection on the major cellular deadenylases, including poly(A) RNase (PARN), plus two primary deadenylase complexes consisting of Caf1/Ccr4 and Pan2/Pan3. Immunoblot analysis of endogenous PARN showed no effect on protein integrity during viral infection (Fig. 7A). To examine the other deadenylase components, cells expressing V5-tagged proteins were infected with PV, followed by immunoblot analysis of the V5-tagged proteins. V5-tagged Ccr4, Caf1, and Pan2 showed little or no change in protein levels throughout the 6-h virus infection (Fig. 7B). However, Pan3 displayed a marked decrease in protein level starting at 4 h postinfection and was undetectable by 6 h postinfection. A faint potential cleavage product that did not accumulate in vivo was also detected (Fig. 7B, arrow). Immunoblot analysis of endogenous Pan3 also revealed the disappearance of detectable protein by 6 hpi and production of a putative cleavage product in many assays (Fig. 8A and C). When cells expressing Pan3 tagged at the N terminus with hemagglutinin (HA) were infected, the HA-Pan3 was also degraded with similar kinetics as endogenous protein (Fig. 8B). To determine if proteasome activity contributed to Pan3 depletion, we performed infections in the presence of MG132, which did not prevent the decay of Pan3 but may have lessened the overall level of degradation slightly. A potential Pan3 cleavage product accumulated with time and was partly stabilized with MG132 treatment (Fig. 8C). Continued accumulation of Pan3 cleavage product in the absence of proteasome activity suggested that other cellular proteases or perhaps viral 3CD may directly or indirectly promote cleavage. Pan3 was determined to have a long half-life, much greater than 6 h, when translation was blocked by cycloheximide treatment (Fig. 8D); however, the half-life during virus infection was shortened dramatically to about 2 h. To analyze the involvement of PV proteinases in Pan3 degradation, we expressed Pan3-V5 in HeLa cells and incubated cytoplasmic lysates with purified PV 2A and 3C proteinases individually to assess cleavage of Pan3. As shown in Fig. 8E, 2A proteinase had no effect on V5-tagged Pan3 expressed in the HeLa cells. Incubation with 3Cpro resulted in a modest 40% decrease in Pan3 in the lysates, but potential cleavage products were not detected.
FIG. 7.
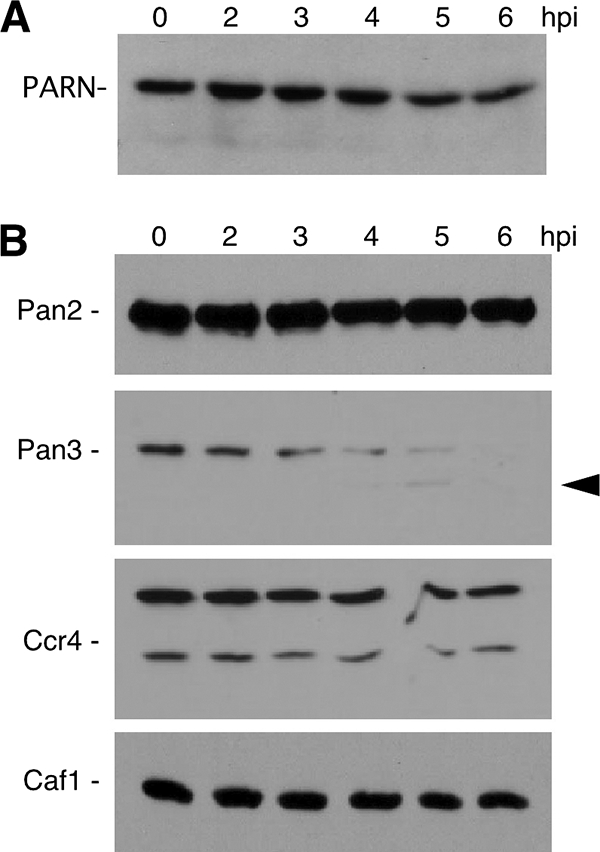
Effect of PV infection on deadenylase complex factors. (A) Infected HeLa cell lysates taken at the indicated time points were subjected to immunoblot analysis with anti-PARN antibody. (B) Lysates from infected HeLa cells expressing V5-tagged Caf1, Ccr4, Pan2, or Pan3 were analyzed by V5-specific immunoblotting. Arrowhead denotes a potential transient cleavage product.
FIG. 8.

Pan3 degradation during PV infection. (A) Lysates taken from infected cells at the indicated time points were analyzed by immunoblotting with anti-V5 or anti-Pan3 antibody. The top panel shows cleavage of Pan3-V5, middle panel shows endogenous Pan3, and bottom panel shows GAPDH as loading control. (B) Lysates harvested from infected 293T and HeLa cells expressing Pan3-HA at indicated time points were analyzed by immunoblotting with anti-HA antibody. (C) Infected cells treated with dimethyl sulfoxide (DMSO) diluent or MG132 (50 μM) were taken at indicated time points and analyzed by immunoblot analysis with anti-Pan3 antibody. (D) Pan3 protein turnover in uninfected cells treated with cycloheximide (CHX) or infected with PV. Immunoblot analysis of cell lysates incubated for time periods indicated was quantitated by densitometry. Results shown are taken from three experiments. (E) Cell lysates from Pan3-V5-transfected cells incubated with viral proteinases (50 ng/μl) for indicated times followed by immunoblot analysis.
DISCUSSION
This study demonstrates two novel host interactions by an RNA virus that have not been reported previously: disruption of cytoplasmic P bodies and degradation or destabilization of several critical components of the cellular mRNA decay machinery. As early as 2 hpi, there are reductions in P body size and number, and by mid-phase of the infectious cycle P bodies are not detectable in cells using several markers. The disruption of P bodies is more rapid and more complete than disruption of stress granules (data not shown) (85), suggesting that the virus has evolved a more effective mechanism of disrupting P body formation. One might actually expect to see an increase in P body size and number as infection progresses due to PV translation inhibition driving an increased pool of silenced mRNAs and thus increasing the nontranslating pool found in P bodies (17, 59). The disparity between the observed absence of P bodies and the expected increase in P bodies suggests that a mechanism distinct from virus-induced translation shutoff actively blocks P body formation.
Dispersal of P bodies required relatively pronounced expression of viral gene products (Fig. 2), and kinetics of P body disruption between 2 and 4 h was consistent with the typical cleavage of several cell proteins by 3Cpro, e.g., G3BP (Fig. 4A), PABP, and PCBP2 by 4 hpi (6, 41, 42, 63, 85). When viral RNA replication was blocked by inclusion of 2 mM guanidine-HCl (62), P bodies were mostly stabilized even though translation of virus proteins from input virion RNA was sufficient to modestly reduce P body numbers by 6 hpi. These infections still resulted in rapid cleavage of eIF4GI and partial translation shutoff (89); thus, eIF4GI cleavage is not directly linked to P body disruption, and the resultant 2-fold inhibition of host translation is not sufficient to induce major changes in P bodies in infected cells. Partial translation inhibition may produce enough silenced mRNPs to drive the modest increase in P body size seen in guanidine-treated infected cells; however, higher levels of virus gene expression, possibly 3CD or 3Cpro specifically, are required to completely disrupt their formation. Guanidine treatment prevents production of sufficient 3Cpro to cause cleavage of G3BP by PV and the subsequent disruption of arsenite-induced stress granules (85). Taken together, the results leave open the possibility that disruption of stress granules and P bodies could be mechanistically linked.
To search for mechanistic clues for the disruption of P bodies, we analyzed the effect of PV infection on several cellular proteins involved in mRNA decay that are also constituents of P bodies. Surprisingly, three factors thought to be required for P body formation, EDC3, EDC4, and GW182, remained unchanged throughout the course of the viral infection (Fig. 4). EDC3 is a component of the eukaryotic mRNA decapping complex and interacts with Dcp1a, Dcp2, and Lsm proteins in P bodies. These interactions have been suggested to play a role in mRNP aggregation and the formation of visible P bodies (15, 82). EDC4 (HEDLS/Ge-1) interacts with Dcp1a, and Dcp2, and its knockdown results in loss of P bodies (88). GW182 plays an integral role in forming a translation-silencing complex on the 3′ untranslated region (UTR) of mRNA in conjunction with bound microRNA/Ago2 that causes deadenylation (4, 19-21, 46, 68).
We identified three proteins involved in both 5′ end and 3′ end mRNA decay, Xrn1, Dcp1a, and Pan3, that undergo degradation during infection or cleavage by 3Cpro (Fig. 4 and 7). Degradation of Xrn1 is significant since it is the major 5′ exonuclease that degrades mRNA after decapping (2, 11). Xrn1 was not cleaved by incubation with either viral protease in vitro; thus, depletion of Xrn1 during infection is likely a combined result of virus-induced translation shutoff and subsequent protein turnover. However, the Xrn1 half-life was significantly decreased in infected cells (Fig. 5C), suggesting that PV may stimulate Xrn1 degradation via an unknown mechanism. Published reports of Xrn1 knockdown in cells did not result in alteration of P bodies or block recruitment of poly(A) RNA to P bodies (13); thus, rapid Xrn1 depletion by PV may stabilize uncapped mRNAs in cells but may not contribute to P body dispersal.
We also identified Dcp1a as a likely substrate of PV 3Cpro. Dcp1a contains a putative 3C protease recognition site (S-V-F-Q/-Q or V-F-Q-Q/-T) near its carboxy-terminal end that is consistent in location with the observed change in Dcp1a migration after incubation with 3Cpro. It is unclear why Dcp1a cleavage products are not detected during infection in cells. MG132 experiments indicated that proteasomal degradation of intact Dcp1a is significant during infection; thus, degradation of 3Cpro cleavage fragments may be further stimulated. Interestingly, it was recently reported that the carboxyl-terminal end of Dcp1a mediates its own trimerization (81). This trimerization was shown to stimulate efficient decapping of mRNA transcripts and the incorporation of Dcp1a into active decapping complexes with Dcp2 and Edc4 and to mediate Dcp1a localization to P bodies (81). Destabilization of Dcp1 during infection did not cause compensatory degradation of Dcp2 and EDC4 (Fig. 4). The putative 3Cpro recognition and cleavage site in Dcp1a is approximately 20 amino acids upstream of the alpha-helical trimerization region, and cleavage could remove this domain. Interestingly, reported small interfering RNA (siRNA) depletion of Dcp1a or Dcp2 did not result in decreased P body size or number (17).
We also identified the proteasome-independent degradation or cleavage of the Pan3 subunit of poly(A) nuclease (PAN) as a result of PV infection; however, other components of poly(A) nucleases, PARN, Pan2, Ccr4, and Caf1, remained unchanged (Fig. 7). The degradation of Pan3 was not blocked by MG132 treatment during infection; however, Pan3-V5 was mostly unmodified by incubation with viral 3Cpro in vitro. This could result from failure of Pan3-V5 to associate with the proper cellular complexes that allow 3Cpro cleavage. For instance, it is known that Pan3-V5 does not enter P bodies, whereas Pan3-HA does (91), and differential interactions in complexes could change Pan3 conformation or mask a cleavage site. Similar incubation of HA-Pan3 or endogenous Pan3 with 3Cpro resulted in no obvious cleavage of the endogenous Pan3 (Fig. 8; also data not shown). Alternatively, Pan3 may be more readily cleaved by the 3Cpro precursor 3CD, which is an abundant active protease in virus-infected cells, or another type of cellular proteinase. 3CD and 3Cpro have similar, but not identical, substrate specificities (87). It is also possible that Pan3 must undergo cycling transient modifications prior to recognition and cleavage by PV proteases. Work continues to elucidate the mechanism of Pan3 cleavage/degradation in infected cells.
How does PV infection disrupt P bodies? Translation block and release of mRNA from ribosomes by puromycin increases P body numbers in cells; thus, PV-mediated translation inhibition that also releases most cell mRNA from ribosomes would be expected to increase P body size and number. Many P body components have been depleted with siRNA approaches, resulting in no change or slight increases in P body size and number. Knockdown of only a few components is associated with P body disruption, including Rck/p54, GW182, Lsm1, EDC4 (also HEDLS or Ge-1), Rap55, and 4E-T (17). Several of these components were not examined in this study; however, Rck/p54, EDC4, and GW182 were stable during infection. Pan3 depletion has also been shown to block P body formation by inhibiting the deadenylation step that precedes inclusion of mRNA into these foci (91); thus, PV-mediated cleavage of this protein may play a major role in P body dispersal.
Though viruses obviously benefit from blocking degradation of their mRNAs, we report a novel mechanism in which an RNA virus attacks multiple components of the cellular RNA decay machinery. Viral interference in RNA decay mechanisms is a relatively unexplored area. Alphaviruses, representing another RNA virus family, evade mammalian deadenylases by recruitment of a protective 38-kDa protein to a motif in the 3′ UTR of virus mRNA (28). In contrast, several other RNA and DNA viruses promote mRNA decay through cap stealing, expression of nucleases, or other mechanisms (76).
Early work in the PV field demonstrated that cellular mRNA was mostly intact in infected cells. The poly(A) segments (40) and cap structures (22) remained intact and similar to those in uninfected cells, and mRNA could be extracted and translated in vitro (55). However, most of the key experiments in these studies were performed using guanidine-infected cells, which block P body disruption and inhibits viral gene expression; thus, it is unknown if PV directly affects cellular mRNA decay in cells. More recently, expression of 2Apro was shown to stabilize PV RNA during infection (36). A cellular substrate of 2Apro that mediated this instability was not identified in that report, and in this study, we demonstrated that 2Apro does not cleave Xrn1, Dcp1a, or Pan3; thus, another potential destabilizing agent of viral RNA remains unidentified.
PV disruption of poly(A) nucleases could be beneficial for replication by increasing the half-life of viral genomic RNA and mRNA in cells. Similarly, PV may attack Xrn1 to stabilize viral mRNA in cells, which contains an unprotected, monophosphate 5′ end (22, 31, 57). The free 5′ monophosphate resulting from decapping activity of the Dcp1a/Dcp2 complex is the most efficient substrate of Xrn1 exonucleolytic activity (11, 27, 67, 78). It is unclear how cleavage of Dcp1a may benefit virus replication.
P bodies are known as sites for both RNA storage and decay; however, the functional impact of P body disruption on virus replication or RNA decay is unclear. Many reports indicate that though RNA decay takes place in P bodies, which are concentrated with decay enzymes, the bulk of decay may occur outside P bodies (59). Despite the concentrated nucleases in P bodies, HIV and some yeast retrotransposons have been reported to use some P body components to aid viral assembly (12, 16, 26). P bodies are known to decrease in size and number in bronchial epithelial Beas-2B cells and in a mouse model in response to activation with proinflammatory cytokines and thus were implicated in upper airway inflammation (90). We have shown that PV and CVB3 infection of Beas-2B and A549 cells also results in disruption of P bodies (data not shown); thus, innate immune responses during virus infection may be affected by P body function. We have also discovered that degradation of Xrn1, Dcp1a, and Pan3 also occurs during infection with coxsackievirus B3; thus, these key host interactions are conserved within different enterovirus species (data not shown). Continued investigation of PV disruption of P bodies may yield clues into the mechanism of P body formation in cells, which remains a poorly understood process.
Poly(A) nuclease is composed of two subunits, Pan2 and Pan3, in both yeast and human and is PABP dependent for its activity. To access mRNA PAN interacts with the C-terminal domain of PABP through a PAM motif that is found on the Pan3 component and is conserved in other PABP-interacting proteins such as Paip-2, Tob2, and eRF3 (73, 74). Further work will be required to determine if PAN3 cleavage by PV abrogates PABP interaction and deadenylase activity. Interestingly, during deadenylation, PAN shortens mammalian poly(A) in the first phase of deadenylation to about 100 nucleotides (nt), whereupon Caf1/Ccr4 remove the remaining adenylate residues (86, 91). This raises questions concerning the role of Pan3 cleavage in infection since the average length of enterovirus poly(A) tails is less than 100 nt (84). However, during the course of infection unknown mechanisms regulate the length of viral poly(A) tails, which have been variably reported much longer than the corresponding poly(U) segment on negative-strand templates or even shorter (77, 84). Modulation of cellular poly(A) nucleases could play an unanticipated role in these processes. We are currently investigating if abrogation of deadenylase function through Pan3 cleavage has the effect of lengthening the PV RNA half-life or increasing virus replication. The potentially pleiotropic roles of RNA decay machinery and cytoplasmic RNA bodies in virus systems require additional investigation.
Acknowledgments
The authors are grateful to Ann-Bin Shyu for helpful comments and providing constructs for expression of Dcp1a, Pan-2, Pan3, Ccr4, and Caf1 and E. Wahle for the gift of anti-PARN antibody.
This work was supported by NIH Public Health Service grants AI 50237 and GM 59803. J.D. and J.W. were supported by NIH training grant T32 AI07471.
Footnotes
Published ahead of print on 20 October 2010.
REFERENCES
- 1.Aizer, A., Y. Brody, L. W. Ler, N. Sonenberg, R. H. Singer, and Y. Shav-Tal. 2008. The dynamics of mammalian P body transport, assembly, and disassembly in vivo. Mol. Biol. Cell 19:4154-4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amrani, N., S. Dong, F. He, R. Ganesan, S. Ghosh, S. Kervestin, C. Li, D. A. Mangus, P. Spatrick, and A. Jacobson. 2006. Aberrant termination triggers nonsense-mediated mRNA decay. Biochem. Soc. Trans. 34:39-42. [DOI] [PubMed] [Google Scholar]
- 3.Anderson, P., and N. Kedersha. 2008. Stress granules: the Tao of RNA triage. Trends Biochem. Sci. 33:141-150. [DOI] [PubMed] [Google Scholar]
- 4.Behm-Ansmant, I., J. Rehwinkel, T. Doerks, A. Stark, P. Bork, and E. Izaurralde. 2006. mRNA degradation by miRNAs and GW182 requires both CCR4:NOT deadenylase and DCP1:DCP2 decapping complexes. Genes Dev. 20:1885-1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blumenthal, J., L. Behar, E. Elliott, and I. Ginzburg. 2009. Dcp1a phosphorylation along neuronal development and stress. FEBS Lett. 583:197-201. [DOI] [PubMed] [Google Scholar]
- 6.Blyn, L. B., K. M. Swiderek, O. Richards, D. C. Stahl, B. L. Semler, and E. Ehrenfeld. 1996. Poly(rC) binding protein 2 binds to stem-loop IV of the poliovirus RNA 5′ noncoding region: identification by automated liquid chromatography-tandem mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 93:11115-11120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonderoff, J. M., J. L. Larey, and R. E. Lloyd. 2008. Cleavage of poly(A)-binding protein by poliovirus 3C proteinase inhibits viral internal ribosome entry site-mediated translation. J. Virol. 82:9389-9399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borman, A. M., R. Kirchweger, E. Ziegler, R. E. Rhoads, T. Skern, and K. M. Kean. 1997. elF4G and its proteolytic cleavage products: effect on initiation of protein synthesis from capped, uncapped, and IRES-containing mRNAs. RNA 3:186-196. [PMC free article] [PubMed] [Google Scholar]
- 9.Brengues, M., and R. Parker. 2007. Accumulation of polyadenylated mRNA, Pab1p, eIF4E, and eIF4G with P-bodies in Saccharomyces cerevisiae. Mol. Biol. Cell 18:2592-2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brengues, M., D. Teixeira, and R. Parker. 2005. Movement of eukaryotic mRNAs between polysomes and cytoplasmic processing bodies. Science 310:486-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caponigro, G., and R. Parker. 1996. Mechanisms and control of mRNA turnover in Saccharomyces cerevisiae. Microbiol. Rev. 60:233-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Checkley, M. A., K. Nagashima, S. J. Lockett, K. M. Nyswaner, and D. J. Garfinkel. 2010. P body components are required for Ty1 retrotransposition during assembly of retrotransposition-competent virus-like particles. Mol. Cell. Biol. 30:382-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cougot, N., S. Babajko, and B. Seraphin. 2004. Cytoplasmic foci are sites of mRNA decay in human cells. J. Cell Biol. 165:31-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dasgupta, A., P. Yalamanchili, M. Clark, et al. 2002. Effects of picornavirus proteinases on host cell transcription, p. 321-335. In B. L. Semler and E. Wimmer (ed.), Molecular biology of picornaviruses. ASM Press, Washington, DC.
- 15.Decker, C. J., D. Teixeira, and R. Parker. 2007. Edc3p and a glutamine/asparagine-rich domain of Lsm4p function in processing body assembly in Saccharomyces cerevisiae. J. Cell Biol. 179:437-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dutko, J. A., A. E. Kenny, E. R. Gamache, and M. J. Curcio. 2010. 5′ To 3′ mRNA decay factors colocalize with Ty1 Gag and human APOBEC3G and promote Ty1 retrotransposition. J. Virol. 84:5052-5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eulalio, A., I. Behm-Ansmant, and E. Izaurralde. 2007. P bodies: at the crossroads of post-transcriptional pathways. Nat. Rev. Mol. Cell Biol. 8:9-22. [DOI] [PubMed] [Google Scholar]
- 18.Eulalio, A., I. Behm-Ansmant, D. Schweizer, and E. Izaurralde. 2007. P body formation is a consequence, not the cause, of RNA-mediated gene silencing. Mol. Cell. Biol. 27:3970-3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eulalio, A., S. Helms, C. Fritzsch, M. Fauser, and E. Izaurralde. 2009. A C-terminal silencing domain in GW182 is essential for miRNA function. RNA 15:1067-1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eulalio, A., E. Huntzinger, and E. Izaurralde. 2008. GW182 interaction with Argonaute is essential for miRNA-mediated translational repression and mRNA decay. Nat. Struct. Mol. Biol. 15:346-353. [DOI] [PubMed] [Google Scholar]
- 21.Eulalio, A., F. Tritschler, R. Buttner, O. Weichenrieder, E. Izaurralde, and V. Truffault. 2009. The RRM domain in GW182 proteins contributes to miRNA-mediated gene silencing. Nucleic Acids Res. 37:2974-2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fernandez-Munoz, R., and J. E. Darnell. 1976. Structural difference between the 5′ termini of viral and cellular mRNA in poliovirus-infected cells: possible basis for the inhibition of host protein synthesis. J. Virol. 18:719-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Franch, H. A., S. Sooparb, J. Du, and N. S. Brown. 2001. A mechanism regulating proteolysis of specific proteins during renal tubular cell growth. J. Biol. Chem. 276:19126-19131. [DOI] [PubMed] [Google Scholar]
- 24.Franks, T. M., and J. Lykke-Andersen. 2008. The control of mRNA decapping and P body formation. Mol. Cell 32:605-615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Franks, T. M., and J. Lykke-Andersen. 2007. TTP and BRF proteins nucleate processing body formation to silence mRNAs with AU-rich elements. Genes Dev. 21:719-735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Furtak, V., A. Mulky, S. A. Rawlings, L. Kozhaya, K. Lee, V. N. Kewalramani, and D. Unutmaz. 2010. Perturbation of the P body component Mov10 inhibits HIV-1 infectivity. PLoS One 5:e9081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gallie, D. R. 1991. The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency. Genes Dev. 5:2108-2116. [DOI] [PubMed] [Google Scholar]
- 28.Garneau, N. L., K. J. Sokoloski, M. Opyrchal, C. P. Neff, C. J. Wilusz, and J. Wilusz. 2008. The 3′ untranslated region of Sindbis virus represses deadenylation of viral transcripts in mosquito and Mammalian cells. J. Virol. 82:880-892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gustin, K. E., and P. Sarnow. 2001. Effects of poliovirus infection on nucleo-cytoplasmic trafficking and nuclear pore complex composition. EMBO J. 20:240-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hambidge, S. J., and P. Sarnow. 1992. Translational enhancement of the poliovirus 5′ noncoding region mediated by virus-encoded polypeptide-2A. Proc. Natl. Acad. Sci. U. S. A. 89:10272-10276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hewlett, M. J., J. K. Rose, and D. Baltimore. 1976. 5′ terminal structure of poliovirus polyribosomal RNA is pUp. Proc. Natl. Acad. Sci. U. S. A. 73:327-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holmes, L. E., S. G. Campbell, S. K. De Long, A. B. Sachs, and M. P. Ashe. 2004. Loss of translational control in yeast compromised for the major mRNA decay pathway. Mol. Cell. Biol. 24:2998-3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ingelfinger, D., D. J. Arndt-Jovin, R. Luhrmann, and T. Achsel. 2002. The human LSm1-7 proteins colocalize with the mRNA-degrading enzymes Dcp1/2 and Xrnl in distinct cytoplasmic foci. RNA 8:1489-1501. [PMC free article] [PubMed] [Google Scholar]
- 34.Johnston, M., M. C. Geoffroy, A. Sobala, R. Hay, and G. Hutvagner. 2010. HSP90 protein stabilizes unloaded Argonaute complexes and microscopic P-bodies in human cells. Mol. Biol. Cell 21:1462-1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones, C., and E. Ehrenfeld. 1983. The effect of poliovirus infection on the translation in vitro of VSV messenger ribonucleoprotein particles. Virology 129:415-430. [DOI] [PubMed] [Google Scholar]
- 36.Jurgens, C. K., D. J. Barton, N. Sharma, B. J. Morasco, S. A. Ogram, and J. B. Flanegan. 2006. 2Apro is a multifunctional protein that regulates the stability, translation and replication of poliovirus RNA. Virology 345:346-357. [DOI] [PubMed] [Google Scholar]
- 37.Kedersha, N., and P. Anderson. 2007. Mammalian stress granules and processing bodies. Methods Enzymol. 431:61-81. [DOI] [PubMed] [Google Scholar]
- 38.Kedersha, N., G. Stoecklin, M. Ayodele, P. Yacono, J. Lykke-Andersen, M. J. Fitzler, D. Scheuner, R. J. Kaufman, D. E. Golan, and P. Anderson. 2005. Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 169:871-884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koch, F., and G. Koch. 1985. The molecular biology of poliovirus. Springer-Verlag, New York, NY.
- 40.Koschel, K. 1974. Poliovirus infection and poly(A) sequences of cytoplasmic cellular RNA. J. Virol. 13:1061-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuyumcu-Martinez, M., G. Belliot, S. V. Sosnovtsev, K. O. Chang, K. Y. Green, and R. E. Lloyd. 2004. Calicivirus 3C-like proteinase inhibits cellular translation by cleavage of poly(A)-binding protein. J. Virol. 78:8172-8182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuyumcu-Martinez, N. M., M. Joachims, and R. E. Lloyd. 2002. Efficient cleavage of ribosome-associated poly(A)-binding protein by enterovirus 3C protease. J. Virol. 76:2062-2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuyumcu-Martinez, N. M., M. E. Van Eden, P. Younan, and R. E. Lloyd. 2004. Cleavage of poly(A)-binding protein by poliovirus 3C protease inhibits host cell translation: a novel mechanism for host translation shutoff. Mol. Cell. Biol. 24:1779-1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lamphear, B. J., R. Q. Yan, F. Yang, D. Waters, H. D. Liebig, H. Klump, E. Kuechler, T. Skern, and R. E. Rhoads. 1993. Mapping the cleavage site in protein synthesis initiation factor-eIF-4g of the 2A proteases from human coxsackievirus and rhinovirus. J. Biol. Chem. 268:19200-19203. [PubMed] [Google Scholar]
- 45.Leung, A. K., J. M. Calabrese, and P. A. Sharp. 2006. Quantitative analysis of Argonaute protein reveals microRNA-dependent localization to stress granules. Proc. Natl. Acad. Sci. U. S. A. 103:18125-18130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu, J., F. V. Rivas, J. Wohlschlegel, J. R. Yates, 3rd., R. Parker, and G. J. Hannon. 2005. A role for the P body component GW182 in microRNA function. Nat. Cell Biol. 7:1161-1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu, J., F. V. Rivas, J. Wohlschlegel, J. R. Yates, 3rd., R. Parker, and G. J. Hannon. 2005. A role for the P body component GW182 in microRNA function. Nat. Cell Biol. 7:1261-1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu, J., M. A. Valencia-Sanchez, G. J. Hannon, and R. Parker. 2005. MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nat. Cell Biol. 7:719-723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lloyd, R. 2006. Translational control by viral proteinases. Virus Res. 119:76-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lykke-Andersen, J. 2002. Identification of a human decapping complex associated with hUpf proteins in nonsense-mediated decay. Mol. Cell. Biol. 22:8114-8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marissen, W. E., A. Gradi, N. Sonenberg, and R. E. Lloyd. 2000. Cleavage of eukaryotic translation imitation factor 4GII correlates with translation inhibition during apoptosis. Cell Death Differ. 7:1234-1243. [DOI] [PubMed] [Google Scholar]
- 52.Marissen, W. E., and R. E. Lloyd. 1998. Eukaryotic translation initiation factor 4G is targeted for proteolytic cleavage by caspase 3 during inhibition of translation in apoptotic cells. Mol. Cell. Biol. 18:7565-7574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mokas, S., J. R. Mills, C. Garreau, M. J. Fournier, F. Robert, P. Arya, R. J. Kaufman, J. Pelletier, and R. Mazroui. 2009. Uncoupling stress granule assembly and translation initiation inhibition. Mol. Biol. Cell 20:2673-2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mollet, S., N. Cougot, A. Wilczynska, F. Dautry, M. Kress, E. Bertrand, and D. Weil. 2008. Translationally repressed mRNA transiently cycles through stress granules during stress. Mol. Biol. Cell 19:4469-4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Munoz, A., R. Harvey, and L. Carrasco. 1983. Cellular RNA is not degraded in interferon-treated HeLa cells after poliovirus infection. FEBS Lett. 160:87-92. [DOI] [PubMed] [Google Scholar]
- 56.Nissan, T., and R. Parker. 2008. Analyzing P-bodies in Saccharomyces cerevisiae. Methods Enzymol. 448:507-520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nomoto, A., Y. F. Lee, and E. Wimmer. 1976. The 5′ end of poliovirus RNA is not capped with m7G(5′)ppp(5′)Np. Proc. Natl. Acad. Sci. U. S. A. 73:375-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ohn, T., N. Kedersha, T. Hickman, S. Tisdale, and P. Anderson. 2008. A functional RNAi screen links O-GlcNAc modification of ribosomal proteins to stress granule and processing body assembly. Nat. Cell Biol. 10:1224-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Parker, R., and U. Sheth. 2007. P bodies and the control of mRNA translation and degradation. Mol. Cell 25:635-646. [DOI] [PubMed] [Google Scholar]
- 60.Pelletier, J., and N. Sonenberg. 1989. Internal binding of eucaryotic ribosomes on poliovirus RNA: translation in HeLa cell extracts. J. Virol. 63:441-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pelletier, J., and N. Sonenberg. 1988. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature 334:320-325. [DOI] [PubMed] [Google Scholar]
- 62.Penman, S., and D. Summers. 1965. Effects on host cell metabolism following synchronous infection with poliovirus. Virology 27:614-620. [DOI] [PubMed] [Google Scholar]
- 63.Perera, R., S. Daijogo, B. L. Walter, J. H. Nguyen, and B. L. Semler. 2007. Cellular protein modification by poliovirus: the two faces of poly(rC)-binding protein. J. Virol. 81:8919-8932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pillai, R. S., S. N. Bhattacharyya, C. G. Artus, T. Zoller, N. Cougot, E. Basyuk, E. Bertrand, and W. Filipowicz. 2005. Inhibition of translational initiation by Let-7 microRNA in human cells. Science 309:1573-1576. [DOI] [PubMed] [Google Scholar]
- 65.Pincus, S. E., D. C. Diamond, E. A. Emini, and E. Wimmer. 1986. Guanidine-selected mutants of poliovirus: mapping of point mutations to polypeptide 2C. J. Virol. 57:638-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pincus, S. E., and E. Wimmer. 1986. Production of guanidine-resistant and -dependent poliovirus mutants from cloned cDNA: mutations in polypeptide 2C are directly responsible for altered guanidine sensitivity. J. Virol. 60:793-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Poole, T. L., and A. Stevens. 1995. Comparison of features of the RNase activity of 5′-exonuclease-1 and 5′-exonuclease-2 of Saccharomyces cerevisiae. Nucleic Acids Symp. Ser. 33:79-81. [PubMed] [Google Scholar]
- 68.Rehwinkel, J., I. Behm-Ansmant, D. Gatfield, and E. Izaurralde. 2005. A crucial role for GW182 and the DCP1:DCP2 decapping complex in miRNA-mediated gene silencing. RNA 11:1640-1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reijns, M. A., R. D. Alexander, M. P. Spiller, and J. D. Beggs. 2008. A role for Q./N-rich aggregation-prone regions in P body localization. J. Cell Sci. 121:2463-2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Scadden, A. D. 2007. Inosine-containing dsRNA binds a stress-granule-like complex and downregulates gene expression in trans. Mol. Cell 28:491-500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sheth, U., and R. Parker. 2003. Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 300:805-808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sheth, U., and R. Parker. 2006. Targeting of aberrant mRNAs to cytoplasmic processing bodies. Cell 125:1095-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Siddiqui, N., G. Kozlov, I. D'Orso, J.-F. Trempe, and K. Gehring. 2003. Solution structure of the C-terminal domain from poly(A)-binding protein in Trypanosoma cruzi: a vegetal PABC domain. Protein Sci. 12:1925-1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Siddiqui, N., D. A. Mangus, T. C. Chang, J. M. Palermino, A. B. Shyu, and K. Gehring. 2007. Poly(A) nuclease interacts with the C-terminal domain of polyadenylate-binding protein domain from poly(A)-binding protein. J. Biol. Chem. 282:25067-25075. [DOI] [PubMed] [Google Scholar]
- 75.Sivan, G., N. Kedersha, and O. Elroy-Stein. 2007. Ribosomal slowdown mediates translational arrest during cellular division. Mol. Cell. Biol. 27:6639-6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sokoloski, K. J., C. J. Wilusz, and J. Wilusz. 2006. Viruses: overturning RNA turnover. RNA Biol. 3:140-144. [DOI] [PubMed] [Google Scholar]
- 77.Steil, B. P., B. J. Kempf, and D. J. Barton. 2010. Poly(A) at the 3′ end of positive-strand RNA and VPg-linked poly(U) at the 5′ end of negative-strand RNA are reciprocal templates during replication of poliovirus RNA. J. Virol. 84:2843-2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stevens, A., and T. L. Poole. 1995. 5′-exonuclease-2 of Saccharomyces cerevisiae. Purification and features of ribonuclease activity with comparison to 5′-exonuclease-1. J. Biol. Chem. 270:16063-16069. [DOI] [PubMed] [Google Scholar]
- 79.Teixeira, D., U. Sheth, M. A. Valencia-Sanchez, M. Brengues, and R. Parker. 2005. Processing bodies require RNA for assembly and contain nontranslating mRNAs. RNA 11:371-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tritschler, F., J. E. Braun, A. Eulalio, V. Truffault, E. Izaurralde, and O. Weichenrieder. 2009. Structural basis for the mutually exclusive anchoring of P body components EDC3 and Tral to the DEAD box protein DDX6/Me31B. Mol. Cell 33:661-668. [DOI] [PubMed] [Google Scholar]
- 81.Tritschler, F., J. E. Braun, C. Motz, C. Igreja, G. Haas, V. Truffault, E. Izaurralde, and O. Weichenrieder. 2009. DCP1 forms asymmetric trimers to assemble into active mRNA decapping complexes in metazoa. Proc. Natl. Acad. Sci. U. S. A. 106:21591-21596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tritschler, F., A. Eulalio, V. Truffault, M. D. Hartmann, S. Helms, S. Schmidt, M. Coles, E. Izaurralde, and O. Weichenrieder. 2007. A divergent Sm fold in EDC3 proteins mediates DCP1 binding and P body targeting. Mol. Cell. Biol. 27:8600-8611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.van Dijk, E., N. Cougot, S. Meyer, S. Babajko, E. Wahle, and B. Seraphin. 2002. Human Dcp2: a catalytically active mRNA decapping enzyme located in specific cytoplasmic structures. EMBO J. 21:6915-6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.van Ooij, M. J., C. Polacek, D. H. Glaudemans, J. Kuijpers, F. J. van Kuppeveld, R. Andino, V. I. Agol, and W. J. Melchers. 2006. Polyadenylation of genomic RNA and initiation of antigenomic RNA in a positive-strand RNA virus are controlled by the same cis-element. Nucleic Acids Res. 34:2953-2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.White, J. P., A. M. Cardenas, W. E. Marissen, and R. E. Lloyd. 2007. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2:295-305. [DOI] [PubMed] [Google Scholar]
- 86.Yamashita, A., T. C. Chang, Y. Yamashita, W. Zhu, Z. Zhong, C. Y. Chen, and A. B. Shyu. 2005. Concerted action of poly(A) nucleases and decapping enzyme in mammalian mRNA turnover. Nat. Struct. Mol. Biol. 12:1054-1063. [DOI] [PubMed] [Google Scholar]
- 87.Ypma-Wong, M. F., P. G. Dewalt, V. H. Johnson, J. G. Lamb, and B. L. Semler. 1988. Protein 3CD is the major poliovirus proteinase responsible for cleavage of the P1 capsid precursor. Virology 166:265-270. [DOI] [PubMed] [Google Scholar]
- 88.Yu, J. H., W. H. Yang, T. Gulick, K. D. Bloch, and D. B. Bloch. 2005. Ge-1 is a central component of the mammalian cytoplasmic mRNA processing body. RNA 11:1795-1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zamora, M., W. E. Marissen, and R. E. Lloyd. 2002. Multiple eIF4GI-specific protease activities present in uninfected and poliovirus-infected cells. J. Virol. 76:165-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhai, Y., Z. Zhong, C. Y. Chen, Z. Xia, L. Song, M. R. Blackburn, and A. B. Shyu. 2008. Coordinated changes in mRNA turnover, translation, and RNA processing bodies in bronchial epithelial cells following inflammatory stimulation. Mol. Cell. Biol. 28:7414-7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zheng, D., N. Ezzeddine, C. Y. Chen, W. Zhu, X. He, and A. B. Shyu. 2008. Deadenylation is prerequisite for P body formation and mRNA decay in mammalian cells. J. Cell Biol. 182:89-101. [DOI] [PMC free article] [PubMed] [Google Scholar]