

Abstract
The genus β human papillomavirus (HPV) type 8 is associated with nonmelanoma skin cancer in patients with epidermodysplasia verruciformis, and evidence for its protumorigenic potential in the general population increases. To date, strategies to suppress genus β HPV infections are limited. Interferon regulatory factors IRF-3 and IRF-7 play key roles in the activation of the innate immune response to viral infections. In this study, we show for the first time that both IRF-3 and IRF-7 regulate transcription of a papillomavirus, but with opposing effects. IRF-7, expressed in the suprabasal layers of human epidermis, increased HPV8 late promoter activity via direct binding to viral DNA. UV-B light-induced activation of the HPV8 promoter involved IRF-7 as a downstream effector. In contrast, IRF-3, expressed in all layers of human epidermis, induced strong HPV8 suppression in primary keratinocytes. IRF-3-mediated suppression prevailed over IRF-7-induced HPV8 transcription. Unlike the E6 oncoprotein of the mucosal high-risk HPV16, the HPV8 E6 protein did not bind to IRF-3 and only weakly antagonized its activity. Strong antiviral activity was also observed, when keratinocytes were treated with potent IRF-3 activators, poly(I:C) or RNA bearing 5′ phosphates. In conclusion, we show that IRF-3 activation induces a state of cell-autonomous immunity against HPV in primary human keratinocytes. Our study suggests that local application of IRF-3-activating compounds might constitute an attractive novel therapeutic strategy against HPV8-associated diseases, particularly in epidermodysplasia verruciformis patients.
Human papillomaviruses (HPVs) are small DNA viruses that infect epithelial cells of skin and mucosa. Depending on the oncogenic potential of particular HPV types and specific sites of infection, lesions induced by HPVs range from benign warts to invasive carcinoma. High-risk mucosal HPV types 16 and 18 have a well-established role in anogenital carcinogenesis, in particular in the development of cervical carcinoma (47). Anogenital condylomas are caused by HPV types 6 and 11. Infection with cutaneous genus β HPV (i.e., type 5 or 8) is involved in the development of nonmelanoma skin cancer at sites exposed to the sun. This was first discovered in patients with the inherited disease epidermodysplasia verruciformis (EV). However, there is increasing evidence that genus β HPV types play a role in the development of squamous and basal cell carcinoma in immunocompromised individuals and in the general population (16a, 34). The carcinogenic potential of HPV8 has been documented in a HPV8-transgenic mouse model (40). In the murine epidermis, the HPV8 E6 protein represents the major oncoprotein necessary and sufficient to induce spontaneous tumor development (25).
The life cycle of HPVs is tightly linked to the differentiation program of epithelial cells. To indicate the sequence of viral gene expression during the HPV life cycle, viral gene products have been classified as early (E) and late (L) proteins. Their transcription is initiated from the HPV early or late promoter, respectively. Viral DNA replication and transcription are controlled by the noncoding control region (NCR) located between the 3′ end of the late gene region and the 5′ end of the early gene region (43). The NCR of EV-associated HPVs differs from that of other HPVs and is characterized by its small size of about 400 bp. The HPV8 late promoter (P7535) has been mapped to position 7535 and the early promoter (P175) to position 175 of the viral genome. Binding of several cellular transcription factors has been detected within the HPV8 NCR. Some of them, such as AP-1, NF1, RUNX1, and p53, induce HPV8 transcription, while PBF, IRF-5.2, and YY1 suppress the HPV8 NCR activity (43).
Viral transcription is an important target of innate immune defense mechanisms. Type I interferons (IFNs) are a family of structurally and functionally related cytokines which inhibit cell proliferation and interfere with viral replication and transcription. Best characterized are IFN-α and IFN-β, but the type I IFN family comprises also IFN-ω and the more recently discovered IFN-ɛ, IFN-κ, and IFN-λ. All type I IFNs exert immunostimulatory activities and thus constitute the first line of antiviral defense (41). Due to their potent antiviral properties, type I IFNs have also been used in the therapy of HPV infection. While treatment of HPV6 or -11-related genital warts with IFN-α has been effective, inconsistent clinical outcomes were documented for lesions induced by high-risk mucosal HPVs (6). In vitro, type I IFNs inhibit growth of HPV-immortalized cervical epithelial cells and suppress transcription of viral E6 and E7 oncogenes in HPV16 or -18-immortalized cells. However, this effect was not observed in all HPV-harboring cervical carcinoma cell lines (41). These studies indicate that the response to IFN treatment depends on the HPV type and possibly also on the integration status of the HPV genome (20).
Activation of endogenous IFN-α and IFN-β gene expression is primarily induced by two cellular transcription factors, interferon regulatory factor 3 (IRF-3) and IRF-7. IRF-3 plays a central role in the initiation of the type I IFN response. It is constitutively expressed in various cell types. IRF-3 activation is triggered by pathogen recognition receptors that sense the presence of viral nucleic acids. IRF-7 is expressed at low baseline levels but efficiently induced by type I IFNs. Recent data show that IRF-7 is activated by UV light in HeLa cells (19). IRF-3 and IRF-7 form homodimeric and heterodimeric complexes with each other and act differentially on type I IFN gene family members. While IRF-7 induces both IFN-α and IFN-β, IRF-3 potently activates IFN-β rather than IFN-α genes (45). IRF-3 is strongly activated by immunostimulatory nucleic acids, such as poly(I:C), a synthetic analog of double-stranded RNA (dsRNA), or RNA bearing 5′ phosphates (5′pppRNA) (18, 35).
Little is known about the impact of type I IFN signaling on the EV-associated HPVs. In this study, we investigated how type I IFNs and their key activators, IRF-3 and IRF-7, influence the transcriptional activity of the high-risk cutaneous HPV8. We demonstrate that both IRF-3 and IRF-7 induce type I IFN in human keratinocytes. Type I IFNs, as well as IRF-3, suppressed the transcriptional activity of HPV8. Unexpectedly, IRF-7 activated HPV8, and our data show that this is mediated through direct interactions of IRF-7 with the HPV8 NCR. IRF-3 suppression prevailed over the IRF-7-mediated activation of HPV8 transcription. Strong suppression of HPV8 was also observed in keratinocytes treated with either of the IRF-3 activators poly(I:C) and 5′pppRNA. This suggested that IRF-3 activators might be of clinical use for the treatment of HPV8-expressing lesions.
MATERIALS AND METHODS
Plasmid constructs and reagents.
pALuc-HPV8NCR encompassed the noncoding region of HPV8 (nucleotide [nt] fragment 7077 to 558, according to GenBank accession no. M12737.1) cloned in front of the firefly luciferase gene. The pALuc-HPV8A reporter construct contained the late HPV8 P7535 promoter (nt 7396 to 7) of the HPV8 genome (44), and pALuc-HPV8B contained the early HPV8 P175 promoter (nt 15 to 195) of the HPV8 genome. All three reporter constructs were kindly provided by H. Pfister, Cologne, Germany. pALuc-HPV8Adel lacking the IRF binding site (nt 7382 to 7389) of the HPV8 NCR was constructed by deleting nucleotide fragment 7371 to 7392 of HPV8 with the Quick-Change XL site-directed mutagenesis kit (Stratagene, La Jolla, CA) and 5′-GGGCTCCGTCAGGGGCTAGATTGTACCGTTTTCGG-3′ mutagenic complementary primers according to the manufacturer's instructions. The reporter vector comprising the human RANTES promoter cloned upstream of the firefly luciferase gene in the pGL3-basic vector was previously described (11).
IRF-3 and constitutively active IRF-3 [IRF-3(5D)] were cloned in the pCMVβL expression vector, while the constitutively active form IRF-7 (IRF-7Δ247-467) was cloned in pFlag-CMV2 (21, 24). A dominant-negative form of IRF-7 (IRF-7Δ12-101) was expressed from pEGFP-C1 (23). The pCAGGS-GFP-IRF7A plasmid, expressing wild-type IRF-7 fused to green fluorescent protein (GFP), was provided by Luis Martinez. Glutathione S-transferase (GST) fusion constructs were generated by recloning different IRF fragments from pCMVβL and pFlag-CMV2, respectively, into the pGEX-2T vector (Amersham Pharmacia, Freiburg, Germany). HPV16 and HPV8 E6 proteins containing the Flag epitope were expressed from pXJ41 vectors kindly provided by G. Steger. pCMV-EGFP, encoding the enhanced green fluorescent protein (EGFP) gene under the control of the human cytomegalovirus (CMV) immediate-early promoter, was used to monitor transfection efficiencies. All constructs obtained by in vitro mutagenesis and by cloning of PCR products were verified by sequencing.
Recombinant human IFN-α2b (Intron A) and IFN-β1b (Betaferon) were obtained from Essex Pharma and Bayer Schering Pharma, respectively. poly(I:C) was purchased from Amersham Biosciences (Freiburg, Germany). 5′ triphosphate RNA (5′pppRNA) was generated from an 89-bp oligonucleotide comprising the T7 promoter and 5′ untranslated region (UTR) region of vesicular stomatitis virus (VSV) by in vitro transcription with a MEGAscript T7 kit (Ambion, Austin, TX) and purified with the RNeasy MinElute cleanup kit (Qiagen, Hilden, Germany).
Cell culture, transient transfection, UV light irradiation, and luciferase assay.
Primary human foreskin keratinocytes (NHK cells) (BioWhittaker, Vervier, Belgium) were cultured in KBM-2 medium (Lonza, Verviers, Belgium) supplemented with insulin, epidermal growth factor, hydrocortisone, epinephrine, and bovine pituitary extract according to the supplier's instructions. The HPV-negative skin squamous cell carcinoma-derived RTS3b cell line (37) was cultivated as previously described (14). The HPV-negative cervical carcinoma cell line C33A (HTB-31; American Type Culture Collection, Manassas, VA) was grown in Dulbecco's modified Eagle's medium with Glutamax I supplemented with 10% fetal calf serum, 100 U/ml penicillin, 0.1 mg/ml streptomycin, and 1 mM sodium pyruvate (all from Invitrogen, Karlsruhe, Germany).
Transfections of NHK cells with TransFast (Promega, Southampton, United Kingdom) were performed as previously reported (31). RTS3b cells were transfected with FuGene 6 (Roche, Mannheim, Germany) or Lipofectamine 2000 (Invitrogen) transfection reagents according to the manufacturers' guidelines. In all RTS3b reporter assays, pEGFP-C1 was coexpressed to measure transfection efficiencies. C33A cells were transfected by the calcium phosphate method according to a standard procedure (8). NHK cells were irradiated with 15 mJ/cm2 of UV-B light using a UVP CL-1000 UV cross-linker with F8T5 bulbs. Untransfected NHK cells were harvested 6 h after UV treatment, transfected NHK cells were irradiated 8 h after transfection.
Luciferase activity was quantified 24 h posttransfection. The cells were detached with trypsin-EDTA solution (Invitrogen), and transfection efficiencies were determined by measuring the EGFP fluorescence by flow cytometry (FACSCalibur, Becton Dickinson, Heidelberg, Germany). Cellular lysates were prepared and assayed for luciferase activity as described previously (31). Transfections were conducted in triplicate. The values of luciferase activity were normalized to the number of EGFP-positive cells and the protein content of the respective lysates.
Expression and purification of GST fusion proteins.
GST, GST-IRF-3, GST-IRF-3N (containing the amino terminus of IRF-3), and GST-IRF-7 fusion proteins were expressed in Escherichia coli and purified with glutathione-Sepharose (13). Protein concentrations were determined by Bradford assay.
EMSA.
Electrophoretic mobility shift assays (EMSAs) were performed with purified GST, GST-IRF-3, GST-IRF-3N, and GST-IRF-7 fusion proteins or nuclear extracts isolated from RTS3b cells or NHK and double-stranded oligonucleotides (nt 7369 to 7398 or nt 7365 to 7391) comprising the IRF-BS within the HPV8 NCR (IRF-BS nt 7382 to7389), according to GenBank accession no. M12737.1. Oligonucleotides were radiolabeled with [γ-32P]ATP, using T4 polynucleotide kinase (New England Biolabs, United Kingdom). Binding reactions were performed for 20 min at room temperature in a 20-μl volume containing GST fusion proteins and 250 pg of labeled oligonucleotides, 0.1 M HEPES, pH 7.9, 20 mM MgCl2, 45 mM KCl, 1 mM EDTA, 1 mM dithiothreitol, 2% glycerine, 0.025 μg/μl poly(dI:dC) (Amersham Pharmacia, Piscataway, NJ), 0.3 μg/μl aprotinin, and 3 mM phenylmethylsulfonyl fluoride (Sigma-Aldrich). Reaction products were separated in a 5% polyacrylamide gel in 0.5× Tris-borate-EDTA. The gels were dried and exposed to autoradiographic films.
Co-IP and Western blot analysis.
C33A cells were seeded at a density of 3.2 × 106 cells onto a 15-cm plate. After 24 h, 7.5 μg of Flag-tagged HPV8 E6, HPV16 E6 in pXJ41, or the respective control vectors was cotransfected with either 7.5 μg of pCMVβL-IRF-3 or pCMVβL-IRF-3(5D). Cell lysis was performed 24 h posttransfection. Coimmunoprecipitation (Co-IP), SDS-PAGE, and Western blotting were performed as previously described (14). Cell lysates were incubated with mouse anti-Flag M5 antibody (Sigma-Aldrich, Taufkirchen, Germany) and precipitated with protein G-Sepharose (Santa Cruz Biotechnology, Heidelberg, Germany). IRF-3 and IRF-3(5D) were detected in the Western blot with rabbit polyclonal antibody sc-9082 (Santa Cruz Biotechnology). The Flag epitope and high-mobility group box protein 1 (HMGB1) were identified using mouse anti-Flag (clone M5; Sigma-Aldrich) and monoclonal mouse anti-HMGB1 (clone 115603; R&D Systems, Wiesbaden-Nordenstadt, Germany) antibodies, respectively. Primary antibodies were detected with horseradish peroxidase-conjugated goat anti-rabbit (Sigma-Aldrich) or goat anti-mouse (Dianova, Hamburg, Germany) secondary antibodies. Blots were developed using a chemiluminescence detection system (Roche) according to the manufacturer's instructions.
Immunohistochemistry.
Skin specimens fixed in 4% neutral buffered formalin, processed routinely, and embedded in paraffin were taken from the local Dermatology archive. The retrospective study was conducted with completely anonymized samples according to the Statement of the National Ethics Council on Biobanks for Research, Berlin, Germany. Tissue sections were pretreated with heat-induced epitope retrieval solution (Dako Cytomation target retrieval solution, pH 6.0, Dako Cytomation, Glostrup, Denmark). Sections were stained with rabbit polyclonal antibody against IRF-3 (ab25950; 10 μg/ml [Abcam, Cambridge, United Kingdom]) and rabbit polyclonal antibody against IRF-7 (H-246, sc-9083; 50 μg/ml [Santa Cruz Biotechnology]). Stainings were performed using the Dako instrument Autostainer Plus and stained with the Dako REAL detection system, with alkaline phosphatase/RED-conjugated rabbit anti-mouse antibody (K5005). Sections were examined by light microscopy.
RNA isolation and real-time PCR.
RTS3b cells were seeded onto six-well plates (3.7 × 105 cells per well) and transfected with 5 μg of pCMVβL-IRF-3(5D), IRF-7Δ247-467 in pFlag-CMV2, or the respective control vectors. Twenty-four hours after transfection, total RNA was isolated from the cells with the RNeasy minikit (Qiagen), according to the manufacturer's protocol. cDNA was generated from 1 μg of RNA with a first-strand cDNA synthesis kit (Fermentas, St. Leon-Rot, Germany). Real-time PCR was performed with the LightCycler 2.0 instrument (Roche, Mannheim, Germany). PCR primers (Operon, Cologne, Germany) and probes (Roche Universal Probe Library; Roche) were designed using the Probe Finder software (Roche). For human IFN-α2 quantitative PCR (qPCR), primer pair 5′-CTGCTTGAAGGACAGACA-3′ and 5′-TTTCAGCCTTTTGGAACTGG-3′ and probe no. 63 were used. For human IFN-β qPCR, primer pair 5′-GCCAGGAGGTTCTCAACAAT-3′ and 5′-CTTTGCTATTTTCAGACAAGATTCA-3′ and probe no. 20 were used. Finally, for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) qPCR, primer pair 5′-CTGTAGCCAAATTCGTTGT-3′ and 5′-CTGACTTCAACAGCGACACC-3′ and probe no. 25 were used. Expression values for IFN-α2 and -β were normalized to GAPDH. Mean values from two independent experiments performed in duplicate were calculated.
ChIP assay.
RTS3b cells were seeded onto 10-cm dishes (1 × 106), transfected with 1.3 μg pALuc-HPV8NCR and 12 μg IRF-7Δ247-467 in pFlag-CMV2 or the respective control vectors, using Fugene 6 transfection reagent (Roche) according to the manufacturer's protocol, and used for chromatin immunoprecipitation (ChIP) analysis. After 24 h, cells were trypsinized and then pelleted at 240 × g for 5 min, and ChIP was performed according to the protocol of the ChIP assay kit from Upstate Biotechnology (Lake Placid, NY), as described previously (30). The cleared supernatant was incubated overnight at 4°C with 2 μg of anti-IRF-7 (H-246) (sc-9083; Santa Cruz) or rabbit IgG (Chromopure, Dianova, Germany) antibodies. The immune complexes were precipitated with 25 μl of protein A/G Sepharose in the presence of salmon sperm DNA for 1 h at 4°C. After elution of the protein-DNA complexes and reversing the cross-links, DNA was isolated and the HPV8 NCR containing IRF-BS sequence from nt 7382 to 7389 was detected and quantified by real-time PCR. Promoter-specific forward (5′-TTCAAGCAGGTTTGCAACAG-3′) and reverse (5′-TGCGGTCGGTTTCTTAGGTA-3′) primers and Human Universal Probe Library system (Light Cycler; Roche) probe no. 4 were used. The 186-bp PCR product was detected by electrophoresis on a 2.5% agarose gel.
Statistical analysis.
To evaluate the statistical differences between analyzed groups, a two-sided unpaired t test was applied.
RESULTS
Type I interferons are potently induced by IRF-3 and IRF-7 and suppress transcriptional activity of HPV8 in human keratinocytes.
The impact of IFNs on HPV8 was investigated in primary human keratinocytes, natural host cells for HPV infection. Cells transfected with a reporter vector expressing luciferase under the control of the complete noncoding regulatory region of HPV8 were treated with 100 U/ml IFN-α or IFN-β for 18 h and assayed for luciferase activity. Both cytokines led to a marked decrease in HPV8 NCR activity. IFN-α reduced the HPV8 NCR activity by 50% and that of IFN-β by 57% compared with the level in the control untreated cells (Fig. 1A).
FIG. 1.

Transcriptional activity of HPV8 is suppressed by type I interferons. (A) NHK were transfected with an HPV8 NCR luciferase construct (0.5 μg) and treated with or without IFN-α or IFN-β (100 U/ml). After 24 h, the luciferase activity was measured and normalized to the protein concentration of the respective luciferase extracts. The value of the untreated control cells was set at 1. Shown are the values averaged from three transfections performed in triplicate ± standard deviations (SD). (B) RTS3b cells were transfected with IRF-3(5D) or IRF-7Δ247-467 expression vectors and the respective control vectors. After 24 h of expression of IFN-α2 (left panel) and IFN-β (right panel), mRNA was quantified by real-time PCR in relation to GAPDH. Shown are the averaged values from two experiments performed in duplicate.
Having found that exogenous type I IFNs strongly suppress the transcriptional activity of HPV8, we sought to determine if endogenous IFNs can be induced in human epidermal cells resulting in HPV8 suppression. As IRF-3 and IRF-7 are key activators of type I IFN genes, vectors encoding constitutively active IRF-3 [IRF-3(5D)] and IRF-7 (IRF-7Δ247-467) were transfected into the HPV-negative RTS3b cell line. mRNA expression levels of type I IFNs were measured by quantitative PCR. As previously reported for other cell types, expression of IFN-α2 could be detected neither in cells transfected with IRF-3(5D) nor in those transfected with the respective control vectors, while it was strongly upregulated after transfection with IRF-7Δ247-467 (Fig. 1B, left panel). Expression of IFN-β was increased in RTS3b cells transfected with either IRF-7Δ247-467 or IRF-3(5D). Constitutively active IRF-7 induced three times higher levels of IFN-β compared with constitutively active IRF-3 (Fig. 1B, right panel). These data demonstrate that in a human epidermal cell line, IRF-7 potently activates both IFN-α and -β genes, while IRF-3 induces primarily the IFN-β gene, as previously documented in other cell types.
IRF-3 suppresses transcriptional activity of HPV8, also in the presence of HPV8 oncoprotein expression.
Next, we investigated the effect of IRF-3 on the activity of HPV8. Primary human keratinocyte cells were cotransfected with the HPV8 NCR-containing reporter construct and increasing amounts of constitutively active IRF-3. Similarly to the case in cells stimulated with type I IFNs, expression of IRF-3(5D) led to a dose-dependent inhibition of HPV8 NCR activity. Strong IRF-3-mediated suppression of the HPV8 NCR was observed. The transcriptional activities of HPV8 were reduced by 70% and 83% in keratinocytes transfected with 0.1 and 0.4 μg of IRF-3(5D), respectively (Fig. 2A).
FIG. 2.

IRF-3 suppresses transcriptional activity of HPV8, also in the presence of HPV8 oncoprotein expression. (A) NHK cells were transfected with the HPV8 NCR luciferase construct (0.5 μg) and IRF-3(5D) expression vectors as indicated. The total amount of DNA was adjusted with control vectors, as appropriate. (B) RTS3b cells were transfected with the HPV8 NCR luciferase construct (0.5 μg) and cotransfected with the IRF-3(5D) (0.4 μg) and HPV8 E6 (0.4 μg) expression vectors. After 24 h, luciferase activities were measured and normalized to protein concentrations of the respective luciferase extracts, and in the case of RTS3b cells, also the percentages of EGFP-positive cells. The normalized luciferase activity of the control transfection was set at 1. Shown are the values averaged from at least two transfections performed in triplicate ± SD. The asterisk represents statistical significance (P = 0.001). (C) C33A cells were transfected with Flag-tagged HPV8 E6, Flag-tagged HPV16 E6, or control pFlag-CMV2 constructs and cotransfected with either wild-type IRF-3 or IRF-3(5D) expression vectors. After 24 h, cell lysates were prepared and precipitated with anti-Flag agarose beads (IP). The precipitates (P) and input (I; 10% of the lysates) were analyzed for IRF-3, IRF-3(5D), Flag, and HMGB1 proteins by Western blotting (WB).
The question arose of whether IRF-3 retains its suppressive property in the presence of the HPV8 E6 protein, which is the major oncoprotein of the virus (25). Ronco et al. (39) previously reported that IRF-3 is bound by HPV16 E6, and this interaction interferes with the IRF-3-mediated transcriptional activation of IFN-β. To assess whether the HPV8 E6 protein affects the IRF-3-mediated suppression of the HPV8 NCR, transfections with IRF-3(5D) and HPV8 E6 expression vectors were performed in RTS3b cells. In this cell line, IRF-3(5D) alone (0.4 μg) suppressed the HPV8 NCR activity by 44%, while HPV8 E6 did not influence the HPV8 NCR activity. In the presence of E6, IRF-3(5D) still suppressed the HPV8 NCR by 29% (Fig. 2B). Similar results were obtained with the HPV8 E7 oncoprotein (data not shown).
To determine whether IRF-3 can physically interact with HPV8 E6, coimmunoprecipitation assays were performed in RTS3b cells coexpressing Flag-tagged HPV8 E6 and either wild-type or constitutively active IRF-3. Cells cotransfected with Flag-tagged HPV16 E6 or empty pFlag-CMV2 vector served as controls. After 24 h, the cells were lysed and Flag-E6 protein was precipitated from the extracts with anti-Flag antibody. The precipitates and 10% of the lysates used for immunoprecipitation (input) were then tested for the presence of IRF-3 and Flag-tagged protein, respectively. HMGB1, a different nuclear factor, was investigated as a control.
As shown in Fig. 2C, IRF-3 and IRF-3(5D) proteins were present in all input nuclear extracts. Control experiments with the anti-Flag antibody verified expression of HPV8 or HPV16 E6. Coimmunoprecipitation confirmed an interaction between HPV16 E6 and wild-type IRF-3 but not HMGB1. These data were in line with the results previously published by Ronco et al. (39). HPV16 E6 also precipitated the constitutively active form of IRF-3, indicating that the 5-amino-acid exchange within IRF-3(5D) does not affect its ability to bind HPV16 E6. On the contrary, HPV8 E6 precipitated neither the wild-type IRF-3 nor its constitutively active form, IRF-3(5D), nor HMGB1 (Fig. 2C). This suggested that HPV8 E6 does not physically interact with the wild-type or constitutively active form of IRF-3 and has only a weak albeit significant effect on the IRF-3-mediated transcriptional repression.
IRF-7 activates the HPV8 NCR.
As shown in Fig. 1B, IRF-7, strongly induced both type I IFNs (IFN-α2 and IFN-β), whereas IRF-3 only led to IFN-β induction at lower levels. Therefore, it was hypothesized that IRF-7 might be a stronger suppressor of HPV8 than IRF-3. Unexpectedly, when the IRF-7-expressing vector was cotransfected with the HPV8 NCR, the activity of HPV8 was not reduced but enhanced in a dose-dependent manner. At the highest concentration, transfection of IRF-7Δ247-467 led to a 10-fold increase in the HPV8 NCR activity in primary human keratinocytes (Fig. 3A). This was in strong contrast to IRF-3(5D).
FIG. 3.

IRF-7 activates the HPV8 NCR. (A) NHK cells were transfected with the HPV8 NCR luciferase construct (0.5 μg) and IRF-7Δ247-467 expression vectors as indicated. The total amount of DNA was adjusted with the pFlag-CMV2 control vector. (B) RTS3b cells were transfected with a RANTES promoter luciferase construct (0.5 μg) and increasing amounts of the IRF-3(5D) (left panel) or IRF-7Δ247-467 (right panel) expression vectors as indicated. After 24 h, luciferase activities were measured and normalized to protein concentrations of the respective luciferase extracts and, in the case of RTS3b cells, also the percentages of EGFP-positive cells. The normalized luciferase activity of the control transfection was set at 1. Shown are the values averaged from two transfections performed in triplicate ± SD.
To investigate whether IRF-3 and IRF-7 also display opposing effects on a well-characterized IRF target gene in a human epidermal cell line, reporter assays with a construct expressing luciferase under the control of the human RANTES promoter were performed. The 5′ regulatory region of the RANTES gene comprises binding sites for IRFs, and both IRF-3 and IRF-7 have been previously shown to enhance its activity in immune cells (11). In line with these data, constitutively active forms of both IRF-3 and IRF-7 potently activated the human RANTES promoter also in RTS3b cells (Fig. 3B).
These data demonstrated that IRF-3 is not a general suppressor of transcription in human keratinocytes or an epidermal cell line. It activates the RANTES promoter as well as IFN-β transcription but suppresses HPV8 activity similar to exogenous type I IFNs. Moreover, in transient transfections it did not influence the expression levels of coexpressed EGFP (data not shown). IRF-7, in contrast, stimulates HPV8 NCR activity. This was surprising, since IRF-7 is a more potent inducer of type I IFNs than IRF-3. These data suggested that IRF-7 can overcome the IFN-mediated inhibition of HPV8. In fact, cells expressing constitutively active IRF-7 were less sensitive to the suppressive effect of IFN-β (data not shown).
IRF-7 activates the HPV8 late promoter through direct interactions with the virus regulatory region.
To further examine the mechanisms of IRF-7-induced activation of the HPV8 NCR, reporter constructs containing either the late P7535 (HPV8A) or the early P175 (HPV8B) promoter of HPV8 were used. In RTS3b cells, the late P7535 promoter of HPV8 was induced by IRF-7Δ247-467 about 4-fold. The extent of activation was even stronger than that of the full-length HPV8 NCR reporter construct in this cell line (data not shown). The activity of the early P175 promoter remained unchanged in the presence of IRF-7Δ247-467 (Fig. 4A). Thus, IRF-7 selectively stimulated only one of the HPV promoters. This suggested that specificity of IRF-7 may depend on its direct interactions with the HPV8 regulatory region.
FIG. 4.

IRF-7 activates the HPV8 late promoter through direct DNA interactions. (A) Localization of late P7535 and early P175 promoters within the HPV8 noncoding region (NCR) and schematic representation of the luciferase reporter constructs comprising the 5′ and 3′ fragments of HPV8 NCR (HPV8A and HPV8B) (upper panel). RTS3b cells were transfected with luciferase constructs containing either the late (pALuc-HPV8A) or the early (pALuc-HPV8B) HPV8 promoter and the IRF-7Δ247-467 expression vector (0.1 or 0.4 μg) or the respective control vector. The EGFP expression vector (0.25 μg) was cotransfected. Luciferase activity was measured after 24 h and normalized to the protein concentration of the respective luciferase extracts and the percentage of EGFP-positive cells. The normalized luciferase activity of the control transfection was set at 1. Shown are the values averaged from two transfections performed in triplicate ± SD (lower panel). (B) 32P-labeled oligonucleotides (nt 7369 to 7398 of HPV8) containing the analyzed IRF-BS nt 7382 to 7389 (capital letters) were incubated with decreasing amounts of GST or GST-IRF-7 fusion proteins (600, 300, 150, or 75 ng) and analyzed by EMSA on the same gel. The arrow indicates complexes corresponding to IRF-7 DNA binding activity. (C) Chromatin immunoprecipitation was performed using RTS3b cells transfected with the pALuc-HPV8NCR and IRF-7Δ247-467 expression vectors. For precipitation, anti-IRF-7 antibody was used. DNA was isolated and amplified by real-time PCR with primers specific for the region from nt 7369 to 7398 of HPV8. The HPV8 region from nt 7369 to 7398 comprising the PCR product (arrow) was visualized on an agarose gel (upper panel) and quantified (lower panel). The amount of target DNA precipitated with the control antibody was set at 1. Measurements were conducted in duplicate. (D) For transfection of RTS3b cells, the pALuc-HPV8A reporter constructs either with an intact IRF-BS (pALuc-HPV8A) or deleted IRF-BS (pALuc-HPV8Adel) were used. A schematic representation of the deleted fragment is shown in the upper panel. The cells were cotransfected with IRF-7Δ247-467 (0.1 μg) or the respective control vector and the EGFP expression vector (0.25 μg). After 24 h, the luciferase activity was measured and normalized to the protein concentration of the respective luciferase extracts and the percentage of EGFP-positive cells. The normalized luciferase activity of the control transfection was set at 1. Shown are the values averaged from three transfections performed in triplicate ± SD (lower panel). The asterisk represents statistical significance (P < 0.0001).
IRF-7 was first described as a factor binding and regulating the promoter region of an Epstein-Barr virus (EBV) gene containing an IRF binding site (29). This prompted us to investigate the DNA sequence of the HPV8 NCR for a potential IRF-7 binding site. Using the Genomatix MatInspector software (38), we found a 5′-AACGGAAA-3′ IRF recognition sequence (IRF-BS nt 7382 to 7389) (9) located 153 to 146 bp upstream from the HPV8 late P7535 promoter (Fig. 4B).
To test IRF-7 binding to the late promoter region in vitro, EMSAs were performed with labeled oligonucleotides (nt 7369 to 7398) comprising the putative HPV8 IRF binding site and GST-IRF-7 fusion protein. The experiments revealed that IRF-7 recognizes the analyzed DNA sequence and strongly binds to it in a dose-dependent manner. No DNA binding was detected in samples containing only the GST protein (Fig. 4B).
Chromatin immunoprecipitation analysis was performed to investigate binding to the DNA region encompassing the HPV8 IRF-BS in vivo. DNA containing the HPV8 NCR was cotransfected with IRF-7Δ247-467 into the RTS3b cell line and precipitated with an anti-IRF-7 antibody. This led to a 2-fold enrichment of DNA comprising IRF-BS nt 7382 to 7389 compared with the control antibody (Fig. 4C).
To elucidate the functional significance of this IRF-BS in the regulation of HPV8 late promoter activity, a 22-bp fragment encompassing the analyzed binding site was deleted from the HPV8 NCR-containing reporter constructs, and luciferase assays were performed. IRF-7Δ247-467 activated the pALuc-HPV8A reporter construct containing the P7535 late promoter (pALuc-HPV8A). However, the stimulatory ability of IRF-7 was significantly reduced in the construct lacking the IRF-BS (pALuc-HPV8Adel) (Fig. 4D; P < 0.0001). Comparable results were obtained for the pALuc-HPV8 NCR reporter construct containing both HPV8 promoters and deletion of the IRF-BS (data not shown). Taken together, these findings demonstrated that sequence-specific DNA binding of IRF-7 contributes to its HPV8 NCR-activating ability.
IRF-7 expression in human keratinocytes is differentiation specific and involved in UV light-mediated activation of the HPV8 NCR.
In a previous study, activation of IRF-7 has been detected in response to UV light in human cervical adenocarcinoma HeLa cells (19). UV-B irradiation, on the other hand, was shown to induce transcriptional activity of HPV8 (2). So far, it had been unclear whether or not UV irradiation involves IRF-7 for activation of the HPV8 NCR.
First, the ability of UV-B to activate IRF-7 in NHK cells was examined. EMSAs were performed with nuclear extracts isolated from NHK cells 6 h after irradiation with 15 mJ/cm2 UV-B. These experiments revealed an increase in DNA binding activity at the IRF-BS in HPV8 (oligonucleotide positions 7369 to 7398) (Fig. 5A).
FIG. 5.

UV light activates HPV8 via IRF-7 in human keratinocytes. (A) Nuclear extracts isolated from NHK cells were prepared at 6 h after irradiation with UV-B (15 mJ/cm2) and analyzed by EMSA using 32P-labeled oligonucleotides (nt 7369 to 7398) comprising the IRF-BS. The arrow indicates IRF-7 binding to the IRF-BS within HPV8. (B) NHK cells were cotransfected with the HPV8 NCR luciferase construct (0.5 μg) and the IRF-7 dominant-negative construct (EGFP-IRF-7Δ12-101) (1.0 μg). The total amount of DNA was adjusted with respective control vectors. Eight hours later, the cells were treated with UV-B (15 mJ/cm2), and 16 h later, the luciferase activity was measured and normalized to protein concentrations of the respective luciferase extracts. The normalized luciferase activity of the control transfection was set at 1. Shown are the values averaged from two transfections performed in duplicate ± SD. The asterisk represents statistical significance (P = 0.01). (C) Expression of IRF-7 in healthy skin (left panel) and sunburned skin (middle panel) and IRF-3 in healthy skin (right panel) was analyzed by immunohistochemistry. Bar, 100 μm.
To test the functional significance of UV-B-induced IRF-7, the transcriptional activity of HPV8 was investigated in luciferase assays in response to UV-B and in the presence or absence of a dominant-negative mutant of IRF-7 (IRF-7Δ12-101). NHK cells were transfected with the HPV8 NCR reporter vector and irradiated with 15 mJ/cm2. UV-B displayed a more than 2-fold increase in HPV8 transcriptional activity compared to the nonirradiated cells. This corresponds to data from previous results (2). In the presence of a dominant-negative form of IRF-7, the UV-B-induced activation of HPV8 was reduced to its basal values. These experiments demonstrated that IRF-7 is a downstream effector of UV-B-mediated activation of HPV8 NCR (Fig. 5B).
These findings prompted us to investigate the expression of IRF-7 in vivo in human epidermis. Immunohistochemical analysis of healthy skin biopsy specimens showed a diffuse cytoplasmic staining of IRF-7 confined almost exclusively to the suprabasal layers. Expression was detected in the spinous layers, a decrease observed a short distance below the stratum granulosum with a strong increase in intensity in the stratum granulosum and stratum corneum. In the basal layer, IRF-7 expression was almost undetectable (Fig. 5C, left panel). In acute dermatitis solaris lesional skin, IRF-7 staining intensity was strongly increased and the IRF-7 expression pattern was even more shifted to the upper differentiating layers of the epidermis. Of note, nuclear staining of IRF-7 was observed in the uppermost differentiated layers, indicating that IRF-7 was activated and translocated to the nucleus (Fig. 5C, middle panel). Thus, the expression pattern of IRF-7 is differentiation specific in human epidermis and apparently UV inducible in vivo. In contrast to that, expression of IRF-3 was almost uniformly distributed within all epidermal cell layers, including the basal layer (Fig. 5C, right panel).
IRF-3-induced suppression prevails over IRF-7-mediated activation of the HPV8 NCR.
As IRF-3 was expressed throughout the whole human epidermis (Fig. 5C, right panel) and unlike IRF-7, suppressed HPV8 NCR activity (Fig. 2A), we were interested how this transcription factor influences the function of IRF-7 with regard to HPV8. EMSA analysis demonstrated that IRF-3 also recognizes and binds to oligonucleotides containing the IRF-BS within HPV8 (Fig. 6A). To evaluate the impact of IRF-3 on IRF-7-mediated activation of HPV8 NCR, luciferase reporter assays were performed.
FIG. 6.

Suppression of IRF-3 dominates over the IRF-7 stimulatory effect. (A) Purified GST and GST-IRF-3 fusion proteins were incubated with 32P-labeled oligonucleotides (nt 7365 to 7391) comprising the IRF-BS and analyzed by EMSA. The IRF-3 band is indicated by an arrow. (B) RTS3b cells were transfected with the HPV8 NCR luciferase construct (0.5 μg) and increasing amounts of IRF-3(5D) (0.025, 0.1, and 0.4 μg) in the presence or absence of the IRF-7Δ247-467 (0.4 μg) expression vector. The total amount of DNA was adjusted with respective control vectors. The EGFP expression vector (0.25 μg) was cotransfected. After 24 h, the luciferase activity was measured and normalized to the protein concentration of the respective luciferase extracts and the percentage of EGFP-positive cells. The normalized luciferase activity of the control transfection was set at 1. Shown are the values averaged from two transfections performed in triplicate ± SD. (C) Nuclear extracts were isolated from RTS3b cells transfected with the pCAGGS control vector or GFP-IRF-7 expression vector and incubated with 32P-labeled oligonucleotides (nt 7365 to 7391). Purified GST or GST-IRF-3N proteins were used for competition in EMSA. An arrow indicates the IRF-7-specific band, which is reduced in the presence of GST-IRF-3N (asterisk).
As shown in Fig. 6B, transfection of 0.4 μg of IRF-7Δ247-467 alone activated the reporter construct by 3.3-fold in RTS3b cells, while 0.025 μg of IRF-3(5D) suppressed the HPV8 NCR activity by 35%. Increasing the amount of IRF-3(5D) to 0.1 and 0.4 μg resulted in about 60% suppression. In parallel, the HPV8 NCR activity was tested in cells transfected with IRF-7 and increasing amounts of IRF-3. Interestingly, the smallest amount of IRF-3(5D) (0.025 μg) was sufficient to revert the activating effect of IRF-7, reducing the HPV8 NCR activity to its basal values. Higher doses of IRF-3(5D) (0.1 and 0.4 μg) even suppressed the HPV8 NCR by 35% and 44%, respectively, despite the presence of the activating IRF-7Δ247-467 (Fig. 6B). These results indicated that the suppressive properties of IRF-3(5D) prevail over the activating properties of IRF-7Δ247-467, leading to HPV8 NCR repression.
To elucidate the underlying mechanism, we investigated whether or not IRF-3 interfered with binding of IRF-7 to the analyzed IRF-BS in HPV8. To better discriminate IRF-3 from IRF-7 binding in EMSA, nuclear extracts containing EGFP-IRF-7 were generated from transiently transfected RTS3b cells. A deletion mutant of GST-IRF-3 (GST-IRF-3N) still comprising the N-terminal DNA-binding site was used as competitor. Gel retardation assays showed that the complex corresponding to IRF-7 bound to IRF-BS (arrow) was displaced from DNA by an excess of GST-IRF-3N but not by GST alone (Fig. 6C). The binding of GST-IRF-3N to the probe is indicated by an asterisk in Fig. 6C. Thus, large amounts of IRF-3 were able to displace IRF-7 from binding to IRF-BS within HPV8. This mechanism of action may at least partially explain the IRF-3 dominance over IRF-7 on the HPV8 NCR.
Poly(I:C) is a strong repressor of HPV8 transcription in primary human keratinocytes.
Poly(I:C) is a synthetic double-stranded RNA and a known activator of IRF-3. It has previously been demonstrated that in human keratinocytes poly(I:C) pathways are operative and IRF-3 becomes activated in response to poly(I:C) (18). We therefore hypothesized that treatment with poly(I:C) might affect the HPV8 NCR activity. The ability of poly(I:C) to induce IRF-3 binding activity in human keratinocytes was confirmed in EMSA. Nuclear extracts isolated from NHK cells stimulated with 10 or 100 μg/ml poly(I:C) were incubated with labeled oligonucleotides containing the IRF-3 consensus binding site. As expected, a dose-dependent increase in DNA binding activity corresponding to IRF-3 was observed (data not shown).
Treatment of RTS3b cells with 10 μg/ml poly(I:C) resulted in suppression of the HPV8 NCR. Also similar to IRF-3, poly(I:C) dominated over the activating effect of IRF-7 on HPV8 transcription (Fig. 7A). Of note, poly(I:C) reduced the activity of both the late (Fig. 7B, left panel) and the early (Fig. 7B, right panel) HPV8 promoters. Suppression of the late promoter was independent of the presence of the IRF binding site investigated in this study (Fig. 7B, left panel). These data were replicated by IRF-3(5D), which also suppressed both the late and the early HPV8 promoters (data not shown).
FIG. 7.

Poly(I:C) suppresses the activity of HPV8 late and early promoters independently of the IRF binding site. (A) RTS3b cells were transfected with the HPV8 NCR luciferase construct (0.5 μg) and IRF-7Δ247-467 expression vectors (0.4 μg). The total amount of DNA was adjusted with empty pFlag-CMV2 control vector. The EGFP expression vector (0.25 μg) was cotransfected. After 6 h, the cells were stimulated with poly(I:C) (10 μg/ml) or medium as a control. After 24 h posttransfection, the luciferase activity was measured and normalized to the protein concentration of the respective luciferase extracts and the percentage of EGFP-positive cells. The normalized luciferase activity of the control transfection was set at 1. Shown are the values averaged from two transfections performed in triplicate ± SD. (B) RTS3b cells were transfected with either the pALuc-HPV8A reporter construct containing an intact IRF-BS (pALuc-HPV8A) or with IRF-BS deleted (pALuc-HPV8Adel) (left panel) or the pALuc-HPV8B reporter construct (right panel). After 6 h, the cells were stimulated with 10 μg/ml poly(I:C). Twenty-four hours posttransfection, luciferase activity was measured and normalized to the protein concentration. The normalized luciferase activity of the control transfection was set at 1. Shown are the values averaged from three transfections performed in triplicate ± SD.
To study the natural host cells for HPV infection, the effects of IRF-3 activators were investigated in primary human keratinocytes. Stimulation with 1 to 100 μg/ml poly(I:C) resulted in 70% repression of the HPV8 NCR activity (Fig. 8A). Similar results were obtained with RNA bearing 5′ phosphates, which displayed a dose-dependent effect. Transfection of 260 ng 5′pppRNA suppressed the HPV8 NCR activity by more than 80% (Fig. 8B).
FIG. 8.
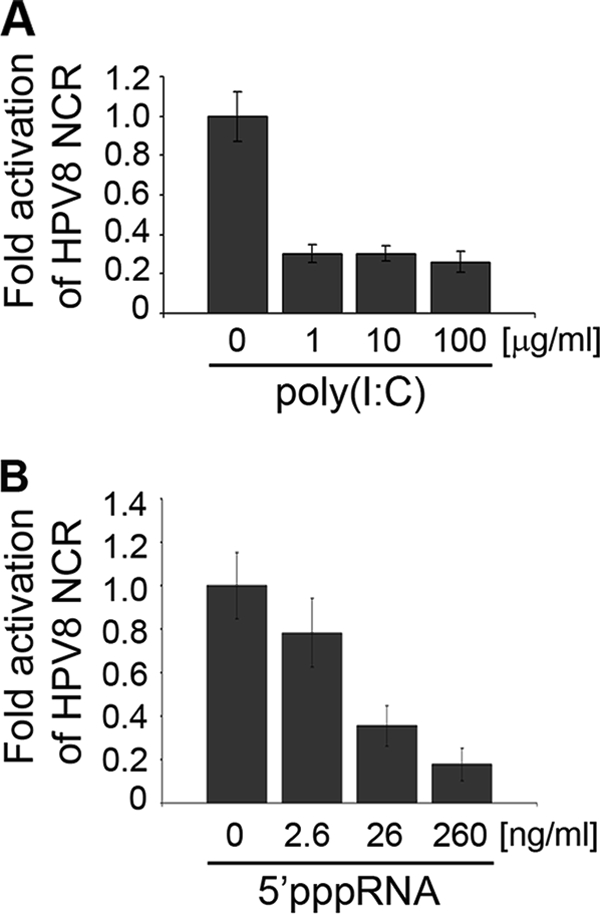
IRF-3 inducers poly(I:C) and 5′pppRNA strongly suppress HPV8 activity in primary human keratinocytes. NHK cells were transfected with the HPV8 NCR luciferase construct (0.5 μg) and after 6 h stimulated with poly(I:C) (1, 10, and 100 μg/ml) (A) or transfected with 5′pppRNA (2.6, 26, or 260 ng/ml) (B). Twenty-four hours posttransfection, luciferase activity was measured and normalized to protein concentration. The normalized luciferase activity of the control transfection was set at 1. Shown are the values averaged from three transfections performed in triplicate ± SD.
DISCUSSION
This study shows for the first time that IRF-3 and IRF-7, key inducers of type I IFNs, are differentially expressed in human epidermis and regulate the transcriptional activity of a human papillomavirus in primary human keratinocytes. While IRF-7 stimulated HPV8 transcription, IRF-3 as well as the IRF-3-activating compounds poly(I:C) and 5′pppRNA turned out to be strong suppressors of the cutaneous HPV. IRF-3 suppression was observed in the presence of viral oncogene expression and prevailed over IRF-7-mediated activation.
In ChIP analysis, we have identified an IRF recognition sequence located within the HPV8 late promoter region. Functional studies showed that this binding site was indispensable for the IRF-7-induced activation of the HPV8 NCR, thus revealing IRF-7 as a direct transcriptional activator of HPV8. Detailed analysis demonstrated that IRF-7 exclusively acts on the late but not on the early promoter of HPV8.
IRF-7 plays a central role in type I IFN immune responses. Type I IFNs induce IRF-7, which in turn directly stimulates type I IFN expression. Functional activity of IRF-7 is achieved upon phosphorylation, which enables nuclear translocation and DNA binding. IRF-7 is activated in response to Toll-like receptor (TLR) signaling via IkappaB kinase-alpha (IKK-α) (15) or in response to UV light via the Jun N-terminal protein kinase (JNK) signaling pathway (19). UV is a major driver of genus β HPV-associated carcinogenesis. Interestingly, UV (2) and the JNK targets AP-1 (44) and IRF-7 (present study) are activators of the HPV8 NCR. Our data shed new light on IRF-7 in human epidermis in vivo. We demonstrate for the first time that IRF-7 is expressed in human epidermis in a differentiation-specific manner, which is further accentuated in sunburned skin. Nuclear IRF-7 was detected exclusively in the uppermost differentiated layers of human epidermis. In vitro, we show that UV-mediated activation of the HPV8 NCR involved IRF-7. This is most interesting in view of the fact that IRF-7 only activates the late promoter responsible for late gene expression, which is confined to differentiated layers of human epidermis. Thus, the IRF-7 expression pattern and its activation converge spatially within epidermal cell layers, in which the HPV productive life cycle takes place and the viral late promoter becomes active. Furthermore, these data suggest that UV light is an important activator of IRF-7 in human skin, underlining the physiological relevance of the in vitro findings demonstrated in this study. Our functional data also suggest that IRF-7 constitutes a functional link between the two cocarcinogens, UV-B light and HPV8 activation.
Activation of a tumor virus by IRF-7 is not unprecedented. IRF-7 has been first identified within the biological context of Epstein-Barr virus (EBV) (29). It directly interacts with the viral DNA and activates the promoter of the viral oncogene latent membrane protein 1 (LMP1). LMP1 in turn induces and activates IRF-7 (28). This positive regulatory circuit is considered to potentiate the oncogenic effects of EBV (27). Whereas IRF-7 regulation by EBV is well documented, no data had been available on IRF-7 regulation by HPVs so far.
In contrast to IRF-7, IRF-5 has recently emerged as a negative regulator of EBV, which breaks the IRF-7/LMP1 regulatory circuit (7, 28). Interestingly, IRF-5.2, a splice variant of IRF-5, was also shown to repress the transcriptional activity of genus β HPVs (1). IRF-5.2 directly interacts with HPV8 at two binding sites, the M29 motif, representing the core element of the HPV8 early promoter, and the negative regulatory region of the HPV8 late promoter (1). The latter IRF-5.2 binding site encompasses nt 7398 to 7411 of the HPV8 genome. It is located downstream of and separated by 9 bp from the IRF binding site at positions 7382 to 7389 identified in the present study. Thus, these two binding sites, although in close proximity to each other, do not overlap and may have independent effects on the virus. IRF-7, IRF-5.2, and IRF-3 (data not shown) represent, thus far, the only interferon regulatory factors that have been documented to directly interact with the NCR of cutaneous EV-associated HPVs.
Previous investigations have demonstrated a crucial role of IRF-3 and IRF-7 in the expression of type I IFN genes and some chemokine genes, such as RANTES, in different cell systems (4, 11, 22). Our study revealed that a similar regulatory program takes place in human keratinocytes. Both IRF-3 and IRF-7 concordantly induced IFN-β expression and the activity of the RANTES promoter, and neither IRF-3 nor IRF-7 influenced expression levels of coexpressed EGFP in transient transfections. In contrast, they displayed opposing effects on HPV8 transcription both in primary human keratinocytes and the epidermal cell line RTS3b. IRF-7 stimulated the viral regulatory region, and IRF-3 suppressed its activity. When coexpressed, the suppressive properties of IRF-3 on HPV8 prevailed over the activating effect of IRF-7. Interestingly, recent data demonstrate that IRF-3 and IRF-7 can also have opposing activities on the expression of some IFN-α genes. While IRF-7 induces these IFN-α genes, IRF-3 can inhibit IRF-7-mediated activation of their promoters (12). In this study, we demonstrate that high levels of IRF-3 may be able to displace IRF-7 from our novel IRF-BS within the HPV8 NCR. This may at least partially explain the IRF-3 dominance over IRF-7 with respect to HPV8 NCR regulation.
The HPV8 E6 protein is considered the major oncoprotein of HPV8 (25). Both E6 proteins from HPV8 and HPV16 have transforming properties (42). They exert their function primarily by interacting with key cellular regulators, a prerequisite for oncogenic transformation. High-risk mucosal HPVs were also found to employ several strategies to impair the IFN and IRF defense system, allowing their persistence in the epithelium. HPV18 E6 and HPV16 E7 were shown to interact with downstream molecules of the type I IFN signaling pathway, suppressing their activity (41). The HPV16 E7 protein interacts with IRF-1, while HPV16 E6 binds to IRF-3. As a consequence, both viral proteins inhibit the activities of these cellular transcription factors (32, 33, 39). Whether genus β HPVs can counteract IRF signaling has not been investigated. Data presented in this study suggest that HPV8 E6 is unable to bind to IRF-3, indicating a difference between HPV8 and HPV16 E6 proteins.
Corresponding to its deficiency in binding to the HPV8 E6 oncoprotein, IRF-3 displayed inhibitory activity toward HPV8 also in the presence of the HPV8 oncoprotein. Moreover, IRF-3 suppression dominated over the activating effects of IRF-7. This suggested that activation of IRF-3 might be of practical use in the treatment of HPV8-expressing lesions. Looking for possible exogenous activators of IRF-3, we have focused on analogues of double-stranded RNA, such as poly(I:C) and 5′pppRNA.
Human keratinocytes respond to poly(I:C) with an antiviral defense program. In these cells, poly(I:C) is sensed by TLR3 or retinoic acid-inducible gene I/melanoma differentiation-associated gene 5 (RIG-I/MDA5) pattern recognition molecules or directly activates the double-stranded RNA (dsRNA)-dependent protein kinase R. In primary human keratinocytes, poly(I:C)-mediated activation of IRF-3 preferentially takes place via RIG-I/MDA5 receptors (18). The potent IRF-3 activator 5′pppRNA is recognized by RIG-I only (36). We confirmed that IRF-3 binds to the novel IRF-BS in HPV8 and that treatment with poly(I:C) increases DNA binding activity corresponding to IRF-3 in keratinocytes (data not shown). Stimulation of keratinocytes with low doses of poly(I:C) or 5′pppRNA led to a potent suppression of HPV8 NCR activity. Suppression was specific, since both compounds led to activation of a cellular promoter in primary human keratinocytes and did not affect their viability (data not shown). Similar to IRF-3, poly(I:C)-mediated suppression was not affected by IRF-7. In contrast to IRF-7, the suppressive effect of IRF-3 and poly(I:C) was independent of the HPV8 IRF-BS found in this study. This pointed to a much broader role of IRF-3 and poly(I:C), which was apparently not restricted to the IRF-BS regulating the late promoter. These data were supported by the fact that IRF-3 similar to poly(I:C) also suppressed the early HPV8 promoter, while IRF-7 did not affect the early promoter. The differences in function corresponded to differential expression of IRF-3 and -7 in human epidermis. While IRF-7 is expressed in a strict differentiation-specific manner, IRF-3 is found in keratinocytes of all epidermal layers, including basal proliferating keratinocytes. Apart from direct displacement of IRF-7 from binding to the HPV8-NCR, it can only be speculated whether IRF-3 displays its suppressive activity also indirectly via induction of other suppressive cellular factors. Interestingly, aside from its effects on HPV transcription, poly(I:C) also suppresses HPV18 DNA replication through induction of the interferon-inducible protein p56 (46). Thus, poly(I:C) displays broad antiviral activity against human papillomaviruses.
A nonsurgical option for the treatment of (pre)cancerous skin lesions includes topical application of imiquimod, a TLR7 agonist, which has immunomodulatory properties. Its application in EV patients led to inconsistent results (5, 10, 16, 17). It is supposed that the antiviral effect of imiquimod is mediated indirectly by activation of the immune system, since TLR7 is constitutively expressed on immune cells (26) but absent in normal human keratinocytes. Recently, it has recently been reported that stimulation with poly(I:C) induces expression of TLR7 on primary human keratinocytes (18).
Our study suggests that local application of poly(I:C), 5′pppRNA, or derivates thereof might represent an attractive strategy to treat HPV8-expressing (precancerous) lesions, particularly in EV patients. As previously reported, poly(I:C) inhibits HPV replication (46). As shown in this study, it acts directly on keratinocytes and potently suppresses HPV8 transcription. Thus, poly(I:C) induces a state of cell-autonomous immunity that enables keratinocytes to efficiently target HPV infection. Due to its effect on TLR7 expression in keratinocytes, it may also increase the effectiveness of imiquimod therapy.
Acknowledgments
We thank Alexandra van Mil, Anne Kerber, and Barbara Best for excellent technical assistance.
This work was supported by grants from the DFG through the SFB670 and the Saarland University HOMFOR Program, both granted to S. Smola.
Footnotes
Published ahead of print on 27 October 2010.
REFERENCES
- 1.Akgul, B., M. Curten, H. Haigis, I. Rogosz, and H. Pfister. 2006. Interferon regulatory factor 5.2 acts as a transcription repressor of epidermodysplasia verruciformis-associated human papillomaviruses. Arch. Virol. 151:2461-2473. [DOI] [PubMed]
- 2.Akgul, B., W. Lemme, R. Garcia-Escudero, A. Storey, and H. J. Pfister. 2005. UV-B irradiation stimulates the promoter activity of the high-risk, cutaneous human papillomavirus 5 and 8 in primary keratinocytes. Arch. Virol. 150:145-151. [DOI] [PMC free article] [PubMed]
- 3.Reference deleted.
- 4.Au, W. C., and P. M. Pitha. 2001. Recruitment of multiple interferon regulatory factors and histone acetyltransferase to the transcriptionally active interferon a promoters. J. Biol. Chem. 276:41629-41637. [DOI] [PubMed] [Google Scholar]
- 5.Baskan, E. B., S. Tunali, S. B. Adim, A. Turan, and S. Toker. 2006. A case of epidermodysplasia verruciformis associated with squamous cell carcinoma and Bowen's disease: a therapeutic challenge. J. Dermatolog. Treat. 17:179-183. [DOI] [PubMed] [Google Scholar]
- 6.Beglin, M., M. Melar-New, and L. Laimins. 2009. Human papillomaviruses and the interferon response. J. Interferon Cytokine Res. 29:629-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen, H., J. Huang, F. Y. Wu, G. Liao, L. Hutt-Fletcher, and S. D. Hayward. 2005. Regulation of expression of the Epstein-Barr virus BamHI-A rightward transcripts. J. Virol. 79:1724-1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dole, V. S., H. Smola, S. Dooley, H. M. Said, M. Oldak, H. J. Pfister, and S. Smola. 2009. The adenoviral E1A oncoprotein activates the Smad7 promoter: requirement of a functional E-box. Int. J. Oncol. 35:1493-1498. [DOI] [PubMed] [Google Scholar]
- 9.Fujii, Y., T. Shimizu, M. Kusumoto, Y. Kyogoku, T. Taniguchi, and T. Hakoshima. 1999. Crystal structure of an IRF-DNA complex reveals novel DNA recognition and cooperative binding to a tandem repeat of core sequences. EMBO J. 18:5028-5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garland, S. M., R. Waddell, A. Mindel, I. M. Denham, and J. C. McCloskey. 2006. An open-label phase II pilot study investigating the optimal duration of imiquimod 5% cream for the treatment of external genital warts in women. Int. J. STD AIDS 17:448-452. [DOI] [PubMed] [Google Scholar]
- 11.Genin, P., M. Algarte, P. Roof, R. Lin, and J. Hiscott. 2000. Regulation of RANTES chemokine gene expression requires cooperativity between NF-kappa B and IFN-regulatory factor transcription factors. J. Immunol. 164:5352-5361. [DOI] [PubMed] [Google Scholar]
- 12.Genin, P., A. Vaccaro, and A. Civas. 2009. The role of differential expression of human interferon-a genes in antiviral immunity. Cytokine Growth Factor Rev. 20:283-295. [DOI] [PubMed] [Google Scholar]
- 13.Habig, M., H. Smola, V. S. Dole, R. Derynck, H. Pfister, and S. Smola-Hess. 2006. E7 proteins from high- and low-risk human papillomaviruses bind to TGF-beta-regulated Smad proteins and inhibit their transcriptional activity. Arch. Virol. 151:1961-1972. [DOI] [PubMed] [Google Scholar]
- 14.Hadaschik, D., K. Hinterkeuser, M. Oldak, H. J. Pfister, and S. Smola-Hess. 2003. The papillomavirus E2 protein binds to and synergizes with C/EBP factors involved in keratinocyte differentiation. J. Virol. 77:5253-5265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoshino, K., T. Sugiyama, M. Matsumoto, T. Tanaka, M. Saito, H. Hemmi, O. Ohara, S. Akira, and T. Kaisho. 2006. IkappaB kinase-alpha is critical for interferon-alpha production induced by Toll-like receptors 7 and 9. Nature 440:949-953. [DOI] [PubMed] [Google Scholar]
- 16.Hu, W., G. Nuovo, M. Willen, and S. Somach. 2004. Epidermodysplasia verruciformis in two half brothers with HIV infection. J. Cutan. Med. Surg. 8:357-360. [DOI] [PubMed] [Google Scholar]
- 16a.IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. 2007. Human papillomaviruses. IARC Monogr. Eval. Carcinog. Risks Hum. 90:1-636. [PMC free article] [PubMed] [Google Scholar]
- 17.Janssen, K., G. P. Lucker, R. H. Houwing, and R. van Rijssel. 2007. Epidermodysplasia verruciformis: unsuccessful therapeutic approach with imiquimod. Int. J. Dermatol. 46(Suppl. 3):45-47. [DOI] [PubMed] [Google Scholar]
- 18.Kalali, B. N., G. Kollisch, J. Mages, T. Muller, S. Bauer, H. Wagner, J. Ring, R. Lang, M. Mempel, and M. Ollert. 2008. Double-stranded RNA induces an antiviral defense status in epidermal keratinocytes through TLR3-, PKR-, and MDA5/RIG-I-mediated differential signaling. J. Immunol. 181:2694-2704. [DOI] [PubMed] [Google Scholar]
- 19.Kim, T. K., T. Kim, T. Y. Kim, W. G. Lee, and J. Yim. 2000. Chemotherapeutic DNA-damaging drugs activate interferon regulatory factor-7 by the mitogen-activated protein kinase kinase-4-cJun NH2-terminal kinase pathway. Cancer Res. 60:1153-1156. [PubMed] [Google Scholar]
- 20.Koromilas, A. E., S. Li, and G. Matlashewski. 2001. Control of interferon signaling in human papillomavirus infection. Cytokine Growth Factor Rev. 12:157-170. [DOI] [PubMed] [Google Scholar]
- 21.Lin, R., P. Genin, Y. Mamane, and J. Hiscott. 2000. Selective DNA binding and association with the CREB binding protein coactivator contribute to differential activation of alpha/beta interferon genes by interferon regulatory factors 3 and 7. Mol. Cell. Biol. 20:6342-6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin, R., C. Heylbroeck, P. Genin, P. M. Pitha, and J. Hiscott. 1999. Essential role of interferon regulatory factor 3 in direct activation of RANTES chemokine transcription. Mol. Cell. Biol. 19:959-966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin, R., Y. Mamane, and J. Hiscott. 2000. Multiple regulatory domains control IRF-7 activity in response to virus infection. J. Biol. Chem. 275:34320-34327. [DOI] [PubMed] [Google Scholar]
- 24.Lin, R., Y. Mamane, and J. Hiscott. 1999. Structural and functional analysis of interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains. Mol. Cell. Biol. 19:2465-2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marcuzzi, G. P., M. Hufbauer, H. U. Kasper, S. J. Weissenborn, S. Smola, and H. Pfister. 2009. Spontaneous tumour development in human papillomavirus type 8 E6 transgenic mice and rapid induction by UV-light exposure and wounding. J. Gen. Virol. 90:2855-2864. [DOI] [PubMed] [Google Scholar]
- 26.Miller, R. L., T. C. Meng, and M. A. Tomai. 2008. The antiviral activity of Toll-like receptor 7 and 7/8 agonists. Drug News Perspect. 21:69-87. [DOI] [PubMed] [Google Scholar]
- 27.Ning, S., A. M. Hahn, L. E. Huye, and J. S. Pagano. 2003. Interferon regulatory factor 7 regulates expression of Epstein-Barr virus latent membrane protein 1: a regulatory circuit. J. Virol. 77:9359-9368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ning, S., L. E. Huye, and J. S. Pagano. 2005. Interferon regulatory factor 5 represses expression of the Epstein-Barr virus oncoprotein LMP1: braking of the IRF7/LMP1 regulatory circuit. J. Virol. 79:11671-11676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nonkwelo, C., I. K. Ruf, and J. Sample. 1997. Interferon-independent and -induced regulation of Epstein-Barr virus EBNA-1 gene transcription in Burkitt lymphoma. J. Virol. 71:6887-6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oldak, M., R. B. Maksym, T. Sperling, M. Yaniv, H. Smola, H. J. Pfister, J. Malejczyk, and S. Smola. 2010. Human papillomavirus type 8 E2 protein unravels JunB/Fra-1 as an activator of the beta4-integrin gene in human keratinocytes. J. Virol. 84:1376-1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oldak, M., H. Smola, M. Aumailley, F. Rivero, H. Pfister, and S. Smola-Hess. 2004. The human papillomavirus type 8 E2 protein suppresses beta4-integrin expression in primary human keratinocytes. J. Virol. 78:10738-10746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park, J. S., E. J. Kim, H. J. Kwon, E. S. Hwang, S. E. Namkoong, and S. J. Um. 2000. Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J. Biol. Chem. 275:6764-6769. [DOI] [PubMed] [Google Scholar]
- 33.Perea, S. E., P. Massimi, and L. Banks. 2000. Human papillomavirus type 16 E7 impairs the activation of the interferon regulatory factor-1. Int. J. Mol. Med. 5:661-666. [DOI] [PubMed] [Google Scholar]
- 34.Pfister, H. 2003. Human papillomavirus and skin cancer. J. Natl. Cancer Inst. Monogr. 31:52-56. [DOI] [PubMed] [Google Scholar]
- 35.Pichlmair, A., and C. Reis e Sousa. 2007. Innate recognition of viruses. Immunity 27:370-383. [DOI] [PubMed] [Google Scholar]
- 36.Pichlmair, A., O. Schulz, C. P. Tan, T. I. Naslund, P. Liljestrom, F. Weber, and C. Reis e Sousa. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314:997-1001. [DOI] [PubMed] [Google Scholar]
- 37.Purdie, K. J., C. J. Sexton, C. M. Proby, M. T. Glover, A. T. Williams, J. N. Stables, and I. M. Leigh. 1993. Malignant transformation of cutaneous lesions in renal allograft patients: a role for human papillomavirus. Cancer Res. 53:5328-5333. [PubMed] [Google Scholar]
- 38.Quandt, K., K. Frech, H. Karas, E. Wingender, and T. Werner. 1995. MatInd and MatInspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res. 23:4878-4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ronco, L. V., A. Y. Karpova, M. Vidal, and P. M. Howley. 1998. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 12:2061-2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schaper, I. D., G. P. Marcuzzi, S. J. Weissenborn, H. U. Kasper, V. Dries, N. Smyth, P. Fuchs, and H. Pfister. 2005. Development of skin tumors in mice transgenic for early genes of human papillomavirus type 8. Cancer Res. 65:1394-1400. [DOI] [PubMed] [Google Scholar]
- 41.Smola-Hess, S., and H. Pfister. 2006. Immune evasion in genital papillomavirus infection and cervical cancer: role of cytokines and chemokines, p. 324-339. In M. S. Campo (ed.), Papillomavirus research from natural history to vaccines and beyond. Caister Academic Press, Norfolk, United Kingdom.
- 42.Smola-Hess, S., and H. J. Pfister. 2002. Interaction of papillomaviral oncoproteins with cellular factors, p. 431-450. In E. B. Andreas Holzenburg (ed.), Structure-function relationships of human pathogenic viruses. Kluwer Academic/Plenum Publishers, New York, NY.
- 43.Steger, G., and H. Pfister. 2007. Transcription and gene expression of epidermodysplasia-verruciformis associated human papillomavirus types, p. 183-201. In B. Norrild (ed.), Human papillomavirus gene regulation and transformation. Transworld Research Network, Kerala, India.
- 44.Stubenrauch, F., J. Malejczyk, P. G. Fuchs, and H. Pfister. 1992. Late promoter of human papillomavirus type 8 and its regulation. J. Virol. 66:3485-3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takaoka, A., T. Tamura, and T. Taniguchi. 2008. Interferon regulatory factor family of transcription factors and regulation of oncogenesis. Cancer Sci. 99:467-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Terenzi, F., P. Saikia, and G. C. Sen. 2008. Interferon-inducible protein, P56, inhibits HPV DNA replication by binding to the viral protein E1. EMBO J. 27:3311-3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.zur Hausen, H. 2009. Papillomaviruses in the causation of human cancers—a brief historical account. Virology 384:260-265. [Epub ahead of print 8 January 2009.] [DOI] [PubMed] [Google Scholar]