

Abstract
The protein kinase found in the short region of alphaherpesviruses, termed US3 in herpes simplex virus type 1 (HSV-1) and pseudorabies virus (PRV) and ORF66 in varicella-zoster virus (VZV), affects several viral and host cell processes, and its specific targets remain an area of active investigation. Reports suggesting that HSV-1 US3 substrates overlap with those of cellular protein kinase A (PKA) prompted the use of an antibody specific for phosphorylated PKA substrates to identify US3/ORF66 targets. HSV-1, VZV, and PRV induced very different substrate profiles that were US3/ORF66 kinase dependent. The predominant VZV-phosphorylated 125-kDa species was identified as matrin 3, one of the major nuclear matrix proteins. Matrin 3 was also phosphorylated by HSV-1 and PRV in a US3 kinase-dependent manner and by VZV ORF66 kinase at a novel residue (KRRRT150EE). Since VZV-directed T150 phosphorylation was not blocked by PKA inhibitors and was not induced by PKA activation, and since PKA predominantly targeted matrin 3 S188, it was concluded that phosphorylation by VZV was PKA independent. However, purified VZV ORF66 kinase did not phosphorylate matrin 3 in vitro, suggesting that additional cellular factors were required. In VZV-infected cells in the absence of the ORF66 kinase, matrin 3 displayed intranuclear changes, while matrin 3 showed a pronounced cytoplasmic distribution in late-stage cells infected with US3-negative HSV-1 or PRV. This work identifies phosphorylation of the nuclear matrix protein matrin 3 as a new conserved target of this kinase group.
All herpesviruses studied to date encode proteins predicted to be protein kinases, based on the presence of conserved signature domains common to cellular kinases. Considerable interest in their roles and functions has developed, because they influence multiple cellular processes by controlling the phosphorylation of both cellular and viral proteins. Two serine/threonine (S/T)-specific protein kinases are found in alphaherpesvirus genomes: the UL13 kinase of herpes simplex virus types 1 and 2 (HSV-1 and HSV-2) (open reading frame 47 [ORF47] in varicella-zoster virus [VZV]) has recognizable homologues in all known herpesviruses, whereas the US3 kinase (ORF66 in VZV) is found in the short genomic region of members of the alphaherpesvirus subfamily. Both alphaherpesvirus kinases are genetically dispensable for viral growth in culture, but kinase-deficient viruses invariably show impaired replication in specific cell types and/or attenuation in animal models of disease (22, 37, 45, 57, 62, 64, 65, 73, 79), suggesting important roles that are dependent on the host cell environment.
The 393-residue ORF66 protein kinase of VZV has such cell-specific importance. VZV causes chickenpox upon primary infection and herpes zoster upon reactivation from a prolonged neuronal latent state. VZVs expressing ORF66 that is translationally halted or kinase inactive show only modest growth impairment in cell cultures used for VZV propagation and in human skin in the SCID-hu mouse model but show severe growth impairment in T cells (46, 65, 73) and in primary corneal stromal fibroblasts (22). This phenotype suggests that ORF66-deficient VZV could be suitable for an improved vaccine candidate. The VZV ORF66 kinase has one known target, the VZV major regulatory transcriptional activator immediate-early 62 (IE62) protein (20). IE62 stimulates viral transcription in the nucleus by interacting with the host basal transcriptional machinery, activators, the mediator complex, and viral promoters (61). However, direct IE62 phosphorylation by ORF66 in VZV-infected cells results in nuclear exclusion of IE62, which then partially relocates to the trans-Golgi network and becomes abundantly incorporated into the assembling VZV tegument (20, 22, 39, 40). Interestingly, the VZV ORF66 protein kinase gene is one of six viral genes found to be expressed during the VZV neuronal latent state in human sensory ganglia, and activity on IE62 may explain the predominantly cytoplasmic location of IE62 in neurons (17). VZV ORF66 also directs the phosphorylation of histone deacetylases (HDACs) 1 and 2 to enhance a protranscriptional environment (79), a feature also shared with HSV-1 US3 and PRV US3 (34, 47, 55, 80). ORF66 also contributes to immune evasion tactics through the blocking of apoptosis (64, 65), the modulation of gamma interferon (IFN-γ) signaling pathways in infected human tonsil T cells (65), and the inhibition of surface antigen presentation by major histocompatibility complex class I (1, 21).
Various members of the alphaherpesvirus US3 kinases have been shown to affect additional host cell processes. HSV and PRV US3s modulate components of the nuclear membrane (33, 41, 49, 50, 81) to facilitate nucleocapsid nuclear egress, and PRV US3 facilitates neuron axonal transport of the virion (18). Roles in the inhibition of apoptosis have been attributed to several members of the alphaherpesviruses (6, 12, 13, 35), as have roles in the restructuring of cytoskeleton networks to aid in intercellular spread (24, 52, 67, 76, 77, 78). It remains to be determined which of these activities are common to VZV and other alphaherpesvirus US3/ORF66 kinases. However, VZV ORF66 has a substrate specificity similar to that of HSV-1 and PRV US3 kinases in that it preferentially targets S/T residues embedded in basophilic motifs. The optimal HSV and PRV US3 kinase motifs, defined by peptide substrate determination with purified kinases, are (R)n-X-(S/T)-Y-Y (where n is >2; S/T is the phosphorylated site; X can be absent or preferably Arg, Ala, Val, Pro, or Ser; and Y cannot be Pro or an acidic residue) (42, 56). The known ORF66 kinase sites on IE62 are similar, but it is notable that neither fully fits the optimal US3 consensus motif at +1 and +2 (RKRKS686QPV and KRRVS722EPV).
More recently, it was recognized that US3 motifs were similar to target motifs of cellular cyclic AMP (cAMP)-dependent protein kinase (PKA), which loosely targets RRX(S/T) (5). Using an antibody that recognizes phosphorylated serine/threonine embedded in this motif, it was shown that multiple host proteins were targeted by both US3 and PKA and that US3 induced PKA activity in HSV-1-infected cells by upregulating PKA expression (5). PKA is ubiquitously expressed and has pleiotropic effects and many substrates, including metabolic enzymes, ion channels, regulatory and structural proteins, and transcription factors (69). PKA is a tetrameric holoenzyme composed of catalytic subunits (C) bound to a regulatory subunit dimer; the enzyme is activated by cooperative binding of cAMP to the regulatory subunits, which then release the catalytic subunits (72). Benetti and Roizman have suggested that US3 activates PKA to induce antiapoptotic functions (5). One VZV study has suggested VZV-induced phosphorylation of some PKA substrates (19).
Here we show that the antibody that recognizes the phosphospecific PKA substrates (anti-PKAps) identifies matrin 3 as a common target of VZV ORF66 and the US3 kinases of HSV and PRV. Matrin 3 is one of 12 major proteins that make up the fibrogranular network of the nuclear matrix, characterized as a proteinaceous subnuclear nonhistone fraction resistant to high-salt, detergent, and nuclease treatment of the interphase cell (53). Additional major components of the matrix are lamins A, B, and C, the nucleolar protein B-23, components of nuclear ribonucleoproteins, and 7 additional nuclear matrins (matrins 4, D, E, F, G, 12, and 13) (53). The nuclear matrix serves as an important structural framework for many nuclear processes, including DNA replication and transcriptional regulation (2, 8, 31). Here we report that the cellular distribution of the nuclear matrix protein matrin 3 is altered by novel phosphorylation induced by the alphaherpesvirus US3 kinase family.
MATERIALS AND METHODS
Cell culture.
MRC-5 cells (human diploid lung fibroblasts), human embryonic kidney (Hek) 293 cells, Hek 293T cells (ATCC, Manassas, VA), SF9 cells (Invitrogen Corp., Carlsbad, CA), and MeWo cells were grown and maintained as described previously (20, 21).
Viruses.
The properties of the three alphaherpesviruses and mutants used in this work are summarized in Table 1 .
TABLE 1.
Properties of the alphaherpesviruses and mutants used in this work
| Alphaherpesvirus | Strain | N-terminal EGFP driven by ORF66/US3 promotera | ORF66/US3 kinase | Kinase activity |
|---|---|---|---|---|
| VZV.GFP66 | pOka | + | ORF66 (full length; aa 1-393) | + |
| VZV.GFP66kd | pOka | + | ORF66kd D206E K208R (aa 1-393) | − |
| VZV.GFP66s | pOka | + | ORF66s (aa 1-84) | − |
| HSV-1 | RE | − | US3 (full length; aa 1-481) | + |
| HSV-EGFP-US3.5 | RE | + | US3.5 (aa 77-481) | + |
| HSV-EGFP-ΔUS3 | RE | + | − | − |
| PRV | Kaplan | − | US3 (full length; aa 1-334) | + |
| PRV.ΔUS3 | Kaplan | + | − | − |
+, present; −, absent.
(i) VZV.
All VZV variants are based on the parent of the Oka vaccine strain and were grown in MeWo cells as described previously (39). The VZV recombinants VZV.GFP-66, VZV.GFP-66kd (kinase dead; D206E K208R), VZV.GFP-66s (with ORF66 truncated at residue 84), and rescuant VZV have been described previously (21, 22) (Table 1).
(ii) HSV-1.
The RE strain and a US3 deletion mutant have been described previously (58, 59). HSV-1 RE making only US3.5 (US3 residues 77 to 481) with an N-terminal green fluorescent protein (GFP) tag was generated using a PCR fragment of HSV-1 RE, made using the GC-Rich PCR proofreading system (Roche, Indianapolis, IN) under hot-start conditions with primers US3.5F (5′-GGGAATTCATGTACGGAAACCAGGACTAC-3′) and US3R (5′-GGAAGATCTTCATTTCTGTTGAAACAGCGGCAA-3′) (restriction sites introduced to facilitate cloning are underlined). The BglII- and EcoRI-digested PCR product was cloned into a BamHI/EcoRI-digested pEGFP-C1 vector in frame with enhanced GFP (EGFP) (pEGFP-US3.5). The cytomegalovirus (CMV) IE promoter was replaced with DNA containing the US3 promoter using a PCR product generated with primers US3PF (5′-GCGCCCTAGGGCTAGCTCGCCGCACCGTGAGTGCCA-3′) and US3PR (5′-GCCATTAATATTAATGCCGCGAACGGCGATCAGAGGGTCAGT-3′). This was digested with AseI and NheI and was then cloned into the pEGFP-US3.5 construct digested with AseI and NheI. Viruses were derived by cotransfecting SspI-linearized constructs with RE DNA and were identified and plaque purified based on gain of EGFP fluorescence, as described previously (59). Correct insertion of GFP at US3 was confirmed by Southern blot analysis of viral DNA.
(iii) PRV.
The PRV US3-null virus (PRV645) and its parent are based on the Kaplan strain and have been described previously (80).
(iv) Baculoviruses.
Baculoviruses expressing glutathione S-transferase (GST)-tagged ORF66 (GST.66) or GST.66kd were derived using the BaculoGold system (BD Pharmingen, San Diego, CA) as described previously (20) and were propagated in SF9 cells.
(v) Adenoviruses.
Replication-incompetent adenoviruses expressing GFP-HA-66 (Ad.GFP-66) or GFP-HA-66kd (Ad.GFP-66kd) under the control of doxycycline-regulated promoters have been described previously (21). Expression was achieved by coinfecting with Adeno.Tet-off (Ad.Toff) expressing a tetracycline-controlled transactivator, as previously described (21).
Preparation for matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) MS.
Confluent Hek 293 cells were infected with Ad.GFP-66 and were harvested when 50% of cells showed signs of cytopathic effect. Phosphate-buffered saline (PBS)-washed cells were lysed with brief sonication on ice in a solubilization buffer of 20 mM Tris-HCl (pH 7.0), 0.5 M KCl, 1 mM EDTA, and 1% NP-40 plus protease inhibitor cocktail (Complete; Roche Diagnostics) and phosphatase inhibitors (20 mM NaF, 1 mM Na4P2O7·10 H2O, 1 mM Na3VO4). Soluble fractions were immunoprecipitated with protein G agarose beads (Sigma, St. Louis, MO) prebound to a rabbit anti (α)-phospho-PKA substrate antibody for 4 h at 4°C, washed with a modified radioimmunoprecipitation assay (RIPA) buffer (20 mM Tris-HCl [pH 7.4], 1% NP-40, 150 mM NaCl, 1 mM EDTA), and finally washed in a buffer of 20 mM Tris-HCl (pH 7.4)-100 mM KCl. Proteins were separated on 6% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels and were stained with Bio-Safe Coomassie G-250 stain (Bio-Rad, Hercules, CA). The region between the 100- and 150-kDa markers was excised and sent for liquid chromatography-tandem mass spectrometry (LC-MS-MS) identification at the University of Pittsburgh Proteomics Core Facility.
Additional plasmids.
Plasmids expressing maltose binding protein (MBP) fused to IE62 residues 571 to 733 have been described previously (20). MBP fused to matrin 3 was generated from a commercial plasmid of the matrin 3 gene (Invitrogen, Carlsbad, CA), which was PCR amplified using oligonucleotides Matrin3Start (5′-GAG CAA TTG TCG ACC ACC ATGTCCAAGTCATTCCAGC-3′) and Matrin3end (5′-GAA GGA TCC TTA ACT AGT TTC CTTCTTCTGTCTGCGTTCTTCTGCCAAT-3′) with the proofreading polymerase Expand (Roche). The PCR fragment was digested with MfeI and BamHI and was cloned into the pMalC2 vector in frame with MBP. Protein was expressed and purified from Escherichia coli BL21 as previously described (20). To express hemagglutinin (HA)-tagged versions of matrin 3 and its mutants under the control of the CMV IE promoter, matrin 3 mutant genes were prepared using splicing by overlap extension (SOE) PCR with Expand proofreading polymerase. The left and right sides were PCR amplified separately with paired oligonucleotides containing the desired mutation, and the complete ORF was subsequently generated using the terminal primers in a second PCR with the left and right PCR substrates. Final PCR products were cut with MfeI and BamHI and were cloned into the EcoRI and BamHI sites in the PGK2-HA vector, described previously (21). The internal primers used to mutate T150 to A and insert a novel silent AscI site for identification were T150Fasc (5′-CTTAAAAGGAGGCGCGCGGAAGAAGGCCCTACC-3′) and T150Rasc (5′-GGTAGGGCCTTCTTCCGCGCGCCTCCTTTTAAG-3′). Those used to mutate S188 to A, S188Fnsi (5′-CACTTTAGAAGAGATGCATTTGATGATCGTGGT-3′) and S188Rnsi (5′-ACCACGATCATCAAATGCATCTCTTCTAAAGTG-3′), also inserted a novel silent NsiI site. Simultaneous mutation of S592, S596, and S598 to A and insertion of a silent BsiWI marker site were effected using nucS123Fbsiw (5′-CTG AAA AAAGATAAAGCCCGAAAAAGAGCG TACGCTCCAGATGGCAAAGAA-3′) and nucS1Rbsiw (5′-TTCTTTGCCATCTGGAGCGTACGCTCTTTTTCGGGCTTTATCTTTTTTCAG-3′). The primers used to mutate S596 and S598 to A and to insert a novel BsiWI site were nucS23Fbsiw (5′-GAT AAATCCCGAAAAAGAGCGTACGCTCCAGATGGCAAAGAA-3′) and nucS23Rbsiw (5′-TTCTTTGCCATCTGGAGCGTACGCTCTTTTTCGGGATTTATC-3′). The primers used to mutate S596 to A and insert a BsiWI site were nucS2FbsiW (5′-GATAAATCCCGAAAAAGACGCTACTCTCCAGATGGCAAAGAA-3′) and nucS2RbsiW (5′-TTCTTTGCCATCTGGAGAGTACGCTCTTTTTCGGGATTTATC-3′).
Drug treatments.
Forskolin (Fisher BioReagents, Fairlawn, NJ) and the PKA inhibitor (PKI) 14-22 amide (EMD Biosciences, San Diego, CA) were reconstituted in dimethyl sulfoxide (DMSO) and minimum essential medium (MEM), respectively. Treatment with PKI 14-22 amide was started 2 h prior to infection and continued until the time of cell harvest. Forskolin treatment was started at the time of infection and continued until the time of cell harvest, as indicated in the figure legends and the text.
Transfections/infection assays.
Transfections of Hek 293T cells seeded onto polylysine-coated 6-well culture dishes used Fugene HD (Roche, Indianapolis, IN) as recommended by the manufacturer. For transfection-infection experiments, MeWo cells set up the day prior were transfected using Fugene HD, incubated for 24 h, and either infected with HSV-1 or PRV at a multiplicity of infection (MOI) of 5 or overlaid with VZV-infected cells at an MOI of 0.3. For adenovirus infections, MRC-5 cells were first incubated under low serum conditions (minimum essential medium with 1% fetal bovine serum [FBS] plus antibiotics) for 1 day prior to infection and were then infected under low serum conditions at an MOI of 5 for Ad.GFP66, Ad.GFP66kd, or Ad.Vector in conjunction with Ad.Toff at an MOI of 2.5. For HSV and PRV infections for immunofluorescent (IF) analyses, virus was bound at 4°C for 1 h, followed by incubation in medium at 37°C until fixation. For VZV IF analyses, cells were infected by overlay of previously titrated stocks of virus in MeWo cells stored in liquid nitrogen and were incubated at 37°C for 36 h prior to fixation.
Antibodies.
Rabbit anti-VZV IE62 protein and anti-ORF4 protein (ORF4p) have been described previously (38, 40). Antibodies to VZV ORF9p (15) were a kind gift of W. T. Ruyechan, SUNY at Buffalo, Buffalo, NY. A monoclonal antibody to the influenza protein HA epitope tag has been described previously (20) and was used at 1:600 for immunoblotting and at 4 μl per immunoprecipitation (IP). Mouse α-tubulin (Sigma-Aldrich, St. Louis, MO) and α-Ku-86 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), were used at 1:8,000 and 1:500, respectively, for immunoblotting. A pool of monoclonal antibodies to HSV-1 gB, a gift from William F. Goins (University of Pittsburgh), was used at 1:6,000 for immunoblotting. Mouse α-ICP4 (Virusys, Taneytown, MD) was used at 1:1,000 for IF. Goat α-PRV UL34 was used at 1:1,000 for immunoblotting and at 1:500 for IF. The rabbit α-phospho-PKA substrate antibody (Cell Signaling Technology, Danvers, MA) was used at 1:800 for immunoblot analysis and 1:50 for IP. The rabbit α-matrin 3 antibody (Bethyl Laboratories, Inc., Montgomery, TX) was used at 1:3,000 for IF, 1:60 for IP, and 1:8,000 for immunoblotting. Goat α-GST (GE Healthcare, Piscataway, NJ) was used at 1:20,000 for immunoblotting. Rabbit α-MBP (New England Biolabs, Inc., Beverly, MA) was used at 1:15,000 for immunoblotting. Species-specific secondary antibodies used in immunoblot analysis were conjugated to horseradish peroxidase (ICN-Cappel, Aurora, OH) and used between 1:20,000 and 1:40,000.
Immunofluorescence.
Immunofluorescent staining procedures were similar to those described previously (21), using cells fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100. For VZV and HSV IF staining, antibodies were detected using a goat α-rabbit immunoglobulin G antibody conjugated to Alexa Fluor 546 or a goat α-mouse immunoglobulin G antibody conjugated to Alexa Fluor 647. For PRV IF staining, a donkey α-rabbit immunoglobulin G antibody conjugated to Alexa Fluor 555 or a donkey α-goat antibody conjugated to Alexa Fluor 647 (Invitrogen Corp, Carlsbad, CA) was used. Immunofluorescence was visualized using an Olympus Fluoview FV1000 confocal microscope equipped with a 60× oil objective. Images are of one slice of Z-stacks acquired in 0.48-μm steps.
Immunoblot analysis and immunoprecipitation.
Protein detection by antibody binding following SDS-PAGE and transfer to polyvinylidene difluoride (PVDF) membranes (Immobilon-P membrane; Millipore, Billerica, MA) has been described previously (20). Where indicated, Immobilon-P membranes were stripped by using 0.2 M NaOH for 5 min and rinsed with distilled H2O prior to a second antibody probing. Digital images of films were acquired using an Epson Perfection 4990 photo scanner with Silver Fast Ai and Adobe Photoshop CS imaging software. Some immunoblot images (see Fig. 1C) were subsequently acquired by using secondary antibodies tagged to infrared-sensitive dyes, followed by image acquisition using an Odyssey infrared imager (LI-COR Inc., Lincoln, NE).
FIG. 1.

Anti-PKAps profiles for cells infected with alphaherpesviruses (VZV, HSV-1, or PRV) with or without expression of their US3 kinases. MeWo cells were either mock infected, infected at an MOI of 0.1 with VZV.GFP-66 or VZV.GFP-66kd (expressing kinase-inactivated ORF66 protein), or infected at an MOI of 1 with HSV-1 (US3), HSV-EGFP-US3.5 (US3.5), HSV-EGFP-ΔUS3 (ΔUS3), PRV (US3), or PRV.ΔUS3 (ΔUS3). Cell extracts were harvested 20 h postinfection and were separated on a 7% SDS-PAGE gel. (A) Immunoblot analysis of proteins is shown following antibody probing with a rabbit α-p-PKA substrate antibody (top) or a mouse α-tubulin (VZV and PRV) or α-Ku-86 (HSV-1) antibody (bottom) as a loading control (LC). (B) Immunoblot analysis of viral proteins to show equal infections (rabbit α-VZV ORF9, mouse α-HSV gB, or goat α-PRV UL34 antibody). (C) Comparisons of the anti-PKAps profiles of MeWo cells or Vero cells infected with HSV-1 or PRV, either with (+) or deleted for (−) the US3 kinase. Profiles of cells infected at two different multiplicities of infection (1 and 5) for each virus, harvested at 18 h postinfection, are shown. The same blot was simultaneously probed with antibodies to tubulin for a loading control. Antibodies in panels A and B were detected by use of secondary antibodies bound to horseradish peroxidase, followed by a fluorescent substrate and collection by film exposure. Antibodies in panel C were simultaneously detected using secondary antibodies linked to IRDye 800CW (anti-mouse) or IRDye 680 (anti-rabbit) and were captured by a LI-COR Odyssey infrared imager. Protein size markers (in kilodaltons) are specified to the left of each panel.
Immunoprecipitations used an equivalent number of cells per condition. Cells were washed with ice-cold 1× PBS and were lysed with brief sonication in a solubilization buffer (20 mM Tris-HCl [pH 7.0], 0.5 M KCl, 1 mM EDTA, and 1% NP-40 plus protease and phosphatase inhibitors) for 30 min on ice. Soluble fractions were reacted with antibody-prebound protein G beads for 3 to 4 h at 4°C; then they were extensively washed in solubilization buffer, given a final wash in 1× PBS, and disrupted by boiling in SDS sample buffer. Where used, 5 μg blocking peptide (Bethyl Laboratories, Inc., Montgomery, TX) against the matrin 3-specific antibody was added to antibody-bound beads for 45 min prior to the addition of soluble fractions.
In vitro kinase assays.
In vitro kinase assays using purified GST, or GST-tagged ORF66 or ORF66kd obtained from baculovirus-infected cells, have been described previously (20, 79). These assays (20) used approximately 2 μg of GST or GST fusion protein and 2 μg of purified MBP or MBP fusion proteins in 70 μl ORF66 kinase assay buffer (20 mM HEPES-KOH [pH 7.5], 50 mM KCl, 10 mM MgCl2, and 5 μg/ml heparin) and 5μCi of [γ-32P]ATP (6,000 Ci/mmol) for 25 min at 35°C. In vitro PKA assays used 2 μg of the PKA catalytic subunit (New England Biolabs, Inc., Beverly, MA) with 2 μg MBP fusion protein, either in the recommended PKA reaction buffer or in ORF66 kinase assay buffer, for 30 min at 30°C. Reactions were stopped by heating in SDS-PAGE sample buffer, and incorporation of 32P into proteins was assessed by SDS-PAGE, transfer to Immobilon-P membranes, and autoradiography. Membranes were also probed with rabbit α-MBP or goat α-GST antibodies to assess protein levels. In vitro PKA assays also used washed protein G beads with immunoprecipitates of HA-tagged matrin 3 proteins under the same conditions.
RESULTS
PKA phosphosubstrate profiles differ for VZV, HSV-1, and PRV.
Using an antibody that recognizes the phosphospecific PKA substrates (referred to here as anti-PKAps), it was shown that HSV-1 US3 kinase substrates partly overlap those of PKA (5). The embedded motifs of the two ORF66 phosphorylation sites in IE62 (S686 and S722) were similar to the optimal consensus motifs for HSV US3 and PRV US3 kinases determined from peptide substrates. As such, the same antibody was predicted to identify a subset of phosphorylated ORF66 kinase-dependent substrates. One previous study reported a small number of unidentified species induced by VZV (19). In our hands, the anti-PKAps antibody identified approximately 9 to 11 protein species in VZV-infected MeWo cells that were not seen in cells infected with VZV kinase-dead (kd) ORF66 (VZV.GFP-66kd) or in uninfected cells. Intriguingly, the profile was unlike that reported by Benetti and Roizman for HSV-1 (5) and showed a predominant 125-kDa species (Fig. 1A). This species was also detected in VZV-infected MRC-5 cells, human foreskin fibroblasts, primary human corneal fibroblasts, and fibroblasts infected with VZV not expressing the ORF47 protein kinase (data not shown). The dissimilarity was confirmed by comparing MeWo cells infected with VZV, HSV-1, or PRV, in which different profiles for each virus were seen that were nevertheless US3 kinase dependent. Species in the 125-kDa region were less obvious in PRV infections in MeWo cells (Fig. 1A). Virus protein-specific antibodies confirmed similar levels of infection for each virus and mutant (Fig. 1B). The striking differences seen in MeWo cell anti-PKAps profiles between PRV and HSV-1 stimulated a further comparison of the anti-PKAps profiles in Vero cells, which are routinely used for HSV and PRV propagation (VZV was not compared, since Vero cells are only semipermissive for VZV growth). In Vero cells, HSV-1 infections with functional US3 kinase stimulated PKA profiles generally similar to those seen in MeWo cells infected with HSV-1. Vero cells infected with PRV yielded a generally more different pattern, in which many individual species appeared cell type specific (Fig. 1C). Like those seen in MeWo cells, the anti-PKAps profiles of PRV and HSV-1 in Vero cells were obviously very different: most of the predominant species seen in HSV-1 infections were not prominently detected in PRV infections, and vice versa. These differences were seen at two different multiplicities of infection. Despite the profile differences, most of the predominant species detected for these two rapidly growing alphaherpesviruses were largely dependent on the functionality of their US3 kinase, since infections with viruses lacking US3 resulted in a loss or reduction of detection of most protein species identified by the anti-PKAps antibody. These data strongly indicate that the three alphaherpesviruses drive the phosphorylation of globally different subsets of proteins through their respective viral short region protein kinases.
The VZV 125-kDa species represents matrin 3, a host cell target.
To determine if the ORF66-induced 125-kDa phosphoprotein species seen in VZV-infected cells was a cellular protein, Hek 293 cells were infected with replication-deficient adenoviruses expressing GFP-tagged ORF66 (Ad.66) or kinase-dead protein (Ad.66kd), along with the Ad.Toff expression regulator virus, in the absence or presence of doxycycline to turn off the expression of ORF66. Anti-PKAps antibodies recognized the 125-kDa species only when functional ORF66 kinase was expressed. The 125-kDa species was absent in all controls, including when ORF66 gene expression was turned off with doxycycline and when kinase-inactive forms were expressed (Fig. 2). This establishes that the 125-kDa species is a host cell protein. An additional 140-kDa species that was not ORF66 dependent was observed in these studies.
FIG. 2.
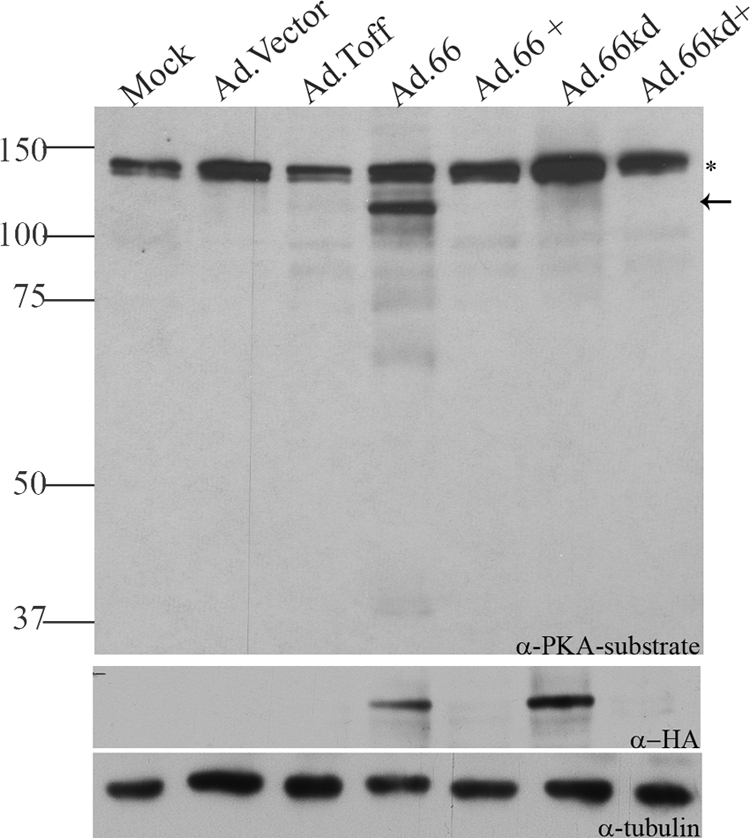
The 125-kDa ORF66-specific PKA substrate protein is a host cell protein. MRC-5 cells were coinfected with replication-deficient adenoviruses: Ad.Toff, which expresses a tetracycline-controlled transactivator, at an MOI of 2.5, and either mock infection or Ad.GFP-66 (expressing functional ORF66) (Ad.66), Ad.GFP-66kd (expressing kinase-inactive ORF66) (Ad.66kd), or Ad.Vector, at an MOI of 5. Doxycycline was added (+) to turn off ORF66/66kd gene expression. Cells were harvested 1 day postinfection, and immunoblot analysis was performed on cell extracts with a rabbit α-p-PKA substrate antibody. The blot was stripped and reprobed with mouse α-HA to analyze for ORF66/66kd expression. Mouse α-tubulin was used to evaluate equal amounts of total protein levels. The star marks a nonspecific protein species that was present under all conditions. The arrow indicates the ORF66-dependent 125-kDA p-PKA substrate band. Protein size markers (in kilodaltons) are specified to the left.
The 125-kDa protein(s) species was identified following scale-up of the adenovirus-ORF66-expressing Hek 293 cells and subsequent immunoprecipitation with the anti-PKAps antibody. The region between 100 and 150 kDa was excised from the gel and sent to the University of Pittsburgh Proteomics facility. Peptides released following trypsin digestion were separated by a capillary C18 high-performance liquid chromatograph (HPLC) (ThermoElectron Surveyor liquid chromatograph) coupled to a nanospray ionization source of an ion trap mass spectrometer (ThermoElectron LCQ Deca XP Plus). The LC-MS-MS spectra were run against a sequence database and analyzed by the BioWorks/SEQUEST program. A candidate protein identified from this analysis was matrin 3. Matrin 3 is an 847-amino-acid (aa) nuclear matrix protein that is not well understood but nevertheless has been implicated in replication, transcription, and RNA processing and quality control (85, 86). The identity of matrin 3 as the VZV ORF66-driven phosphorylated 125-kDa species was confirmed using matrin 3-specific antibodies to immunoprecipitate extracts of Hek 293 cells infected with Ad.66 or Ad.66kd. Immunoblotting of precipitates with anti-PKAps established phosphorylation of matrin 3 only in cells expressing functional ORF66 kinase (Fig. 3A). In a “reverse” experiment in which anti-PKAps immunoprecipitates were similarly blotted and probed for matrin 3, phosphorylated matrin 3 was seen only in the extracts of cells in which functional ORF66 kinase was expressed.
FIG. 3.
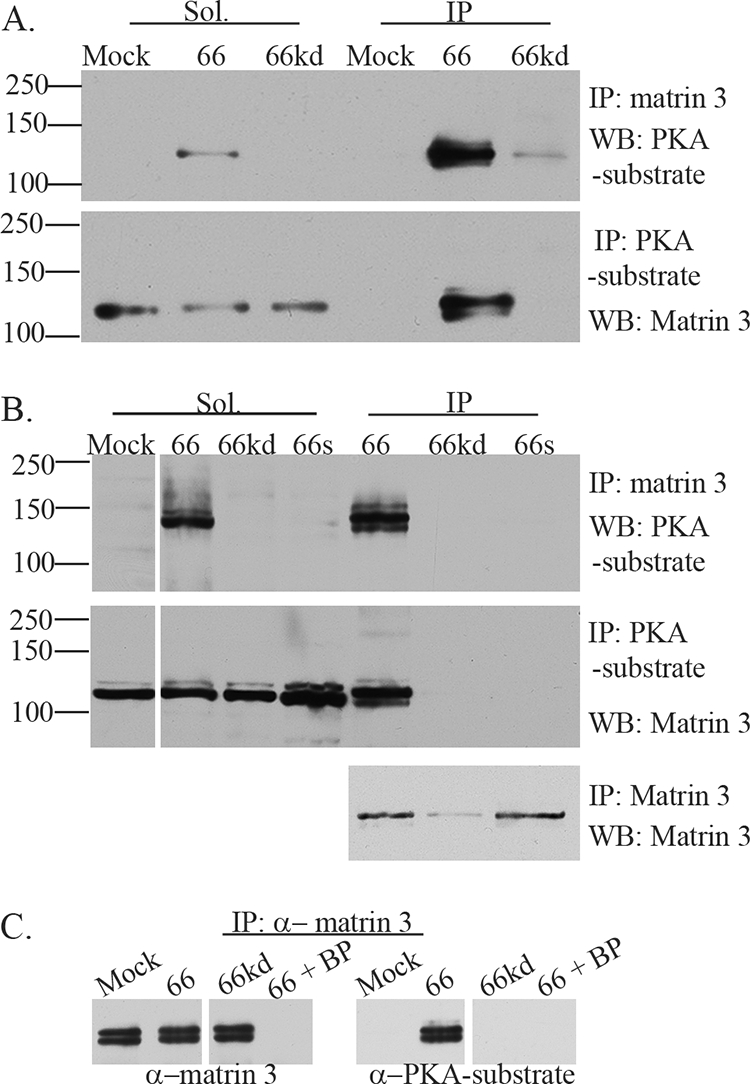
VZV ORF66 kinase activity induces specific matrin 3 phosphorylation. (A) Hek 293 cells were either infected with Ad.GFP-66 or Ad.GFP-66kd at an MOI of 0.3 or mock infected and were then incubated for 24 h. Soluble fractions (Sol) and immunoprecipitation (IP) products were separated on a 7% gel and analyzed by immunoblotting (Western blotting [WB]). IPs were performed using rabbit α-matrin 3, followed by probing of membranes with the rabbit α-PKA substrate antibody (top), or IPs were performed using the rabbit α-PKA substrate antibody, followed by probing of membranes with the rabbit α-matrin 3 antibody (bottom). (B) MRC-5 cells either were infected with VZV.GFP-66 (66), VZV.GFP-66kd (66kd), or VZV.GFP-66s (66s) at an MOI of 0.01 or were mock infected; then they were harvested 2 days postinfection. Soluble extracts (Sol) or immunoprecipitations were separated on a 7% SDS-PAGE gel and were immunoblotted (top and center) as described above. The top membrane was then stripped and reprobed for matrin 3 to analyze levels of matrin 3 in IP fractions (bottom). Protein size markers (in kilodaltons) are specified to the left. (C) MRC-5 cells either were infected with VZV expressing ORF66 or ORF66kd at an MOI of 0.01 or were mock infected; they were harvested at 2 days postinfection. Cleared lysates were then immunoprecipitated using a rabbit α-matrin 3 antibody in addition to matrin 3 blocking peptide (BP) where indicated. Proteins were separated on a 6% SDS-PAGE gel, and membranes were then probed either with a rabbit α-matrin 3 antibody (left), to test for the efficiency of IPs and the blocking peptide, or with a rabbit α-PKA substrate antibody, to analyze for phosphorylation of matrin 3 (right).
We then established that matrin 3 was phosphorylated in an ORF66-dependent fashion in the context of VZV infection. Matrin 3-specific or total anti-PKAps immunoprecipitates of MRC-5 cells infected with VZV or kinase-inactive mutants (VZV.GFP-66kd and VZV.GFP-66s) were recognized by anti-PKAps (Fig. 3B, top) or anti-matrin 3 antibodies (Fig. 3B, center) in immunoblots only if ORF66 kinase was expressed. Matrin 3 was present in the anti-matrin 3 immunoprecipitates for all VZV infections (Fig. 3B, bottom). In addition to a major 125-kDa species, additional matrin 3 forms were detected: these are suspected to reflect additional modified and/or phosphorylated forms of matrin 3, as seen in other studies (29). The specificities of the matrin 3 species and the antibodies used in its detection were established using a blocking peptide for the matrin 3 antibodies, which efficiently blocked anti-matrin 3 from binding matrin 3 in immunoprecipitates (Fig. 3C, left) and also prevented immunoblot detection of species at the 125-kDa size by the anti-PKAps antibody (Fig. 3C, right). These approaches established that at least one phosphorylation event of matrin 3 during VZV infection is ORF66 dependent. The approaches used here resulted in almost complete solubilization of matrin 3, so the findings are not a result of differential solubilization (data not shown). VZV infection did not radically alter global levels of matrin 3 as a result of infection, since similar amounts of matrin 3 were detected in all immunoprecipitates.
The phosphorylation of matrin 3 is conserved in HSV-1 and PRV.
The remarkably different profiles of substrates identified by the anti-PKAps antibody raised the question of whether matrin 3 was a VZV-specific target. However, similar analyses of immunoprecipitates of matrin 3 from HSV-1- and PRV-infected cells with the anti-PKAps antibody revealed matrin 3 phosphorylation only under conditions in which the respective US3 kinase of each virus was expressed. In cells infected with HSV-1 only expressing US3.5, phosphorylation of matrin 3 was induced as efficiently as with wild-type HSV-1. No detectable matrin 3 phosphorylation was seen in cells infected with HSV-1 lacking US3 kinase (Fig. 4A, ΔUS3). The anti-PKAps antibody detected a marginal phosphorylated species in the 125-kDa region in cell extracts that was not matrin 3, and this may reflect the phosphorylation of a similarly sized protein, such as gB (33, 81). For PRV, similar results were found, although PRV ΔUS3 reproducibly showed a small level of matrin 3 phosphorylation (Fig. 4B, top right). This suggests that PRV can induce matrin 3 phosphorylation to a low level in a US3 kinase-independent manner. This mirrors similar results seen with PRV-induced phosphorylation of HDAC 2 (80). Global matrin 3 levels did not change during HSV or PRV infections. We conclude from these data that matrin 3 is a conserved phosphorylation target for the VZV, HSV, and PRV US3 kinases.
FIG. 4.

Matrin 3 is a conserved phosphorylation target for the US3 kinases during infection. MRC-5 cells were either infected with HSV-1 (US3), HSV-EGFP-US3.5 (US3.5), or HSV-EGFP-ΔUS3 (ΔUS3) at an MOI of 1 (A) or with PRV (US3) or PRV.ΔUS3 (ΔUS3) at an MOI of 1 (B) or were mock infected; then they were harvested 1 day postinfection. Cleared lysates were immunoprecipitated using the rabbit α-matrin 3 antibody, and whole-cell extracts (Cell Ext.) or immunoprecipitation (IP) products were separated on a 7% (HSV) or 6% (PRV) SDS-PAGE gel and analyzed by immunoblotting. Membranes were probed with a rabbit α-PKA substrate antibody and were then stripped and reprobed using a rabbit α-matrin 3 antibody as indicated. Cell extracts were also analyzed for viral protein expression using a mouse α-HSV gB (A) or a goat α-PRV UL34 (B) antibody. Protein size markers (in kilodaltons) are specified to the right.
Identification of matrin 3 target residues for the ORF66/US3 kinases.
Screening of matrin 3 for the candidate serine and threonine residues that could be recognized by the anti-PKAps antibody (i.e., serine or threonine preceded by at least two basic residues at −2 and −3) revealed five candidates, including the known phosphorylated residues S188, S596, and S598 (3, 54). N-terminally HA-tagged matrin 3 genes encoding serine-to-alanine mutations at each position were coexpressed in cells with the ORF66 kinase; the proteins were then immunoprecipitated with anti-HA monoclonal antibodies; and the blots were probed with the anti-PKAps antibody. This revealed that each mutant matrin 3 was efficiently expressed at similar levels (Fig. 5A) and that all were detected by the anti-PKAps antibody in the presence of ORF66, except HA-Matr3-T150A. This mutant showed only trace levels of reactivity with the anti-PKAps antibody, even though it was efficiently expressed and immunoprecipitated (Fig. 5A). Matrin 3 with the T150A mutation was subsequently found to show strong nuclear localization similar to that of all other mutants and wild-type protein, indicating that differences in levels of phosphorylation were not due to cellular localization, misfolding, or aggregation (data not shown). These results strongly suggest that T150 is the major ORF66-dependent phosphorylation site on matrin 3. Interestingly, matrin 3 T150-proximal residues (LQLKRRRT150EEGPTL) fit well with the consensus site for PKA and the ORF66 site at IE62 S722, in that the residue following the target is acidic. The HSV-1 and PRV optimal US3 motif predicts that peptides with a +1 acidic residue are not optimal targets (42, 56).
FIG. 5.

Matrin 3 amino acid T150 is the major phosphorylation site for VZV ORF66 and for HSV-1 and PRV US3 kinases. (A) The top two panels show immunoblot analysis of Hek 293T cells transfected with plasmids expressing HA alone, HA-tagged full-length matrin 3 (HA-Matr3), or one of the following five HA-tagged matrin 3 serine/threonine-to-alanine mutants: the S188A, T150A, S592/596/598A, S596/598A, or S596A mutant. The lower two panels are similar analyses of cells cotransfected with one of the HA-tagged proteins and EGFP.66. The next day, cells were harvested, and cleared lysates were immunoprecipitated using a mouse α-HA antibody, followed by separation of IP products on a 6% SDS-PAGE gel and transfer to PVDF membranes. Membranes were probed with a rabbit α-PKA substrate antibody to analyze matrin 3 phosphorylation (top panel of each pair) or with a mouse α-HA antibody to analyze equivalent HA immunoprecipitation (bottom panel of each pair). Protein size markers (in kilodaltons) are specified to the left. (B) Immunoblot analysis of a MeWo cell transfection/infection in which cells were transfected with plasmids expressing either a vector (HA), HA-tagged matrin 3 containing a T150A mutation (T150A), or HA-tagged matrin 3 (HA-Matr3). At 24 h posttransfection, cells either were overlaid with VZV (top) at an MOI of 0.3 or were infected with HSV-1 or PRV at an MOI of 5. Immunoprecipitates from cleared lysates using a mouse α-HA antibody were separated on a 6% SDS-PAGE gel and analyzed by immunoblotting using a rabbit α-PKA substrate.
The T150 mutation was then examined for phosphorylation in the context of wild-type VZV, HSV-1, and PRV infections. MeWo cells expressing HA-Matr3-T150A or HA-Matr3 were infected with VZV, HSV-1, or PRV, and after further incubation for 24 h, the HA-tagged matrin 3 proteins were immunoprecipitated and assessed for specific ORF66-directed phosphorylation by immunoblotting with the anti-PKAps antibody. Matrin 3 T150A displayed much lower levels of phosphorylation than unaltered matrin 3 in VZV, HSV-1, and PRV infections (Fig. 5B), even though parallel blots showed equal precipitation of mutant and wild-type proteins (data not shown). These results are consistent with a conserved ORF66/US3-directed phosphorylation of matrin 3 at residue T150 by different alphaherpesviruses.
VZV ORF66-induced matrin 3 phosphorylation takes place through a non-PKA-dependent pathway.
The HSV-1 US3 kinase has been shown not only to target proteins that overlap those targeted by PKA but also to induce the activation of PKA itself (5). Even though the PKA phosphosubstrate profile for VZV was quite different from that of HSV-1 and PRV, the possibility existed that matrin 3 phosphorylation occurred through VZV activation of PKA. To address this, we examined MRC-5 cells infected with Ad.GFP-66 or Ad.GFP-66kd and determined the sensitivity of matrin 3 ORF66-induced phosphorylation to the PKA inhibitor (PKI) 14-22 amide. Immunoblotting of cell extracts probed with anti-PKAps revealed that infections in 60 μM PKI resulted in the diminishment of most species but efficient recognition of the 125-kDa species. A 50-kDa species not yet identified was also apparent (Fig. 6A). We also addressed the phosphorylation of matrin 3 following induction of PKA activity by using the established PKA activator forskolin, which activates adenylate cyclase and increases intracellular cAMP levels. Assessment of extracts of cells infected with VZV.GFP-66 or VZV.GFP-66kd in the presence of forskolin showed that the anti-PKAps antibody identified several novel protein species, particularly 37- and 60-kDa protein species, but did not result in enhanced phosphorylation of matrin 3 (Fig. 6B). Furthermore, ORF66-induced matrin 3 phosphorylation was unaffected by forskolin treatment (Fig. 6B, lower panels). Taken together with those of the inhibitor studies, these results indicate that PKA did not play a significant role in ORF66-directed matrin 3 phosphorylation. Finally, it has been shown that matrin 3 can be phosphorylated by PKA in neurons, with the consequence of inducing apoptosis (26). In vitro kinase assays were performed using commercial purified PKA in conjunction with immunoprecipitated HA-tagged matrin 3 mutant and wild-type proteins obtained from transfected cells. Wild-type matrin 3 and matrin 3 proteins with an S596A or T150A change showed extensive phosphorylation in vitro with approximately 2 μg of PKA catalytic subunit alpha (Fig. 6C). However, matrin 3 with an S188A mutation showed significantly lower phosphorylation than HA-Matr3, consistent with the conclusion that ORF66-dependent phosphorylation of matrin 3 is not mediated through PKA, which rather appears to preferentially target matrin 3 S188.
FIG. 6.

The 125-kDa matrin 3 is phosphorylated through a non-PKA pathway. (A) MRC-5 cells were pretreated with a PKA substrate inhibitor (PKI), 14-22 amide, 2 h prior to infection. Then cells either were infected with Ad.Toff at an MOI of 2.5 and with Ad.GFP-66 (66) or Ad.GFP-66kd (66kd) at an MOI of 5, with or without PKI, or were mock infected. Doses of PKI at increasing micromolar concentrations are given above the gel. Whole-cell extracts were immunoblotted. The membrane was first probed with a mouse α-HA antibody, stripped, and then reprobed with a rabbit α-PKA substrate antibody. The arrow indicates the 125-kDa PKA substrate (PKA-sub)/matrin 3 protein. The star indicates an undefined protein whose phosphorylation is PKI resistant. (B) MRC-5 cells either were infected with VZV.GFP-66 (66) or VZV.GFP-66kd (66kd) at an MOI of 0.1 or were mock infected. Where indicated, cells were treated with 10 μM forskolin or DMSO at the time of infection. Cells were harvested 1 day postinfection, analyzed by immunoblotting, and probed with a rabbit α-PKA substrate antibody. Viral protein expression was analyzed using a rabbit α-VZV ORF4 antibody. Cleared lysates were immunoprecipitated for matrin 3 and were subsequently analyzed by immunoblotting using rabbit α-PKA substrate; the blot was stripped and reprobed with a rabbit matrin 3 antibody. (C) In vitro kinase assay of immunoprecipitated HA alone, HA-tagged matrin 3 (HA-Matr3), or the HA-tagged S188A, T150A, or S596A matrin 3 mutant, incubated with PKA where specified (+ PKA). HA proteins were derived from transfected Hek 293T cells and were purified by immunoprecipitation using a mouse α-HA antibody. Following the kinase assay, proteins were separated on an SDS-PAGE gel, transferred to a PVDF membrane, and exposed to film. These membranes were then analyzed for HA fusion protein expression by probing with a mouse α-HA antibody. Protein size markers (in kilodaltons) are specified to the left.
Assessment of matrin 3 phosphorylation by ORF66 kinase in vitro.
We have previously established that ORF66 kinase directly phosphorylates IE62 (20). A similar assay was employed to determine if matrin 3 was phosphorylated under the conditions under which ORF66 directly targets IE62 in vitro. Functional ORF66 or the kinase-dead point-inactivated ORF66kd protein fused to GST was expressed from baculovirus vector-infected SF9 insect cells and was partially purified by GST affinity chromatography. Purified MBP-matrin 3 protein or MBP-IE62 expressed in E. coli BL21 was used as the substrate for in vitro kinase assays. In all assays, GST66 showed autophosphorylation, as indicated by [γ-32P]ATP labeling of a 75-kDa species (Fig. 7, lanes 2, 6, 10, and 14). In the same assay, the GST66kd protein showed greatly diminished phosphorylation, although phosphorylation was not fully absent (Fig. 7, lanes 3, 7, 11, and 15). The residual phosphorylation was suspected to be due to low levels of cellular kinases copurifying with and phosphorylating the ORF66 kinase. The MBP-IE62571-733 peptide was extensively phosphorylated by functional GST ORF66 kinase, as expected from previous studies (Fig. 7, lane 10). This region of IE62 contains the ORF66 target sites S686 and S722, mapped previously (20). The same IE62 peptide showed only a comparatively minor level of phosphorylation when combined with GST66kd protein (Fig. 7, lane 11). This authenticates the in vitro kinase assay used (20). MBP-matrin 3 fusion proteins showed two species of 170 kDa and 125 kDa, with the larger form predicted to be full-length MBP-matrin 3 fusion protein. However, neither species showed any appreciable phosphorylation by purified ORF66 kinase under conditions under which IE62 was targeted (Fig. 7, lanes 12 to 15). The two MBP-matrin 3 forms were, however, efficiently phosphorylated by the purified PKA catalytic subunit, in both the specific optimal PKA reaction buffer (Fig. 7, lane 17) and the optimal ORF66 kinase buffer (Fig. 7, lane 18). These data strongly suggest that the matrin 3 substrate obtained from bacterial extracts did not retain sufficient integrity to be a target for the VZV kinase in vitro or that additional cellular components are required for matrin 3 phosphorylation by ORF66 kinase.
FIG. 7.

ORF66 does not phosphorylate matrin 3 under the conditions required for phosphorylation of IE62. Purified MBP fusion proteins and GST kinase were incubated in optimal kinase buffer, separated on a 7% SDS-PAGE gel, transferred to PVDF membranes, and exposed to film for autoradiography. Lanes 1 to 4 contain GST, GST.66, GST66kd, or MBP incubated alone, respectively. Lanes 5 to 7 contain MBP incubated with GST, GST.66, or GST.66kd, respectively. Lane 8 contains MBP62p alone, and lanes 9 to 11 contain MBP62p incubated with GST, GST.66, or GST.66kd, respectively. Lane 12 contains MBP-matrin 3 (MBPmatr3) alone, and lanes 13 to 15 contain MBPmatr3 incubated with GST, GST.66, or GST.66kd, respectively. Images of lanes 12 to 15 are 4 times more exposed in order to show MBPmatr3 proteins. Lane 16 also contains MBPmatr3 alone, and lanes 17 and 18 contain equivalent levels of PKA incubated with MBPmatr3 in PKA reaction buffer (lane 17) or optimal ORF66 kinase buffer (lane 18). Protein levels of substrate and CRF66 protein were analyzed by immunoblotting using rabbit α-MBP and goat α-GST antibodies (lanes 1 to 15). Protein size markers (in kilodaltons) are specified to the left of autoradiographs.
Matrin 3 localization in alphaherpesvirus-infected cells.
The ORF66 kinase-directed phosphorylation of IE62 induces radical changes in subcellular localization, leading to the exclusion of IE62 from the nucleus. The HSV-1 US3 kinase has also been shown to induce the cellular relocalization of nuclear lamins (49). Given these previous observations, we hypothesized that the ORF66-directed phosphorylation of matrin 3 could affect its cellular distribution. In mock-infected MRC-5 cells, matrin 3 shows a dominant speckled nuclear localization with exclusion from nucleoli (Fig. 8Ai and iv and Di), in agreement with previous reports (44, 53). Matrin 3 showed a predominantly nuclear but more diffuse distribution in VZV-infected cells, although it remained excluded from the nucleolus (Fig. 8Dii). Such distribution patterns were apparent in VZV-infected syncytia (Fig. 8Bi and iv). In many VZV-infected cells expressing the inactive ORF66kd protein, matrin 3 remained nuclear but displayed subtle changes in multiple areas of the nucleus that showed reduced levels of matrin 3 or none (Fig. 8Ci and iv and Diii). Similar observations were made with VZV expressing the ORF66 stop mutant (VZV.GFP-66s). These studies suggest that the expression of the kinase has effects on matrin 3 intranuclear localization in VZV infection.
FIG. 8.

The ORF66 kinase influences matrin 3 nuclear distribution in VZV infections. Shown are immunofluorescence analyses of mock-infected MRC-5 cells (A) and of MRC-5 cells infected with VZV.GFP-66 (B) or VZV.GFP-66kd (C) at an MOI of 0.003. Cells were fixed with 4% paraformaldehyde 3 days postinfection and were immunostained either with a rabbit α-matrin 3 antibody (A to C, panels i, and D), detected with α-rabbit Alexa Fluor 546, or with mouse α-IE62 (A to C, panels iii), detected with α-mouse Alexa Fluor 647. ORF66 expression was determined by GFP autofluorescence (A to C, panels ii). The merge images (A to C, panels iv) are overlays of matrin 3 (gray), GFP (green), IE62 (red), and nuclei stained with Hoechst dye (blue). (D) Representative single-cell images depicting matrin 3 localization. Infection conditions are given on the images. Fluorescence images were taken using a 60× objective.
The kinetics of the effects of ORF66 on matrin 3 distribution in VZV-infected cells was difficult to assess further, since single-step VZV infections cannot be easily established. Extension of these studies to HSV-1 and PRV infection of MRC-5 cells revealed that at early stages of HSV-1 infection, matrin 3 showed no apparent changes in cellular/nuclear distribution from that in mock-infected cells (Fig. 9A), but at late stages, matrin 3 showed different phenotypes, depending on the expression of the US3 kinase. Cells expressing HSV US3 (Fig. 9B) or US3.5 (Fig. 9C) at 18 h postinfection displayed matrin 3 predominantly in a diffuse distribution in the nucleus (Fig. 9B and C, panels i and iv), with some instances of nuclear condensation in US3.5-expressing cells. However, cells infected with HSV lacking US3 consistently showed obvious cytoplasmic accumulations of matrin 3 in addition to nuclear forms (Fig. 9Di and iv). HSV-1 ICP4 exhibited nuclear localization in infected cells (Fig. 9B to D, panels iii) and late accumulation of minor cytoplasmic forms that were not US3 dependent. In late-stage PRV-infected cells, matrin 3 showed strong cytoplasmic distribution in more than 50% of infected cells if the US3 kinase was not expressed, and some demonstrated an apparent nuclear exclusion of matrin 3 (Fig. 9Fi and iv), as opposed to the more predominant nuclear localization seen in cells infected with PRV expressing US3 (Fig. 9E). These results indicate that efficient nuclear retention of matrin 3 requires HSV and PRV US3 kinase activity in the context of viral infections.
FIG. 9.

HSV-1 and PRV US3 kinases drive nuclear retention of matrin 3 during infection. Shown are immunofluorescence analyses of MRC-5 cells that were either mock infected (A) or infected with HSV-1 (B), HSV.US3.5 (C), HSV.ΔUS3 (D), PRV (E), or PRV.ΔUS3 (F) at an MOI of 5. Cells were fixed with 4% paraformaldehyde 18 h (HSV) or 11 h (PRV) postinfection; then they were immunostained either with a rabbit α-matrin 3 antibody (A to F, panels i), detected with α-rabbit Alexa Fluor 546 (HSV) or α-rabbit Alexa Fluor 555 (PRV); with mouse α-HSV ICP4 (A to D, panels iii) (magenta), detected with α-mouse Alexa Fluor 647; or with goat α-PRV UL34 (E and F, panels iii) (magenta), detected with α-goat Alexa Fluor 647. GFP-US3.5 expression or US3 promoter activity in HSV.ΔUS3- and PRV.ΔUS3-infected cells was determined using GFP autofluorescence (A to F, panels ii) (green). The merge images (A to F, panels iv) are overlays of matrin 3 (gray) and nuclei stained with Hoechst dye (blue). Fluorescence images were taken using a 60× objective.
DISCUSSION
While the US3/ORF66 protein kinases influence several host cell processes, including actin dynamics, apoptosis, nuclear membrane architecture, and intracellular signaling, the targets through which they act remain poorly defined. Several studies have shown the kinase activities to be critical for effective viral growth in specific cell types relevant to virus-host pathogenesis (22, 46, 64, 65). Here we report that the nuclear matrix protein matrin 3 is a new target of the kinases. We show that the protein profiles recognized by an antibody to phosphosubstrates of PKA are quite different for three different alphaherpesviruses but that most are dependent on their US3/ORF66 kinase. However, all three alphaherpesviruses drive the phosphorylation of matrin 3 at a specific residue, T150, in a US3/ORF66-dependent manner. The activity to matrin 3 in VZV infections is not reliant on the activity of PKA. Finally, our data indicate that the phosphorylation of matrin 3 affects its cellular distribution, particularly for HSV and PRV. This work adds matrin 3 to the limited repertoire of known targets for these kinases.
The difference in the number and size distribution of US3/ORF66 kinase-dependent phosphorylated proteins recognized by the anti-PKAps antibody between the herpesviruses was somewhat unexpected. Even though the species seen reflect only a subset of phosphorylated targets in the cell and include both direct and indirect targets, we predicted that the species detected would be common for all three herpesvirus kinases, because the three herpesvirus kinases target very similar motifs. Furthermore, several phenotypic effects on host cells are common to at least two of the three herpesviruses. For example, HSV US3 (5, 6, 12, 13), PRV US3 (25), and VZV ORF66 (64, 65) contribute to a block of apoptosis of infected cells. Benetti and Roizman argued that HSV-1 US3-induced PKA activity activated antiapoptotic factors and that HSV-1 US3 increased expression of the PKA catalytic subunit, leading to blocks in apoptosis (5). Activation of PKA substrates during viral infection has been reported for hepatitis C virus (23), and activated PKA pathways have been exploited by many viruses, including adenoviruses (75), human immunodeficiency virus (HIV) (43), human cytomegalovirus (HCMV) (36), and HSV (5, 82). One previous study examining VZV found slightly upregulated PKA activity and a few phosphorylated species induced by VZV infection (19). The widely different profiles suggest that each herpesviral US3 kinase has distinct downstream consequences on the phosphorylation state in infected cells and may activate additional pathways. Our work suggested that PKA activity was not greatly activated in VZV-infected cells and ORF66-expressing cells, since few of the multiple species seen in HSV-infected cells were apparent in VZV-infected cells. For VZV, most of the principal proteins recognized by the anti-PKAps antibody remained phosphorylated in the presence of established PKA inhibitors, and PKA activation by forskolin did not lead to enhanced phosphorylation of most of the species detected with the anti-PKAps antibody. Future work will address what additional signaling pathways are activated by each virus through its US3/ORF66 kinase.
We show here that phosphorylation of matrin 3 is common to all three herpesviruses and that for VZV it occurs through an indirect pathway other than PKA activation. Interaction with matrix proteins has been described for many viruses, including HIV, simian virus 40 (SV40), PRV, HCMV, human papillomavirus (7, 27, 51, 63, 66, 74), and HSV-1 (9, 10, 14, 49, 83, 84). At least two forms of matrin 3, which are suspected to reflect additional phosphorylation events, were detected in our studies. Differentially phosphorylated matrin 3 isoforms have been reported in rat cells, with 130-kDa and 123-kDa matrin 3 species localizing to the nucleus and extranuclear compartments, respectively (29). Matrin 3 forms with slightly different isoelectric points have also been isolated in rat neurons (26). VZV ORF66-induced phosphorylation occurs predominantly at the T150 major phosphorylation site (Fig. 5), which is not a known PKA target site and is consistent with the continual phosphorylation of T150 in the presence of PKA inhibitors. The PKA inhibitor 14-22 amide did not block the phosphorylation of matrin 3, and activation of PKA using forskolin also did not increase matrin 3 phosphorylation (Fig. 6). Our studies strongly suggest that most PKA-directed phosphorylation in vitro is rather directed at S188 of matrin 3. Thus, we strongly suspect that the phosphorylation of matrin 3 detected by the phosphospecific antibody at T150 occurs through another ORF66-activated pathway, at least in VZV-infected cells. Unless HSV and PRV infections alter the substrate specificity of PKA, we predict that matrin 3 targeting at T150 is also PKA independent for HSV and PRV. While we were unable to show direct phosphorylation of matrin 3 by ORF66, it is possible that the assay cannot be carried out using by a matrin 3 substrate made in bacteria, due to the conformation that is required for its recognition by the viral kinase. However, VZV kinase and purified PKA recognized bacterially made IE62 peptide and matrin 3, respectively, and we suspect it more likely that matrin 3 phosphorylation by ORF66 requires nuclear proteins; this will be the subject of further investigation. Interestingly, the ORF66-directed phosphorylation of matrin 3 appears similar to that of HDAC 1 and 2, which were recently shown to be phosphorylated by ORF66/US3 expression in a manner that could not be demonstrated using in vitro kinase assays (79, 80).
Despite the different profiles for the alphaherpesviruses, the phosphorylation of the 125-kDa matrin 3 protein appears to be conserved. It is not yet clear how US3/ORF66-driven phosphorylation of matrin 3 plays a role in viral infection. The functions of matrin 3 are only just beginning to come to light; they are indicating that it is critical to many host cell nuclear processes, some of which would be predicted to be important in viral replication. The N terminus contains a large number of free hydroxyl groups (26 of the first 100 residues), as is characteristic of lamin proteins, while the C terminus is acidic, as is found in many nuclear proteins, and contains two tandem RNA recognition motifs in addition to a nuclear localization signal (4). There are reports proposing intriguing roles for matrin 3 as a critical component of functional nuclear microenvironments of chromatin replication and transcription (44). Moreover, matrin 3 has been found to interact with hnRNP-L, an RNA splicing regulator, and scaffold attachment factor B (SAFB), involved in RNA processing and chromatin organization (85). Matrin 3 can bind DNA at scaffold/matrix attachment region (S/MAR) sites that influence chromatin structure by associating with matrix-binding proteins that anchor chromatin into loop domains (28). Additionally, matrin 3 specifically locates in close proximity to nascent DNA replication and transcription zones within the nucleus, and as a major nuclear matrix protein, it is likely closely involved as a platform for genomic functions such as sites of viral replication and transcription. Recently, a comparable role has been proposed for lamin A/C: serving as a scaffold for HSV genomes and nuclear factors that reduce heterochromatin assembly on viral promoters (70). Matrin 3 may also have a role in capsid assembly, since capsid proteins localize to nuclear matrix fractions (9, 14). This would correlate with lines of evidence suggesting that ORF66 is involved in capsid assembly: in VZV ORF66-deficient infected T cells from SCID-hu T cell xenografts, there is greatly reduced intranuclear assembly of nucleocapsids (65). Matrin 3 is also involved as part of a complex that retains hyperedited nuclear double-stranded RNAs (dsRNAs) in the nucleus and prevents their export to the cytoplasm for translation. dsRNAs have been identified for HSV and are likely made by all herpesviruses (32). The ORF66/US3 kinases might be predicted to be involved in regulating the interaction of matrin 3 with the p54nrb-PSF complex and to modulate the balance between the nuclear accumulation of viral dsRNA and its release to the cytoplasm. Lastly, matrin 3 degradation in neurons, leading to neuron death, has been reported following N-methyl-d-aspartic acid (NMDA)-induced phosphorylation of matrin 3, detected using an anti-PKA substrate antibody. ORF66 is a known latency-associated protein (16, 17), and thus, expression during latency and reactivation may lead to changes in matrin 3 turnover during latency.
The effects of phosphorylation on matrin 3 distribution seen in PRV and HSV-1 echo the findings of similar studies that US3/ORF66 kinase affects the distribution of other nuclear proteins. ORF66 kinase in VZV directly regulates the nuclear localization of VZV IE62 to enable the inclusion of IE62 in the virion tegument. HSV US3 kinase causes the redistribution of nuclear lamins A/C so as to breach the laminar network so that nucleocapsid envelopment can proceed (49). Our data show that the lack of phosphorylation of matrin 3 was associated with nuclear regions devoid of matrin 3 that were filled with IE62 protein in VZV ORF66kd-infected cells (Fig. 8). This is reminiscent of the findings for lamin A/C, which normally form a reticular pattern in infected cells, where an absence of US3 kinase activity leads to larger intranuclear regions devoid of lamin A/C (49). Thus, ORF66 may maintain a balance between disruption of a major component of the nuclear matrix and retention of matrin 3 at replication compartments, where viral replication and transcription occur. HSV infection also causes reorganization of the nuclear matrix protein nuclear mitotic apparatus (NuMA) protein, which is excluded from replication compartments, with redistribution dependent on viral DNA synthesis (84). In HSV-1 and PRV immunofluorescence analyses, matrin 3 showed reproducible cytoplasmic accumulation in cells that did not express US3 kinase in HSV-1 and PRV. This suggests a role for the US3 kinases in the efficient retention of matrin 3 in the nucleus. The fact that the phosphorylation site T150 is far from the putative nuclear localization signal (residues 710 to 718) would suggest that the phosphorylation regulates the nuclear import signal indirectly, by inducing conformational changes, affecting association with other proteins, or by enhancing an as yet unmapped nuclear export activity. Notably, the general kinase inhibitor staurosporine also leads to matrin 3 accumulation in the cytoplasm, supporting the likelihood that nuclear localization is regulated by phosphorylation (30). Thus, in the absence of US3, matrin 3 may be lost to the cytoplasm due to the nuclear component reorganization that occurs in HSV and PRV infections (11, 48, 60, 68, 71). Matrin 3 may also be interacting with a viral protein that is also a US3 target and that localizes to the cytoplasm late in infection in the absence of US3 expression. The basis for not seeing a similar cytoplasmic phenotype of matrin 3 in ORF66-deficient VZV-infected cells is as yet unclear, although high-multiplicity synchronous VZV infections cannot be performed in the manner used here for HSV and PRV. These differences in matrin 3 localization may reflect a functional difference between VZV and the two more aggressively replicating alphaherpesviruses.
In summary, the work presented here shows that a consequence of the expression of the US3/ORF66 kinases is the phosphorylation of the essential nuclear matrix protein matrin 3 at a novel site, which, in VZV, occurs through a non-PKA-dependent pathway. The specific phosphorylation affects the intranuclear and nuclear localization of matrin 3, particularly in late HSV and PRV infections. This is the first report of viral manipulation of this nuclear matrix protein.
Acknowledgments
This work was supported by grant NS064022, a CORE Grant for Vision Research (EY08098), funds from Research to Prevent Blindness Inc. and The Eye & Ear Foundation of Pittsburgh (P.R.K.), NIH grant AI48626 to B.W.B., and an NIH predoctoral T32 training grant award (AI060525, to A.E.).
We sincerely thank Kira L. Lathrop of the University of Pittsburgh Department of Ophthalmology core module for technical assistance with confocal imaging.
Footnotes
Published ahead of print on 20 October 2010.
REFERENCES
- 1.Abendroth, A., I. Lin, B. Slobedman, H. Ploegh, and A. M. Arvin. 2001. Varicella-zoster virus retains major histocompatibility complex class I proteins in the Golgi compartment of infected cells. J. Virol. 75:4878-4888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anachkova, B., V. Djeliova, and G. Russev. 2005. Nuclear matrix support of DNA replication. J. Cell. Biochem. 96:951-961. [DOI] [PubMed] [Google Scholar]
- 3.Beausoleil, S. A., M. Jedrychowski, D. Schwartz, J. E. Elias, J. Villen, J. Li, M. A. Cohn, L. C. Cantley, and S. P. Gygi. 2004. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. U. S. A. 101:12130-12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belgrader, P., R. Dey, and R. Berezney. 1991. Molecular cloning of matrin 3. A 125-kilodalton protein of the nuclear matrix contains an extensive acidic domain. J. Biol. Chem. 266:9893-9899. [PubMed] [Google Scholar]
- 5.Benetti, L., and B. Roizman. 2004. Herpes simplex virus protein kinase US3 activates and functionally overlaps protein kinase A to block apoptosis. Proc. Natl. Acad. Sci. U. S. A. 101:9411-9416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benetti, L., and B. Roizman. 2007. In transduced cells, the US3 protein kinase of herpes simplex virus 1 precludes activation and induction of apoptosis by transfected procaspase 3. J. Virol. 81:10242-10248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ben-Porat, T., R. A. Veach, M. L. Blankenship, and A. S. Kaplan. 1984. Differential association with cellular substructures of pseudorabies virus DNA during early and late phases of replication. Virology 139:205-222. [DOI] [PubMed] [Google Scholar]
- 8.Berezney, R., and D. S. Coffey. 1975. Nuclear protein matrix: association with newly synthesized DNA. Science 189:291-293. [DOI] [PubMed] [Google Scholar]
- 9.Bibor-Hardy, V., A. Dagenais, and R. Simard. 1985. In situ localization of the major capsid protein during lytic infection by herpes simplex virus. J. Gen. Virol. 66(Pt. 4):897-901. [DOI] [PubMed] [Google Scholar]
- 10.Bibor-Hardy, V., M. Pouchelet, E. St-Pierre, M. Herzberg, and R. Simard. 1982. The nuclear matrix is involved in herpes simplex virogenesis. Virology 121:296-306. [DOI] [PubMed] [Google Scholar]
- 11.Bjerke, S. L., and R. J. Roller. 2006. Roles for herpes simplex virus type 1 UL34 and US3 proteins in disrupting the nuclear lamina during herpes simplex virus type 1 egress. Virology 347:261-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cartier, A., E. Broberg, T. Komai, M. Henriksson, and M. G. Masucci. 2003. The herpes simplex virus-1 Us3 protein kinase blocks CD8T cell lysis by preventing the cleavage of Bid by granzyme B. Cell Death Differ. 10:1320-1328. [DOI] [PubMed] [Google Scholar]
- 13.Cartier, A., T. Komai, and M. G. Masucci. 2003. The Us3 protein kinase of herpes simplex virus 1 blocks apoptosis and induces phosphorylation of the Bcl-2 family member Bad. Exp. Cell Res. 291:242-250. [DOI] [PubMed] [Google Scholar]
- 14.Chang, Y. E., and B. Roizman. 1993. The product of the UL31 gene of herpes simplex virus 1 is a nuclear phosphoprotein which partitions with the nuclear matrix. J. Virol. 67:6348-6356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cilloniz, C., W. Jackson, C. Grose, D. Czechowski, J. Hay, and W. T. Ruyechan. 2007. The varicella-zoster virus (VZV) ORF9 protein interacts with the IE62 major VZV transactivator. J. Virol. 81:761-774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cohrs, R. J., and D. H. Gilden. 2007. Prevalence and abundance of latently transcribed varicella-zoster virus genes in human ganglia. J. Virol. 81:2950-2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohrs, R. J., D. H. Gilden, P. R. Kinchington, E. Grinfeld, and P. G. Kennedy. 2003. Varicella-zoster virus gene 66 transcription and translation in latently infected human ganglia. J. Virol. 77:6660-6665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coller, K. E., and G. A. Smith. 2008. Two viral kinases are required for sustained long distance axon transport of a neuroinvasive herpesvirus. Traffic 9:1458-1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Desloges, N., M. Rahaus, and M. H. Wolff. 2008. The phosphorylation profile of protein kinase A substrates is modulated during varicella-zoster virus infection. Med. Microbiol. Immunol. 197:353-360. [DOI] [PubMed] [Google Scholar]
- 20.Eisfeld, A. J., S. E. Turse, S. A. Jackson, E. C. Lerner, and P. R. Kinchington. 2006. Phosphorylation of the varicella-zoster virus (VZV) major transcriptional regulatory protein IE62 by the VZV open reading frame 66 protein kinase. J. Virol. 80:1710-1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eisfeld, A. J., M. B. Yee, A. Erazo, A. Abendroth, and P. R. Kinchington. 2007. Downregulation of class I major histocompatibility complex surface expression by varicella-zoster virus involves open reading frame 66 protein kinase-dependent and -independent mechanisms. J. Virol. 81:9034-9049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Erazo, A., M. B. Yee, N. Osterrieder, and P. R. Kinchington. 2008. Varicella-zoster virus open reading frame 66 protein kinase is required for efficient viral growth in primary human corneal stromal fibroblast cells. J. Virol. 82:7653-7665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farquhar, M. J., H. J. Harris, M. Diskar, S. Jones, C. J. Mee, S. U. Nielsen, C. L. Brimacombe, S. Molina, G. L. Toms, P. Maurel, J. Howl, F. W. Herberg, S. C. van Ijzendoorn, P. Balfe, and J. A. McKeating. 2008. Protein kinase A-dependent step(s) in hepatitis C virus entry and infectivity. J. Virol. 82:8797-8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Favoreel, H. W., G. Van Minnebruggen, D. Adriaensen, and H. J. Nauwynck. 2005. Cytoskeletal rearrangements and cell extensions induced by the US3 kinase of an alphaherpesvirus are associated with enhanced spread. Proc. Natl. Acad. Sci. U. S. A. 102:8990-8995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geenen, K., H. W. Favoreel, L. Olsen, L. W. Enquist, and H. J. Nauwynck. 2005. The pseudorabies virus US3 protein kinase possesses anti-apoptotic activity that protects cells from apoptosis during infection and after treatment with sorbitol or staurosporine. Virology 331:144-150. [DOI] [PubMed] [Google Scholar]
- 26.Giordano, G., A. M. Sanchez-Perez, C. Montoliu, R. Berezney, K. Malyavantham, L. G. Costa, J. J. Calvete, and V. Felipo. 2005. Activation of NMDA receptors induces protein kinase A-mediated phosphorylation and degradation of matrin 3. Blocking these effects prevents NMDA-induced neuronal death. J. Neurochem. 94:808-818. [DOI] [PubMed] [Google Scholar]
- 27.Greenfield, I., J. Nickerson, S. Penman, and M. Stanley. 1991. Human papillomavirus 16 E7 protein is associated with the nuclear matrix. Proc. Natl. Acad. Sci. U. S. A. 88:11217-11221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hibino, Y. 2000. Functional arrangement of genomic DNA and structure of nuclear matrix. Yakugaku Zasshi 120:520-533. (In Japanese.) [PubMed] [Google Scholar]
- 29.Hibino, Y., T. Usui, Y. Morita, N. Hirose, M. Okazaki, N. Sugano, and K. Hiraga. 2006. Molecular properties and intracellular localization of rat liver nuclear scaffold protein P130. Biochim. Biophys. Acta 1759:195-207. [DOI] [PubMed] [Google Scholar]
- 30.Hisada-Ishii, S., M. Ebihara, N. Kobayashi, and Y. Kitagawa. 2007. Bipartite nuclear localization signal of matrin 3 is essential for vertebrate cells. Biochem. Biophys. Res. Commun. 354:72-76. [DOI] [PubMed] [Google Scholar]
- 31.Jackson, D. A. 2003. The principles of nuclear structure. Chromosome Res. 11:387-401. [DOI] [PubMed] [Google Scholar]
- 32.Jacquemont, B., and B. Roizman. 1975. RNA synthesis in cells infected with herpes simplex virus. X. Properties of viral symmetric transcripts and of double-stranded RNA prepared from them. J. Virol. 15:707-713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kato, A., J. Arii, I. Shiratori, H. Akashi, H. Arase, and Y. Kawaguchi. 2009. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral envelope glycoprotein B and regulates its expression on the cell surface. J. Virol. 83:250-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kato, A., M. Tanaka, M. Yamamoto, R. Asai, T. Sata, Y. Nishiyama, and Y. Kawaguchi. 2008. Identification of a physiological phosphorylation site of the herpes simplex virus 1-encoded protein kinase Us3 which regulates its optimal catalytic activity in vitro and influences its function in infected cells. J. Virol. 82:6172-6189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kato, A., M. Yamamoto, T. Ohno, H. Kodaira, Y. Nishiyama, and Y. Kawaguchi. 2005. Identification of proteins phosphorylated directly by the Us3 protein kinase encoded by herpes simplex virus 1. J. Virol. 79:9325-9331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keller, M. J., A. W. Wu, J. I. Andrews, P. W. McGonagill, E. E. Tibesar, and J. L. Meier. 2007. Reversal of human cytomegalovirus major immediate-early enhancer/promoter silencing in quiescently infected cells via the cyclic AMP signaling pathway. J. Virol. 81:6669-6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kimman, T. G., N. De Wind, T. De Bruin, Y. de Visser, and J. Voermans. 1994. Inactivation of glycoprotein gE and thymidine kinase or the US3-encoded protein kinase synergistically decreases in vivo replication of pseudorabies virus and the induction of protective immunity. Virology 205:511-518. [DOI] [PubMed] [Google Scholar]
- 38.Kinchington, P. R., D. Bookey, and S. E. Turse. 1995. The transcriptional regulatory proteins encoded by varicella-zoster virus open reading frames (ORFs) 4 and 63, but not ORF 61, are associated with purified virus particles. J. Virol. 69:4274-4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kinchington, P. R., K. Fite, A. Seman, and S. E. Turse. 2001. Virion association of IE62, the varicella-zoster virus (VZV) major transcriptional regulatory protein, requires expression of the VZV open reading frame 66 protein kinase. J. Virol. 75:9106-9113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kinchington, P. R., K. Fite, and S. E. Turse. 2000. Nuclear accumulation of IE62, the varicella-zoster virus (VZV) major transcriptional regulatory protein, is inhibited by phosphorylation mediated by the VZV open reading frame 66 protein kinase. J. Virol. 74:2265-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leach, N., S. L. Bjerke, D. K. Christensen, J. M. Bouchard, F. Mou, R. Park, J. Baines, T. Haraguchi, and R. J. Roller. 2007. Emerin is hyperphosphorylated and redistributed in herpes simplex virus type 1-infected cells in a manner dependent on both UL34 and US3. J. Virol. 81:10792-10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leader, D. P., A. D. Deana, F. Marchiori, F. C. Purves, and L. A. Pinna. 1991. Further definition of the substrate specificity of the alpha-herpesvirus protein kinase and comparison with protein kinases A and C. Biochim. Biophys. Acta 1091:426-431. [DOI] [PubMed] [Google Scholar]
- 43.Li, P. L., T. Wang, K. A. Buckley, A. L. Chenine, S. Popov, and R. M. Ruprecht. 2005. Phosphorylation of HIV Nef by cAMP-dependent protein kinase. Virology 331:367-374. [DOI] [PubMed] [Google Scholar]
- 44.Malyavantham, K. S., S. Bhattacharya, M. Barbeitos, L. Mukherjee, J. Xu, F. O. Fackelmayer, and R. Berezney. 2008. Identifying functional neighborhoods within the cell nucleus: proximity analysis of early S-phase replicating chromatin domains to sites of transcription, RNA polymerase II, HP1γ, matrin 3 and SAF-A. J. Cell. Biochem. 105:391-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meignier, B., R. Longnecker, P. Mavromara-Nazos, A. E. Sears, and B. Roizman. 1988. Virulence of and establishment of latency by genetically engineered deletion mutants of herpes simplex virus 1. Virology 162:251-254. [DOI] [PubMed] [Google Scholar]
- 46.Moffat, J. F., L. Zerboni, M. H. Sommer, T. C. Heineman, J. I. Cohen, H. Kaneshima, and A. M. Arvin. 1998. The ORF47 and ORF66 putative protein kinases of varicella-zoster virus determine tropism for human T cells and skin in the SCID-hu mouse. Proc. Natl. Acad. Sci. U. S. A. 95:11969-11974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morimoto, T., J. Arii, M. Tanaka, T. Sata, H. Akashi, M. Yamada, Y. Nishiyama, M. Uema, and Y. Kawaguchi. 2009. Differences in the regulatory and functional effects of the Us3 protein kinase activities of herpes simplex virus 1 and 2. J. Virol. 83:11624-11634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morris, J. B., H. Hofemeister, and P. O'Hare. 2007. Herpes simplex virus infection induces phosphorylation and delocalization of emerin, a key inner nuclear membrane protein. J. Virol. 81:4429-4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mou, F., T. Forest, and J. D. Baines. 2007. US3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin A/C in infected cells. J. Virol. 81:6459-6470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mou, F., E. Wills, and J. D. Baines. 2009. Phosphorylation of the UL31 protein of herpes simplex virus 1 by the US3-encoded kinase regulates localization of the nuclear envelopment complex and egress of nucleocapsids. J. Virol. 83:5181-5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Muller, W. E., T. Okamoto, P. Reuter, D. Ugarkovic, and H. C. Schroder. 1990. Functional characterization of Tat protein from human immunodeficiency virus. Evidence that Tat links viral RNAs to nuclear matrix. J. Biol. Chem. 265:3803-3808. [PubMed] [Google Scholar]
- 52.Murata, T., F. Goshima, T. Daikoku, H. Takakuwa, and Y. Nishiyama. 2000. Expression of herpes simplex virus type 2 US3 affects the Cdc42/Rac pathway and attenuates c-Jun N-terminal kinase activation. Genes Cells 5:1017-1027. [DOI] [PubMed] [Google Scholar]
- 53.Nakayasu, H., and R. Berezney. 1991. Nuclear matrins: identification of the major nuclear matrix proteins. Proc. Natl. Acad. Sci. U. S. A. 88:10312-10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olsen, J. V., B. Blagoev, F. Gnad, B. Macek, C. Kumar, P. Mortensen, and M. Mann. 2006. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127:635-648. [DOI] [PubMed] [Google Scholar]
- 55.Poon, A. P., Y. Liang, and B. Roizman. 2003. Herpes simplex virus 1 gene expression is accelerated by inhibitors of histone deacetylases in rabbit skin cells infected with a mutant carrying a cDNA copy of the infected-cell protein no. 0. J. Virol. 77:12671-12678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Purves, F. C., A. D. Deana, F. Marchiori, D. P. Leader, and L. A. Pinna. 1986. The substrate specificity of the protein kinase induced in cells infected with herpesviruses: studies with synthetic substrates [corrected] indicate structural requirements distinct from other protein kinases. Biochim. Biophys. Acta 889:208-215. [DOI] [PubMed] [Google Scholar]
- 57.Purves, F. C., R. M. Longnecker, D. P. Leader, and B. Roizman. 1987. Herpes simplex virus 1 protein kinase is encoded by open reading frame US3 which is not essential for virus growth in cell culture. J. Virol. 61:2896-2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ramachandran, S., K. A. Davoli, M. B. Yee, R. L. Hendricks, and P. R. Kinchington. 2010. Delaying the expression of herpes simplex virus type 1 glycoprotein B (gB) to a true late gene alters neurovirulence and inhibits the gB-CD8+ T-cell response in the trigeminal ganglion. J. Virol. 84:8811-8820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramachandran, S., J. E. Knickelbein, C. Ferko, R. L. Hendricks, and P. R. Kinchington. 2008. Development and pathogenic evaluation of recombinant herpes simplex virus type 1 expressing two fluorescent reporter genes from different lytic promoters. Virology 378:254-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reynolds, A. E., L. Liang, and J. D. Baines. 2004. Conformational changes in the nuclear lamina induced by herpes simplex virus type 1 require genes UL31 and UL34. J. Virol. 78:5564-5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ruyechan, W. T., H. Peng, M. Yang, and J. Hay. 2003. Cellular factors and IE62 activation of VZV promoters. J. Med. Virol. 70(Suppl. 1):S90-S94. [DOI] [PubMed] [Google Scholar]
- 62.Sagou, K., T. Imai, H. Sagara, M. Uema, and Y. Kawaguchi. 2009. Regulation of the catalytic activity of herpes simplex virus 1 protein kinase Us3 by autophosphorylation and its role in pathogenesis. J. Virol. 83:5773-5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sanchez, V., P. C. Angeletti, J. A. Engler, and W. J. Britt. 1998. Localization of human cytomegalovirus structural proteins to the nuclear matrix of infected human fibroblasts. J. Virol. 72:3321-3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schaap, A., J. F. Fortin, M. Sommer, L. Zerboni, S. Stamatis, C. C. Ku, G. P. Nolan, and A. M. Arvin. 2005. T-cell tropism and the role of ORF66 protein in pathogenesis of varicella-zoster virus infection. J. Virol. 79:12921-12933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schaap-Nutt, A., M. Sommer, X. Che, L. Zerboni, and A. M. Arvin. 2006. ORF66 protein kinase function is required for T-cell tropism of varicella-zoster virus in vivo. J. Virol. 80:11806-11816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schirmbeck, R., and W. Deppert. 1987. Specific interaction of simian virus 40 large T antigen with cellular chromatin and nuclear matrix during the course of infection. J. Virol. 61:3561-3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schumacher, D., B. K. Tischer, S. Trapp, and N. Osterrieder. 2005. The protein encoded by the US3 orthologue of Marek's disease virus is required for efficient de-envelopment of perinuclear virions and involved in actin stress fiber breakdown. J. Virol. 79:3987-3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Scott, E. S., and P. O'Hare. 2001. Fate of the inner nuclear membrane protein lamin B receptor and nuclear lamins in herpes simplex virus type 1 infection. J. Virol. 75:8818-8830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shabb, J. B. 2001. Physiological substrates of cAMP-dependent protein kinase. Chem. Rev. 101:2381-2411. [DOI] [PubMed] [Google Scholar]
- 70.Silva, L., A. Cliffe, L. Chang, and D. M. Knipe. 2008. Role for A-type lamins in herpesviral DNA targeting and heterochromatin modulation. PLoS Pathog. 4:e1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Simpson-Holley, M., R. C. Colgrove, G. Nalepa, J. W. Harper, and D. M. Knipe. 2005. Identification and functional evaluation of cellular and viral factors involved in the alteration of nuclear architecture during herpes simplex virus 1 infection. J. Virol. 79:12840-12851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Skalhegg, B. S., and K. Tasken. 2000. Specificity in the cAMP/PKA signaling pathway. Differential expression, regulation, and subcellular localization of subunits of PKA. Front. Biosci. 5:D678-D693. [DOI] [PubMed] [Google Scholar]
- 73.Soong, W., J. C. Schultz, A. C. Patera, M. H. Sommer, and J. I. Cohen. 2000. Infection of human T lymphocytes with varicella-zoster virus: an analysis with viral mutants and clinical isolates. J. Virol. 74:1864-1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sreenath, K., L. Pavithra, S. Singh, S. Sinha, P. K. Dash, N. B. Siddappa, U. Ranga, D. Mitra, and S. Chattopadhyay. 2010. Nuclear matrix protein SMAR1 represses HIV-1 LTR mediated transcription through chromatin remodeling. Virology 400:76-85. [DOI] [PubMed] [Google Scholar]
- 75.Suomalainen, M., M. Y. Nakano, K. Boucke, S. Keller, and U. F. Greber. 2001. Adenovirus-activated PKA and p38/MAPK pathways boost microtubule-mediated nuclear targeting of virus. EMBO J. 20:1310-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Van den Broeke, C., M. Deruelle, H. J. Nauwynck, K. E. Coller, G. A. Smith, J. Van Doorsselaere, and H. W. Favoreel. 2009. The kinase activity of pseudorabies virus US3 is required for modulation of the actin cytoskeleton. Virology 385:155-160. [DOI] [PubMed] [Google Scholar]
- 77.Van den Broeke, C., M. Radu, M. Deruelle, H. Nauwynck, C. Hofmann, Z. M. Jaffer, J. Chernoff, and H. W. Favoreel. 2009. Alphaherpesvirus US3-mediated reorganization of the actin cytoskeleton is mediated by group A p21-activated kinases. Proc. Natl. Acad. Sci. U. S. A. 106:8707-8712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Van Minnebruggen, G., H. W. Favoreel, L. Jacobs, and H. J. Nauwynck. 2003. Pseudorabies virus US3 protein kinase mediates actin stress fiber breakdown. J. Virol. 77:9074-9080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Walters, M. S., A. Erazo, P. R. Kinchington, and S. Silverstein. 2009. Histone deacetylases 1 and 2 are phosphorylated at novel sites during varicella-zoster virus infection. J. Virol. 83:11502-11513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Walters, M. S., P. R. Kinchington, B. W. Banfield, and S. Silverstein. 2010. Hyperphosphorylation of histone deacetylase 2 by alphaherpesvirus US3 kinases. J. Virol. 84:9666-9676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wisner, T. W., C. C. Wright, A. Kato, Y. Kawaguchi, F. Mou, J. D. Baines, R. J. Roller, and D. C. Johnson. 2009. Herpesvirus gB-induced fusion between the virion envelope and outer nuclear membrane during virus egress is regulated by the viral US3 kinase. J. Virol. 83:3115-3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xia, K., D. M. Knipe, and N. A. DeLuca. 1996. Role of protein kinase A and the serine-rich region of herpes simplex virus type 1 ICP4 in viral replication. J. Virol. 70:1050-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yamada, H., T. Daikoku, Y. Yamashita, Y. M. Jiang, T. Tsurumi, and Y. Nishiyama. 1997. The product of the US10 gene of herpes simplex virus type 1 is a capsid/tegument-associated phosphoprotein which copurifies with the nuclear matrix. J. Gen. Virol. 78(Pt. 11):2923-2931. [DOI] [PubMed] [Google Scholar]
- 84.Yamauchi, Y., K. Kiriyama, H. Kimura, and Y. Nishiyama. 2008. Herpes simplex virus induces extensive modification and dynamic relocalisation of the nuclear mitotic apparatus (NuMA) protein in interphase cells. J. Cell Sci. 121:2087-2096. [DOI] [PubMed] [Google Scholar]
- 85.Zeitz, M. J., K. S. Malyavantham, B. Seifert, and R. Berezney. 2009. Matrin 3: chromosomal distribution and protein interactions. J. Cell. Biochem. 108:125-133. [DOI] [PubMed] [Google Scholar]
- 86.Zhang, Z., and G. G. Carmichael. 2001. The fate of dsRNA in the nucleus: a p54(nrb)-containing complex mediates the nuclear retention of promiscuously A-to-I edited RNAs. Cell 106:465-475. [DOI] [PubMed] [Google Scholar]