

Abstract
Objective:
Diabetes mellitus has been associated with an increased risk of Alzheimer disease (AD), but how it exerts its effect remains controversial. Possible pathophysiologic mechanisms are glucose toxicity and a direct effect of insulin on amyloid metabolism. Most studies had short follow-up, and longer-term effects of diabetes on AD risk are unknown. We investigated whether fasting glucose and insulin levels and insulin resistance are associated with the risk of AD and whether this risk is constant over time.
Methods:
The study was based on 3,139 participants of the Rotterdam Study, a population-based cohort study. All subjects were free from dementia, did not have a history of diabetes, and had fasting levels of glucose and insulin measured at baseline. Insulin resistance was estimated with the homeostasis model assessment. We investigated how fasting glucose, insulin, and insulin resistance are related to the risk of AD in 3 different strata according to time-to-event, using Cox proportional hazards models.
Results:
During follow-up, 211 participants developed AD, 71 of them within 3 years of baseline. Levels of insulin and insulin resistance were associated with a higher risk of AD within 3 years of baseline. After 3 years, the risk was no longer increased. Glucose was not associated with a higher risk of AD. There was no interaction of APOE ε4 carriership and insulin metabolism on the risk of AD.
Conclusions:
Our findings suggest that insulin metabolism influences the clinical manifestation of AD only within 3 years.
GLOSSARY
- AD
= Alzheimer disease;
- DSM-III-R
= Diagnostic and Statistical Manual of Mental Disorders, 3rd edition, revised;
- HDL
= high-density lipoprotein;
- HOMA
= homeostasis model assessment.
Diabetes and Alzheimer disease (AD) are both common diseases in elderly individuals. Although many studies have suggested that patients with diabetes have a higher risk of developing AD,1,2 the results are conflicting and the real relationship between diabetes and AD remains controversial.
Three characteristics related to type 2 diabetes that might underlie the effect of diabetes on the brain in the development of AD are insulin resistance, hyperinsulinemia, and hyperglycemia.3–5 These mechanisms are thought to act on different pathways that are important in the pathophysiology of AD, either indirectly, through inflammation or the development of vascular disease, or directly, through effects on amyloid and tau metabolism and the formation of advanced glycation end products.1,5–7
We previously reported from the Rotterdam Study, a population-based cohort study, an association between diabetes and a higher risk of AD.2 This study was based on the initial cohort that started in 1990 and had only a short follow-up of on average 2.1 years.1,2 In 1997, at the third survey of the Rotterdam Study, fasting blood samples were drawn. Using this survey as baseline for our current study, we now have levels of fasting glucose and insulin available at baseline and a much longer follow-up, up to 10 years (average, 7.2 years).
To investigate the relationships that might underlie the effect of diabetes on the risk of AD, we determined whether fasting glucose and insulin levels and insulin resistance are associated with the risk of AD in persons without diabetes and whether this risk remains the same over a longer follow-up period.
METHODS
The Rotterdam Study.
The Rotterdam Study is a prospective population-based cohort study that is conducted among all inhabitants aged 55 years and older of Ommoord, a district of Rotterdam, the Netherlands.8 Of 10,274 eligible subjects, 7,983 (78%) participated in the baseline examinations between 1990 and 1993. Follow-up examinations were conducted in 1993–1994, 1997–1999, and 2002–2004. In addition, through linkage with records of general practitioners, the total cohort was continuously monitored for morbidity and mortality.
Study population.
This study was based on the third survey (1997–1999) of the Rotterdam Study in which fasting blood samples were obtained. Of the 5,990 participants of the original cohort who were still alive at that time, 4,797 participated, 3,795 of whom had fasting blood samples drawn. Of these participants, 3,432 were free of dementia and had complete data on glucose and insulin levels, cognitive function, and cardiovascular risk factors, including waist circumference, blood pressure, triglycerides, and high-density lipoprotein (HDL) cholesterol. We excluded 313 participants who had a history of diabetes at the time of the third survey. This resulted in a study population of 3,139 subjects.
Standard protocol approvals, registrations, and patient consents.
The medical ethics committee at Erasmus University of Rotterdam approved the study, and written informed consent was obtained from all participants.
Glucose and insulin assessments.
Fasting blood serum was drawn during the examination at the research center. The blood was stored at −80°C in a number of 5-mL aliquots. Glucose levels were measured within 1 week of sampling using the glucose hexokinase method.9 The remaining serum was kept frozen for later analyses. In 2008, stored serum that had not been thawed previously was used for insulin measurements. Serum insulin was determined by metric assay (Biosource Diagnostics, Camarillo, CA). This assay has no cross-reactivity with either proinsulin or C-peptide. All measurements were done at the clinical chemistry laboratory at Erasmus Medical Center in Rotterdam.
Insulin resistance.
Data on fasting glucose and fasting insulin levels were used to calculate the degree of insulin resistance according to the homeostasis model assessment (HOMA).10 The HOMA index, which is calculated by dividing the product of fasting levels of glucose and insulin by a constant, has been shown to correlate well with the euglycemic hyperinsulinemic clamp method,11 which is the gold standard for measuring insulin resistance.
Dementia case finding.
The diagnosis of dementia was made following a 3-step protocol.12 Two brief tests of cognition (Mini-Mental State Examination13 and Geriatric Mental State schedule14 organic level) were used to screen all subjects. Participants with screen-positive results (Mini-Mental State Examination score <26 or Geriatric Mental State organic level >0) underwent the Cambridge Examination for Mental Disorders of the Elderly.15 Subjects who were suspected of having dementia were, if necessary, examined by a neuropsychologist. In addition, the total cohort was continuously monitored for incident dementia through computerized linkage between the study database and digitized medical records from general practitioners and the Regional Institute for Outpatient Mental Health Care.12 The diagnoses of dementia and AD were made in accordance with internationally accepted criteria for dementia (DSM-III-R),16 and AD (National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer's Disease and Related Disorders Association),17 by a panel of a neurologist, neuropsychologist, and research physician. The follow-up with regard to dementia diagnosis was virtually complete (98.6%) until January 1, 2007.
History of diabetes.
History of diabetes was defined as 1) self-reported history of diabetes (reported use of oral antidiabetes medication, or insulin use, or treatment by diet), 2) registration by a general practitioner as having diabetes, or 3) a previous diagnosis of diabetes during the 1990–1993 or 1993–1994 examinations (based on a random nonfasting plasma glucose level ≥11.1 mmol/L).18
Newly diagnosed diabetes.
Newly diagnosed diabetes was defined as a fasting glucose level ≥7.0 mmol/L in participants without a history of diabetes.
Covariates.
Educational level was assessed during the first interview that took place between 1990 and 1993 and was dichotomized into primary education (with or without an unfinished higher education) vs lower vocational to university education. APOE genotype was assessed on coded DNA samples using PCR without knowledge of the dementia diagnosis. APOE ε4 status was defined as carriership of 1 or 2 ε4 alleles.
Blood pressure was measured at the right brachial artery using a random-zero sphygmomanometer with the participant in a sitting position. The waist circumference was measured in centimeters. HDL cholesterol levels were measured in fasting serum within 1 week of the visit to the research center. Serum levels of triglycerides were measured in 2008 at the same time the insulin levels were measured.
Statistical analyses.
Because of a nonnormal distribution of insulin levels and HOMA, we used a log2 transformation, which means that every unit increase on the logarithmic scale reflects a doubling of the absolute values.
We used Cox proportional hazards models to examine the associations of fasting levels of glucose and insulin, and insulin resistance with the risk of developing AD during follow-up.
All analyses were adjusted for age and sex, and additional adjustments were made for level of education, APOE ε4 status, and the components of the metabolic syndrome (waist circumference, systolic and diastolic blood pressure, triglycerides, and HDL cholesterol).19
We added a time-dependent covariate to the model to test the proportionality assumption. Because this assumption was violated, we performed the analyses in 3 different strata according to follow-up time: 1) short follow-up (maximum 3 years), 2) medium follow-up (3–5.5 years), and 3) long follow-up (5.5–9.7 years). Cutoffs were chosen to have approximately equal numbers of incident cases within the follow-up intervals. We repeated the analyses in strata of APOE ε4 carriership. To test for interaction of APOE ε4 status on the relationship of levels of glucose, insulin, and insulin resistance with the risk of AD, we first added an interaction term with APOE ε4 status to the different models. This is the method that is usually performed to test for interaction and assesses interaction on a multiplicative scale, which, however, measures differences in rate ratios. When biological interaction occurs, one would expect to see interaction on an additive scale, which measures differences in absolute risks.20,21 We used the relative risk due to interaction to test for interaction on an additive scale.22 Although the number of patients with AD was small in the younger age group, we also performed stratified analyses by age median to test whether the associations of levels of glucose and insulin and insulin resistance with the risk of AD are different for younger vs older people.
All analyses were done using the SPSS statistical package 15.0 (SPSS Inc., Chicago, IL) or SAS 9.2 (SAS Institute Inc., Cary, NC).
RESULTS
The baseline characteristics of the study population are shown in table 1. After a follow-up of 22,494 person-years (mean, 7.2 years; SD, 2.1), 211 participants developed AD, 71 of them within 3 years of baseline, 72 between 3 and 5.5 years, and 68 after 5.5 years. When we performed the analyses with the total follow-up period, fasting levels of glucose and insulin and insulin resistance were not associated with the risk of AD (data not shown).
Table 1 Baseline characteristics
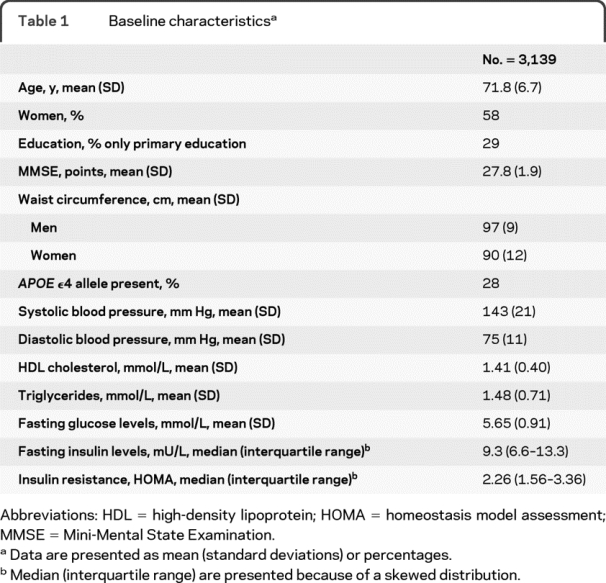
Table 2 shows the associations of fasting insulin and glucose levels and insulin resistance with the risk of AD in the different follow-up strata. Fasting levels of glucose were not associated with the risk of AD after a short or medium follow-up time. There was a relation between higher levels of glucose and a lower risk of AD after 5.5 years (hazard ratio per SD increase, 0.61; 95% confidence interval, 0.41–0.89).
Table 2 Hazard ratios (with corresponding 95% confidence intervals) of the association of fasting glucose and insulin levels and insulin resistance with the risk of Alzheimer disease according to time-to-event
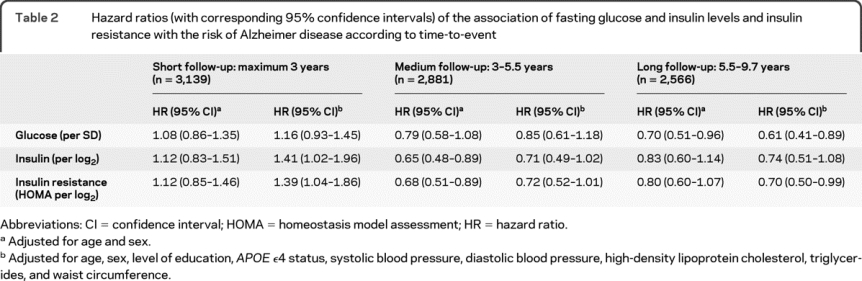
Fasting levels of insulin and insulin resistance were associated with a higher short-term risk of AD with an increase in risk of approximately 40% per doubling of insulin levels and insulin resistance in the fully adjusted model. Higher levels of insulin and insulin resistance were no longer associated with an increased risk of AD after 3 years, and the relation even seemed to invert, reaching borderline significance in some of the fully adjusted models.
Exclusion of 162 participants who had newly diagnosed diabetes based on their fasting glucose levels (≥7.0 mmol/L) did not alter the results.
When we applied the fully adjusted model in strata of APOE ε4 carriership, levels of glucose and insulin and insulin resistance were all associated with the short-term risk of AD in noncarriers of the APOE ε4 allele (table 3). In these noncarriers, doubling of levels of insulin and insulin resistance increased the short-term risk of AD by 75%. In carriers of the APOE ε4 allele, higher levels of glucose and insulin and insulin resistance had no apparent effect on the short-term risk of AD. There was no significant interaction of APOE ε4 status and glucose levels, insulin levels, and insulin resistance on an additive scale and a borderline significant interaction of APOE ε4 status and insulin and insulin resistance on a multiplicative scale.
Table 3 Hazard ratios (with corresponding 95% confidence intervals) of the association of fasting glucose and insulin levels and insulin resistance with the short-term risk of Alzheimer disease according to APOE ε4 carriership, with p values for interaction
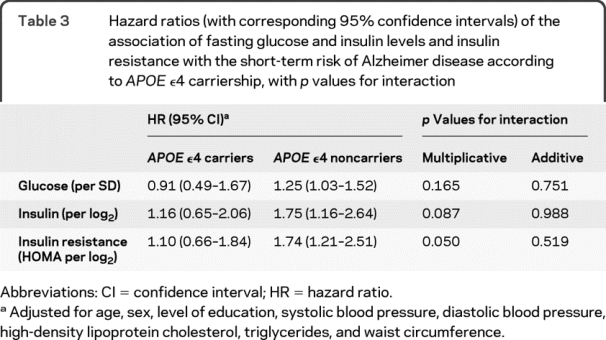
The risk of AD after a medium or long follow-up did not differ between carriers and noncarriers of the APOE ε4 allele (p values for multiplicative and additive interaction >0.25).
In the stratified analyses by age median (71.0 years), there was no effect of glucose levels on the risk of AD, and higher insulin levels and insulin resistance were also not associated with an increased risk of AD after 3 years of follow-up in both age groups.
DISCUSSION
The major strengths of our study are the prospective population-based design, the large number of participants, and the long follow-up period with almost no loss to follow-up. The latter in particular is important because theoretically the strength of a longitudinal association may diminish or even invert if there is selective attrition of people who develop dementia over the follow-up period. We are confident, however, that through our very dense and complete case finding and follow-up, selective attrition cannot explain the findings in our study.
Unfortunately, we had no repeated glucose and insulin measurements over time. Therefore, we could not assess whether changes in glucose and insulin levels have an impact on the longer-term risk of AD, and we might underestimate the longer-term risk of AD.
We considered whether the assessment of insulin levels after 10 years of storage at −80°C might have affected the stability of the samples. Although we are not aware of a study on the stability of insulin levels after long-term storage of serum, there are several studies on the stability of other proteins and hormones that show that long-term storage at −25°C and especially at −80°C does not affect the stability of the measurements and that the expected stability of materials stored at −80°C is at least 10 years.23–25 Furthermore, any change in insulin levels after 10 years of storage should affect all samples, without any relation with our outcome AD.
Because we had no fasting blood samples at the start of the Rotterdam Study, we used the third survey as the baseline for the current study. This was a selection of more healthy participants, with fewer cognitive deficits and less diabetes, who came to the research center and had fasting blood drawn. However, because we excluded patients with prevalent AD and participants with a history of diabetes, we consider it unlikely that this selection will have influenced our results.
Our results show that characteristics related to diabetes, especially higher levels of insulin and insulin resistance, are associated with a higher risk of AD only within 3 years. This supports our previous finding of an association of diabetes with a higher risk of AD with an average follow-up of 2.1 years.2 Another recent study from our cohort showed no association of higher levels of glucose and insulin resistance with cognitive decline after on average 4.6 years in participants without a history of diabetes. In that study, there was selective nonparticipation with regard to the outcome of cognitive decline of those who were diagnosed with dementia in between cognitive examinations and did not visit the research center for the follow-up assessment.26 These are the participants who developed dementia mainly within 3 years of baseline, which is the time period in which we find an increased risk of AD with higher levels of insulin and insulin resistance.
Several other studies have shown associations of the glucose and insulin metabolism with the risk of AD.27–32 Most of these studies had an average follow-up time of around 5 years. The studies that had a longer follow-up time often did not find a higher risk of AD with disturbances in the insulin metabolism. A recent study from the Uppsala Longitudinal Study of Adult Men showed no association of 6 of 7 measures of insulin metabolism including fasting insulin levels and insulin sensitivity, determined by the euglycemic insulin clamp method, with the risk of AD after a median follow-up of 12 years. A low early insulin response to oral glucose challenge was, however, associated with a higher risk of AD.29 In the Framingham Study after a long follow-up period of on average 12.7 years, diabetes did not increase the overall risk of dementia and AD.33 If we look at the risk of AD over the total follow-up period in our study, without dividing the time to event into strata, we also find no association of our 3 measures and the risk of AD.
The higher short-term risk of AD that disappears after 3 years of follow-up might be explained by several mechanisms. First, because the underlying neuropathologic changes that cause AD are already ongoing years before the clinical diagnosis can be made, our results might reflect reversed causality or a common pathophysiology instead of a causal role of insulin metabolism in AD. Second, we hypothesized that diabetes and disturbances in insulin metabolism only increase the risk of AD for a short time period by advancing the onset in those already on the verge of getting AD. This would mean that disturbances in the insulin metabolism are not a cause of the neuropathologic changes that result in AD but do influence and accelerate those changes, leading to an earlier onset of AD.
We found that after 3 years of follow-up the risk of AD was no longer increased and even inverted. It seems unlikely that the inversion of the effect on AD risk reflects a true biological mechanism. It is more likely that competing risks might explain these findings: people with diabetes or prediabetes are at a higher risk of cardiovascular diseases and death, which makes them no longer at risk of AD, especially in the long term.
Although there was a difference in the effect size on the short-term risk between carriers and noncarriers of the APOE ε4 allele, there was no interaction on an additive scale. The borderline significant negative interaction on a multiplicative scale suggests that the differences in risk ratio can be explained by the differences in baseline absolute risks and not by a true interaction between APOE ε4 status and insulin metabolism.20
Different studies reported conflicting results on the influence of the APOE ε4 allele on the relation between diabetes or insulin metabolism and the risk of AD. A number of studies reported higher effect estimates in noncarriers of the APOE ε4 allele, although none of these studies reported whether testing for additive interaction was done.32,34,35 The Uppsala Longitudinal Study of Adult Men showed a stronger effect of a low acute or early insulin response, assessed at midlife28 and at age 71,29 on the risk of AD in noncarriers of the APOE ε4 allele. However, in the same study, higher levels of fasting insulin and HOMA, both assessed at midlife, were associated with the risk of AD only in carriers of the APOE ε4 allele, which conflicts with our results.28 We assessed our measurements at an older age, which might explain these differences. Several other studies also showed a stronger effect of diabetes and hyperinsulinemia on the risk of AD in carriers of the APOE ε4 allele.31,36,37 Investigators from the Kungsholmen Project initially reported an effect of diabetes on the risk of AD only in carriers of the APOE ε4 allele,37 yet in a later study they found an effect of borderline diabetes, defined as a random plasma glucose level of 7.8–11.0 mmol/L, on the risk of AD only in noncarriers.38 These discrepant results fit our finding of no interaction between APOE ε4 status and insulin metabolism on the risk of AD.
AUTHOR CONTRIBUTIONS
Statistical analysis was conducted by Dr. Elisabeth M.C. Schrijvers.
DISCLOSURE
Dr. Schrijvers reports no disclosures. Dr. Witteman receives research support from the Netherlands Organization for Scientific Research, the Netherlands Organization for Health Research and Development, the Netherlands Consortium for Healthy Ageing, and the Netherlands Heart Foundation. Dr. Sijbrands receives research support from Pfizer Inc, Merck Sharp & Dohme, and the Netherlands Heart Foundation. Dr. Hofman has received funding for travel from GlaxoSmithKline; serves as Editor-in-Chief for the European Journal of Epidemiology; receives royalties from the publication of Grondslagen der epidemiologie (Elsevier, 2008), Klinische epidemiologie (Elsevier, 2000), and Investigating Neurological Disease (Cambridge University Press, 1996); and receives research support from the Netherlands Genomics Initiative for the Rotterdam Study and from the Ministry of Health for the Generation R study. Dr. Koudstaal receives royalties from the publication of Neurologie (Elsevier, 4th edition, 2010) and Anamnese en lichamelijk onderzoek (Elsevier, 4th edition, 2006); has received travel and honoraria from SERVIER; and receives research support from the Neurovascular Research Fund Rotterdam. Dr. Breteler serves on editorial advisory boards for Neuroepidemiology and Alzheimer's & Dementia; and receives research support from Pfizer Inc, the Netherlands Organization for Scientific Research, the Alzheimer's Association USA, the NIH (1 R01 AG 033 193-01, PI on subcontract), the Internationale Stichting Alzheimer Onderzoek (ISAO), the Dutch Cancer Society, the Dutch Parkinsonfonds, and the Netherlands Brain Foundation.
Address correspondence and reprint requests to Prof. Dr. Monique M.B. Breteler, Department of Epidemiology, Erasmus MC University Medical Center, PO Box 2040, 3000 CA Rotterdam, the Netherlands m.breteler@erasmusmc.nl
Study funding: Supported by the Alzheimer's Association (IIRG-06-27261) and by the Internationale Stichting Alzheimer Onderzoek. The Rotterdam Study is supported by the Erasmus MC University Medical Center and Erasmus University Rotterdam, the Netherlands Organization for Scientific Research, the Netherlands Organization for Health Research and Development, the Research Institute for Diseases in the Elderly, the Ministry of Education, Culture and Science, the Ministry of Health, Welfare and Sports, the European Commission, and the Municipality of Rotterdam.
Disclosure: Author disclosures are provided at the end of the article.
Received June 4, 2010. Accepted in final form August 20, 2010.
REFERENCES
- 1.Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol 2006;5:64–74. [DOI] [PubMed] [Google Scholar]
- 2.Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: the Rotterdam Study. Neurology 1999;53:1937–1942. [DOI] [PubMed] [Google Scholar]
- 3.Craft S. Insulin resistance syndrome and Alzheimer disease: pathophysiologic mechanisms and therapeutic implications. Alzheimer Dis Assoc Disord 2006;20:298–301. [DOI] [PubMed] [Google Scholar]
- 4.Strachan MW, Reynolds RM, Frier BM, Mitchell RJ, Price JF. The relationship between type 2 diabetes and dementia. Br Med Bull 2008;88:131–146. [DOI] [PubMed] [Google Scholar]
- 5.Luchsinger JA. Adiposity, hyperinsulinemia, diabetes and Alzheimer's disease: an epidemiological perspective. Eur J Pharmacol 2008;585:119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Craft S. The role of metabolic disorders in Alzheimer disease and vascular dementia: two roads converged. Arch Neurol 2009;66:300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sabayan B, Foroughinia F, Mowla A, Borhanihaghighi A. Role of insulin metabolism disturbances in the development of Alzheimer disease: mini review. Am J Alzheimers Dis Other Demen 2008;23:192–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hofman A, Breteler MM, van Duijn CM, et al. The Rotterdam Study: 2010 objectives and design update. Eur J Epidemiol 2009;24:553–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neeley WE. Simple automated determination of serum or plasma glucose by a hexokinase-glucose-6-phosphate dehydrogenase method. Clin Chem 1972;18:509–515. [PubMed] [Google Scholar]
- 10.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985;28:412–419. [DOI] [PubMed] [Google Scholar]
- 11.Bonora E, Targher G, Alberiche M, et al. Homeostasis model assessment closely mirrors the glucose clamp technique in the assessment of insulin sensitivity: studies in subjects with various degrees of glucose tolerance and insulin sensitivity. Diabetes Care 2000;23:57–63. [DOI] [PubMed] [Google Scholar]
- 12.Ott A, Breteler MM, van Harskamp F, Stijnen T, Hofman A. Incidence and risk of dementia: The Rotterdam Study. Am J Epidemiol 1998;147:574–580. [DOI] [PubMed] [Google Scholar]
- 13.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 14.Copeland JR, Kelleher MJ, Kellett JM, et al. A semi-structured clinical interview for the assessment of diagnosis and mental state in the elderly: the Geriatric Mental State Schedule. I. Development and reliability. Psychol Med 1976;6:439–449. [DOI] [PubMed] [Google Scholar]
- 15.Roth M, Tym E, Mountjoy CQ, et al. CAMDEX: a standardised instrument for the diagnosis of mental disorder in the elderly with special reference to the early detection of dementia. Br J Psychiatry 1986;149:698–709. [DOI] [PubMed] [Google Scholar]
- 16.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-III-R. Washington, DC: American Psychiatric Association; 1987. [Google Scholar]
- 17.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 18.Dehghan A, van Hoek M, Sijbrands EJ, Stijnen T, Hofman A, Witteman JC. Risk of type 2 diabetes attributable to C-reactive protein and other risk factors. Diabetes Care 2007;30:2695–2699. [DOI] [PubMed] [Google Scholar]
- 19.Executive Summary of the Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 2001;285:2486–2497. [DOI] [PubMed] [Google Scholar]
- 20.Rothman KJ. Measuring interactions. In: Epidemiology: An Introduction. New York: Oxford University Press; 2002:168–180. [Google Scholar]
- 21.Knol MJ, van der Tweel I, Grobbee DE, Numans ME, Geerlings MI. Estimating interaction on an additive scale between continuous determinants in a logistic regression model. Int J Epidemiol 2007;36:1111–1118. [DOI] [PubMed] [Google Scholar]
- 22.Li R, Chambless L. Test for additive interaction in proportional hazards models. Ann Epidemiol 2007;17:227–236. [DOI] [PubMed] [Google Scholar]
- 23.Bolelli G, Muti P, Micheli A, et al. Validity for epidemiological studies of long-term cryoconservation of steroid and protein hormones in serum and plasma. Cancer Epidemiol Biomarkers Prev 1995;4:509–513. [PubMed] [Google Scholar]
- 24.Henriksen GM, Pedersen MM, Nørgaard I, et al. Minimally processed fresh frozen human reference sera: preparation, testing, and application to international external quality assurance. Scand J Clin Lab Invest 2004;64:293–308. [DOI] [PubMed] [Google Scholar]
- 25.Jellum E, Andersen A, Lund-Larsen P, et al. Experiences of the Janus Serum Bank in Norway. Environ Health Perspect 1995;103(suppl 3):85–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Euser SM, Sattar N, Witteman JC, et al. A prospective analysis of elevated fasting glucose levels and cognitive function in older people: results from PROSPER and the Rotterdam Study. Diabetes 2010;59:1601–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peila R, Rodriguez BL, White LR, Launer LJ. Fasting insulin and incident dementia in an elderly population of Japanese-American men. Neurology 2004;63:228–233. [DOI] [PubMed] [Google Scholar]
- 28.Rönnemaa E, Zethelius B, Sundelöf J, et al. Impaired insulin secretion increases the risk of Alzheimer disease. Neurology 2008;71:1065–1071. [DOI] [PubMed] [Google Scholar]
- 29.Rönnemaa E, Zethelius B, Sundelöf J, et al. Glucose metabolism and the risk of Alzheimer's disease and dementia: a population-based 12 year follow-up study in 71-year-old men. Diabetologia 2009;52:1504–1510. [DOI] [PubMed] [Google Scholar]
- 30.Yaffe K, Blackwell T, Kanaya AM, et al. Diabetes, impaired fasting glucose, and development of cognitive impairment in older women. Neurology 2004;63:658–663. [DOI] [PubMed] [Google Scholar]
- 31.Luchsinger JA, Tang MX, Shea S, Mayeux R. Hyperinsulinemia and risk of Alzheimer disease. Neurology 2004;63:1187–1192. [DOI] [PubMed] [Google Scholar]
- 32.Kuusisto J, Koivisto K, Mykkänen L, et al. Association between features of the insulin resistance syndrome and Alzheimer's disease independently of apolipoprotein E4 phenotype: cross sectional population based study. BMJ 1997;315:1045–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akomolafe A, Beiser A, Meigs JB, et al. Diabetes mellitus and risk of developing Alzheimer disease: results from the Framingham Study. Arch Neurol 2006;63:1551–1555. [DOI] [PubMed] [Google Scholar]
- 34.Craft S, Asthana S, Schellenberg G, et al. Insulin effects on glucose metabolism, memory, and plasma amyloid precursor protein in Alzheimer's disease differ according to apolipoprotein-E genotype. Ann NY Acad Sci 2000;903:222–228. [DOI] [PubMed] [Google Scholar]
- 35.Reger MA, Watson GS, Frey WH 2nd, et al. Effects of intranasal insulin on cognition in memory-impaired older adults: modulation by APOE genotype. Neurobiol Aging 2006;27:451–458. [DOI] [PubMed] [Google Scholar]
- 36.Peila R, Rodriguez BL, Launer LJ. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: the Honolulu-Asia Aging Study. Diabetes 2002;51:1256–1262. [DOI] [PubMed] [Google Scholar]
- 37.Xu WL, Qiu CX, Wahlin A, Winblad B, Fratiglioni L. Diabetes mellitus and risk of dementia in the Kungsholmen project: a 6-year follow-up study. Neurology 2004;63:1181–1186. [DOI] [PubMed] [Google Scholar]
- 38.Xu W, Qiu C, Winblad B, Fratiglioni L. The effect of borderline diabetes on the risk of dementia and Alzheimer's disease. Diabetes 2007;56:211–216. [DOI] [PubMed] [Google Scholar]