

Alzheimer’s disease (AD) is a neurodegenerative disorder and the leading cause of senile dementia. The brains of AD patients show two major pathological hallmarks: amyloid or senile plaques (SPs) and neurofibrillary tangles (NFTs). While NFTs are bundles of protein filaments found in the cytoplasm of neurons, SPs are extracellular aggregates of insoluble protein fibrils. The major component of SPs is a small, hydrophobic peptide termed amyloid β peptide (Aβ), which is derived from a ubiquitous type I transmembrane protein of unknown function named amyloid precursor protein (APP) (1).
AD genes in Aβ production
Generation of Aβ from APP occurs in both secretory and endocytic compartments by regulated intramembrane proteolysis (RIP) (2), a sequential, two-step cleavage of transmembrane proteins with the second cleavage occurring within the transmembrane domain. In the case of APP, RIP is initiated by β-site APP-cleaving enzyme (BACE) (3), an aspartyl proteinase that cleaves APP’s ectodomain and liberates the NH2-terminus of Aβ. Aβ generation is completed by intramembrane cleavage of APP, which requires presenilin-1 (PS1) (4), an unusual aspartic proteinase with eight transmembrane domains. This cleavage can occur at slightly different positions, resulting in two principal forms of Aβ, Aβ40 and Aβ42, with 40 and 42 amino acid residues respectively. Once formed, the Aβ is released outside the cell. While Aβ40 constitutes about 90% of the total Aβ generated, the slightly longer Aβ42 has a higher tendency to form fibrils. Since all known genetic risk factors for AD impact Aβ metabolism, it is believed that the accumulation of Aβ fibrils into amyloid plaques plays a key role in the onset and/or progression of the disease.
As with most degenerative diseases, no single genetic defect is responsible for all cases of AD. To date, six proteins have been genetically linked to AD (Table 1). Mutations in APP (5), PS1 (6), and PS2 (7) are associated with early-onset forms of familial AD. They are transmitted as an autosomal dominant trait and lead to increased production and deposition of Aβ. The ε4 allele of the apoE gene constitutes a major risk factor for late-onset AD (8). AD patients homozygous for the ε4 allele develop the disease about 20 years earlier than patients carrying copies of the other apoE alleles (ε2 or ε3) (8). ApoE associates with lipoprotein particles and facilitates their interaction with lipoprotein receptors.
Table 1.
Genetic factors associated with Alzheimer’s disease

In neurons, the major apoE receptor is the LDL receptor–related protein (LRP), a large endocytic receptor that regulates proteinase and lipoprotein levels by mediating their catabolism. Interestingly, a silent polymorphism in exon 3 of the LRP gene (C776T) is associated with an altered risk for late-onset AD (9). AD patients carry the C allele at a higher frequency than the general population, but since the C-to-T variation does not alter the protein, it has not been clear what effect this polymorphism might have on LRP function. Finally, a polymorphism in another LRP ligand, α2-macroglobulin (α2M), appears to be associated with increased risk for late-onset AD (10). α2M is a circulating proteinase inhibitor that can neutralize proteinases from all four classes. In this process, α2M becomes activated to form α2M*, which can be recognized by LRP.
It is worth noting that, in spite of their diverse nature, all of the molecules thus far linked to AD have two common features. First, they all modulate (directly or indirectly) the metabolism of Aβ. Second, they are all linked to LRP, either as ligands (APP, apoE, α2M*) or as molecules that can affect its expression (PS1, and probably PS2) (11).
Clearance of Aβ from the brain
Despite the fact that Aβ is produced as a normal consequence of APP metabolism (12), Aβ fibrils do not accumulate in large quantities in healthy individuals. This indicates the existence of clearance mechanisms and suggests that Aβ deposition is the net result of a balance between its production and catabolism. Aβ clearance occurs by at least three pathways: extracellular proteolysis, transport across the blood–brain barrier (BBB), and receptor-mediated endocytosis. Two metalloproteinases, insulin-degrading enzyme (13) and a neutral endopeptidase similar or identical to neprilysin (14), have been implicated in the extracellular degradation of Aβ. Infusion of neutral endopeptidase inhibitors in the rat brain results in abnormal deposition of endogenous Aβ (14), highlighting the importance of extracellular degradative pathways in clearing Aβ. Aβ transport across the BBB is less well understood, but gp330/megalin and its ligand apoJ may be involved. ApoJ, the predominant Aβ-binding protein in cerebrospinal fluid, mediates binding of Aβ to gp330 (15). Furthermore, transport of apoJ-Aβ complexes across the BBB appears to be inhibited by anti-gp330 antibodies (16).
Aβ can also be removed by binding to endocytic receptors, probably including the class A and class B scavenger receptors (17), which can bind to and internalize fibrillar forms of Aβ. On the other hand, Aβ can form complexes with LRP ligands such as apoE (18), lactoferrin (19), and α2M* (19), which can then be internalized via LRP. In the current issue of the JCI, Kang et al. (20) address the relevance of LRP-mediated Aβ clearance in AD. First, the authors confirm that addition of α2M* to cell culture medium significantly enhances LRP-mediated uptake of Aβ. More importantly, they shed light on how the silent C776T LRP polymorphism may be involved in AD. The authors find that AD patients with the C/C genotype have significantly lower levels of LRP antigen in their brains, compared with patients with the C/T or T/T genotype. The lower brain LRP levels in the C/C genotype correlate with increased Aβ plasma levels, and a higher number of SPs. Kang et al. (20) also find that the brains of AD patients have significantly lower LRP antigen levels than that of age-matched controls. Moreover, in normal subjects brain LRP levels decrease substantially with age, the major risk factor for nonfamilial AD.
This study (20) is significant in that it provides supporting evidence, although circumstantial, that LRP modulates Aβ deposition by increasing its clearance. If true, this is indeed a surprising finding for several reasons. First, Aβ can be cleared through several pathways, and it is not apparent that a single one would dominate. Second, Aβ does not directly bind LRP; rather, it must first bind to an LRP ligand (e.g., apoE or α2M*) before LRP-mediated clearance can occur. α2M* is present in very low concentrations and is generated only in response to wound repair or inflammatory events. These low levels of α2M* are not likely to saturate LRP-mediated clearance mechanisms.
These findings add to the large body of data suggesting that LRP is involved in the pathobiology of AD. LRP serves as a receptor for APP, apoE, and α2M, all of which have been genetically linked to AD. Moreover, LRP expression seems to be affected by PS1 (11), the major genetic factor associated with familial AD. While in the normal brain LRP expression is mostly restricted to neurons, LRP levels are upregulated in reactive astrocytes and activated microglia (21), both of which are associated with mature amyloid plaques. In addition, many LRP ligands and LRP itself occur in SPs. LRP has also been shown to bind the longer isoforms of APP (APP751 and APP770) (22), which contain a Kunitz-type proteinase inhibitor (KPI) domain, here referred to as APPKPI, and are the most abundant forms of APP in brain. Recently, Ulery et al. (23) found that long-term culture of cells in the presence of the LRP antagonist receptor-associated protein (RAP) significantly reduces Aβ synthesis, while restoring LRP function in LRP-deficient cells substantially increases the amount of Aβ generated. Together, these data indicate that LRP modulates APPKPI processing, leading to increased Aβ production.
While these latter experiments appear to conflict with the results of Kang and colleagues (20), they suggest that LRP influences both the clearance and the production of Aβ and thus plays a dual role in AD pathology (Figure 1). Increased expression of LRP in activated glia in the AD brain is well documented and could lead to increased Aβ production in these cells. At the same time, decreased LRP expression at clearance sites (perhaps neurons or sites along the capillary membranes) could lead to decreased α2M* and/or apoE-promoted Aβ catabolism, resulting in increased Aβ deposition. It is important to test these models in the future. Since deletion of the RAP gene in mice leads to a reduction in LRP levels in brain (24), it should be possible to cross these mice with APP transgenic mice to test the effect of LRP levels on Aβ deposition. While the studies of Kang et al. (20) measured total LRP antigen from extracts of the midfrontal cortex, it is important to determine whether this reduction is uniform, or specific to some brain regions. The mechanism by which the C/C genotype decreases LRP levels remains to be determined, as does the location within the brain where LRP-mediated Aβ clearance and production take place.
Figure 1.
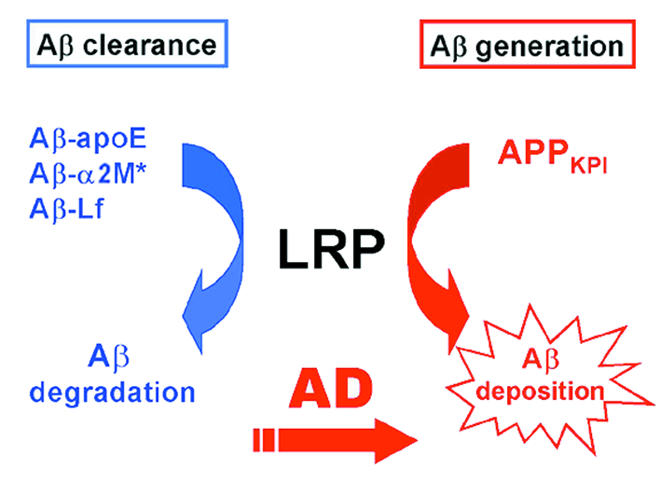
Proposed dual role of LRP in Aβ metabolism. Association of Aβ with LRP ligands (apoE, α2M*, or lactoferrin) results in rapid LRP-mediated endocytosis of the complex. On the other hand, association of APP isoforms containing KPI domains (APPKPI) leads to increased Aβ generation and deposition. In AD, the catabolic pathway may be impaired by decreased levels of LRP at clearance sites (perhaps neurons or sites along the capillary membranes), while Aβ generation may be enhanced by upregulation of LRP in activated glia found in the AD brain.
References
- 1.Tanzi RE, et al. The amyloid β protein gene: cDNA cloning, mRNA distribution, and genetic linkage near the Alzheimer locus. Science. 1987;235:880–994. doi: 10.1126/science.2949367. [DOI] [PubMed] [Google Scholar]
- 2.Brown MS, Ye J, Rawson RB, Goldstein JL. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell. 2000;100:391–398. doi: 10.1016/s0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- 3.Vassar R, et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 4.Li YM, et al. Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature. 2000;405:689–694. doi: 10.1038/35015085. [DOI] [PubMed] [Google Scholar]
- 5.St. George-Hyslop PH, et al. The genetic defect causing familial Alzheimer’s disease maps on chromosome 21. Science. 1987;235:885–890. doi: 10.1126/science.2880399. [DOI] [PubMed] [Google Scholar]
- 6.Sherrington R, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 7.The structure of the presenilin 1 (S182) gene and identification of six novel mutations in early onset AD families. Alzheimer’s Disease Collaborative Group. Nat Genet. 1995;11:219–222. doi: 10.1038/ng1095-219. [DOI] [PubMed] [Google Scholar]
- 8.Corder EH, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 9.Kang DE, et al. Genetic association of the low-density lipoprotein receptor- related protein gene (LRP), an apolipoprotein E receptor, with late-onset Alzheimer’s disease. Neurology. 1997;49:56–61. doi: 10.1212/wnl.49.1.56. [DOI] [PubMed] [Google Scholar]
- 10.Blacker D, et al. Alpha-2 macroglobulin is genetically associated with Alzheimer disease. Nat Genet. 1998;19:357–360. doi: 10.1038/1243. [DOI] [PubMed] [Google Scholar]
- 11.Van Uden E, et al. Aberrant presenilin-1 expression downregulates LDL receptor-related protein (LRP): is LRP central to Alzheimer’s disease pathogenesis? Mol Cell Neurosci. 1999;14:129–140. doi: 10.1006/mcne.1999.0772. [DOI] [PubMed] [Google Scholar]
- 12.Haass C, et al. Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- 13.Qiu WQ, et al. Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J Biol Chem. 1998;273:32730–32738. doi: 10.1074/jbc.273.49.32730. [DOI] [PubMed] [Google Scholar]
- 14.Iwata N, et al. Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med. 2000;6:143–150. doi: 10.1038/72237. [DOI] [PubMed] [Google Scholar]
- 15.Hammad SM, Ranganathan S, Loukinova E, Twal WO, Argraves WS. Interaction of apolipoprotein J-amyloid beta-peptide complex with low density lipoprotein receptor-related protein-2/megalin. A mechanism to prevent pathological accumulation of amyloid beta-peptide. J Biol Chem. 1997;272:18644–18649. doi: 10.1074/jbc.272.30.18644. [DOI] [PubMed] [Google Scholar]
- 16.Zlokovic BV, et al. Glycoprotein 330/megalin: probable role in receptor-mediated transport of apolipoprotein J alone and in a complex with Alzheimer disease amyloid beta at the blood-brain and blood-cerebrospinal fluid barriers. Proc Natl Acad Sci USA. 1996;93:4229–4234. doi: 10.1073/pnas.93.9.4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer’s disease amyloid beta-protein via a scavenger receptor. Neuron. 1996;17:553–565. doi: 10.1016/s0896-6273(00)80187-7. [DOI] [PubMed] [Google Scholar]
- 18.Yang DS, et al. Apolipoprotein E promotes the binding and uptake of beta-amyloid into Chinese hamster ovary cells in an isoform-specific manner. Neuroscience. 1999;90:1217–1226. doi: 10.1016/s0306-4522(98)00561-2. [DOI] [PubMed] [Google Scholar]
- 19.Qiu Z, Strickland DK, Hyman BT, Rebeck GW. Alpha2-macroglobulin enhances the clearance of endogenous soluble beta-amyloid peptide via low-density lipoprotein receptor-related protein in cortical neurons. J Neurochem. 1999;73:1393–1398. doi: 10.1046/j.1471-4159.1999.0731393.x. [DOI] [PubMed] [Google Scholar]
- 20.Kang DE, et al. Modulation of amyloid β-protein clearance and Alzheimer’s disease susceptibility by the LDL receptor–related protein pathway. J Clin Invest. 2000;106:1159–1166. doi: 10.1172/JCI11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rebeck GW, Reiter JS, Strickland DK, Hyman BT. Apolipoprotein E in sporadic Alzheimer’s disease: allelic variation and receptor interactions. Neuron. 1993;11:575–580. doi: 10.1016/0896-6273(93)90070-8. [DOI] [PubMed] [Google Scholar]
- 22.Kounnas MZ, et al. LDL receptor-related protein, a multifunctional apoE receptor, binds secreted β-amyloid precursor protein and mediates its degradation. Cell. 1995;82:331–340. doi: 10.1016/0092-8674(95)90320-8. [DOI] [PubMed] [Google Scholar]
- 23.Ulery PG, et al. Modulation of beta-amyloid precursor protein processing by the low density lipoprotein receptor-related protein (LRP). Evidence that LRP contributes to the pathogenesis of Alzheimer’s disease. J Biol Chem. 2000;275:7410–7415. doi: 10.1074/jbc.275.10.7410. [DOI] [PubMed] [Google Scholar]
- 24.Willnow TE, Armstrong SA, Hammer RE, Herz J. Functional expression of low density lipoprotein receptor-related protein is controlled by receptor-associated protein in vivo. Proc Natl Acad Sci USA. 1995;92:4537–4541. doi: 10.1073/pnas.92.10.4537. [DOI] [PMC free article] [PubMed] [Google Scholar]