

Abstract
There is great concern that one mild traumatic brain injury (mTBI) predisposes individuals to an exacerbated response with a subsequent mTBI. Although no mechanism has been identified, mounting evidence suggests traumatic axonal injury (TAI) plays a role in this process. By using a cell culture system, a threshold of mild TAI was found where dynamic stretch of cortical axons at strains lower than 5% induced no overt pathological changes. However, the axons were found to display an increased expression of sodium channels (NaChs) by 24 hr. After a second, identical mild injury, pathologic increases in [Ca2+]i were observed, leading to axon degeneration. The central role of NaChs in this response was demonstrated by blocking NaChs with tetrodotoxin prior to the second injury, which completely abolished postinjury increases in [Ca2+]i. These data suggest that mild TAI induces a form of sodium channelopathy on axons that greatly exaggerates the pathophysiologic response to subsequent mild injuries.
Keywords: axon trauma, calcium, diffuse axonal injury, repetitive injury, sodium channels, traumatic brain injury
Mild traumatic brain injury (mTBI), also referred to as “concussion,” affects over 1 million individuals in the United States annually. Despite the name, these injuries are by no means mild, with approximately 15% of mTBI patients suffering persistent cognitive dysfunction (National Center for Injury Prevention Control, 2003). Moreover, after subsequent or “repetitive” mTBI, cognitive deficits become far more common and overt. Several studies have demonstrated that athletes with two or more concussions frequently have measurable long-term cognitive deficits, such as delayed speed of processing and memory dysfunction (Collins et al., 1999; Iverson et al., 2004; Moser et al., 2005; Wall et al., 2006; Covassin et al., 2008). Likewise, several studies using animal models have demonstrated a worsened outcome with repetitive TBI (Laurer et al., 2001; Uryu et al., 2002; Raghupathi et al., 2004; Yoshiyama et al., 2005; Huh et al., 2007). However, it is unknown whether this is due simply to a cumulative effect of repeat injuries or if an initial mTBI triggers a physiological change that predisposes individuals to an exacerbated outcome from a subsequent mTBI.
Although it has previously been thought that there were no significant pathological changes associated with mTBI, mounting evidence suggests that mTBI patients suffer from a form of diffuse axonal injury (DAI). DAI has been shown to be responsible for the immediate loss of consciousness after TBI and has been implicated in the classic postconcussive syndrome (Adams et al., 1982, 1989; Graham et al., 1988; Povlishock, 1992; Smith and Meaney, 2000). Moreover, several recent studies using advanced neuroimaging techniques have identified selective white matter abnormalities in mTBI patients consistent with DAI (Inglese et al., 2005; Bazarian et al., 2007; Wilde et al., 2008). Accordingly, it is believed that traumatic axonal injury plays an important role in repetitive mTBI.
Here we utilized an in vitro system to examine the effects of repetitive mild traumatic axonal injury. This model induces dynamic stretch injury of isolated axons spanning two populations of cortical neurons using mechanical loading conditions that occur to axons during real-world TBI.
MATERIALS AND METHODS
Cell Culture
Custom-designed culture wells were prepared as previously described in detail (Smith et al., 1999b; Iwata et al., 2004). Briefly, a deformable substrate (Specialty Manufacturing, Saginaw, MI) was placed over the bottom of steel wells (see Fig. 1). A 2- × 16-mm silicone barrier was placed in the center of the membrane to create a 2-mm “gap” after neuron plating. Rat primary neurons from embryonic day 17 Sprague Dawley rats were plated at 375,000 cells/cm2 in the wells. The cells were maintained with Neurobasal media and B-27 neural supplement (Invitrogen, Gaithersburg, MD), 5% fetal bovine serum (Hyclone, Logan, UT), and 2% L-glutamine (Sigma, St. Louis, MO) and allowed to attach for 24 hr before the barrier was removed. After barrier removal, axons began traversing the gap, forming synapses with neurons on the other side. The experiments were performed at 12 days in vitro (DIV).
Fig. 1.

Top: Axon stretch injury model. Two populations of cortical neurons are spanned by an axon-only gap on a deformable substrate. An air pulse deforms the gap, causing tensile elongation of the axons that can be viewed with a microscope. Bottom: Photomicrographs of axon fascicles showing changes in calcium concentration (calcium fluorescence). Images are representative of intracellular calcium level both before and after injury at each level of strain (strain applies only to the “after-injury” condition). The strain threshold at which no morphologic changes (undulations denoted by arrows) or increases in calcium fluorescence (brighter colors) were induced was found to be below 5%. Scale bar = 20 μm. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Axonal Stretch Injury
As in previous studies, we used a specially designed axon stretch injury apparatus (see Fig. 1) and technique (Smith et al., 1999b; Iwata et al., 2004). This method closely mimics the mechanical loading conditions of DAI in humans by using dynamic uniaxial stretch or “tensile elongation” of axons to induce injury at adjustable strains and strain rates. Stretch injury was induced by placing the culture wells in an air-tight chamber with a stainless-steel bottom plate. A 2- × 18-mm slit in the plate is aligned with the axon-only region on the deformable substrate. The introduction of compressed air into the chamber was used to induce stretch to only the axons traversing the gap by deflecting the membrane downward. The pulse was gated by a solenoid (Parker General Valve, Elyria, OH), and a dynamic pressure transducer (model EPX-V01-25P-/16F-RF; Entran, Fairfield, NJ) recorded the data. The rate at which this strain was applied to the axons was between 20 and 35 sec−1, well within the range for traumatic injury experienced by the human brain during rotational acceleration. Measurement of nominal uniaxial strain (ε) was calculated by determining the center-line membrane deflection (δ) relative to the slit width (w) and substituting into the geometric relationship (Smith et al., 1999b):
Analysis of [Ca2+]i After Injury: Threshold Determination
Intraaxonal Ca2+ levels were determined as previously described in detail (Wolf et al., 2001; Iwata et al., 2004). Briefly, the cells were loaded with 2 μM fluo-4 ester (Invitrogen) solubilized in DMSO (0.05% final) with pluronic F-127 [0.004% (w/v) final] in CSS (120 mM NaCl, 5.4 mM KCl, 0.8 mM MgCl2, 1.8 mM CaCl2, 15 mM glucose, and 25 mM HEPES, pH 7.4, adjusted to 220 mOsm with sorbitol), in which all experiments were run (Takahashi et al., 1999). Because of the small diameter and volume of the axons, fluo-4 AM, an analog of the widely used fluo-3 AM, was used to achieve the maximum fluorescence after binding Ca2+. Fluo-4 is superior in this system because of its increased fluorescence excitation at 488 nm compared with fluo-3 AM. The ion dissociation constant Kd (Ca2+) is reported to be similar for the two dyes under identical conditions (fluo-3, 325 nM; fluo-4, 345 nM; manufacturer’s specifications). In addition, we evaluated fascicles of 5–10 μm to improve total fluorescence. The dye solution was loaded at 37°C for 30 min, after which the cells were rinsed with CSS, allowed to sit for another 30 min, and rinsed again before injury. Pluronic F-127 (Invitrogen) was used to disperse the dye further in the CSS and allow greater access to the cytoplasm. F-127 is a nonionic surfactant used to solubilize hydrophobic dyes such as fluo-3 and fura-2, which are commonly used for cell loading in in vitro studies. Given that F-127 was used at such a low concentration and that the wells were washed several times prior to injury, it is unlikely that it had an effect on membrane permeability or stability. The ionophore 4-bromo-A23187 at 50 μM (Invitrogen) served as a positive control for dye response to Ca2+ influx and was used under all treatment conditions to ensure that proper dye loading had occurred and that the treatments had not altered the Ca2+ affinity of the dye. Fluorescence microscopy was performed on a Nikon inverted microscope equipped with a Cooke Sensicam QE High Performance digital camera (Optical Apparatus, Ardmore, PA). The dye was excited at 488 nm, and the emitted fluorescence was collected at 515 nm. Images were taken at 1 sec intervals for 10 sec, at which time the injury was induced. For the 2-min threshold experiments, sampling continued at 1-sec intervals after injury. Analysis of changes in fluorescence of the fluo-4 dye was performed on five representative axons from each culture and a total of six cultures per condition or injury level (n = 6). Three random regions from each axon were analyzed and then averaged. To account for potential variation in dye loading among axons or experiments, we used a standard procedure for nonratioable indicators in which self-ratios were taken (F/F0) between the measured fluorescence (F) and the initial fluorescence (F0). Background fluorescence subtraction was accomplished by continuously sampling three areas in the field that had no axons in them for the duration of the experiment. The mean of these values was obtained and subtracted from the raw value obtained at each analyzed region of the axon before analysis.
Modulation of [Ca2+]i Changes After Injury
Based on previous studies (Wolf et al., 2001; Iwata et al., 2004), posttraumatic increases in [Ca2+]i could be completely abolished by preinjury treatment with the sodium channel (NaCh) blocker tetrodotoxin (TTX). Here, injuries were performed using either CSS (n = 6 wells; no treatment) or TTX treatment (n = 6; 1 μM, 10 min pre-second injury treatment). To compare experimental groups, mean values for three areas of each axon analyzed in the culture were then averaged to obtain a mean value for each experiment (as described above). (F/F0) values/experimental groups were evaluated by a one-way ANOVA with a post hoc Newman-Keuls multiple-comparisons test.
NaCh Immunocytochemical Analysis
The cultures were fixed in 4% paraformaldehyde (in 0.1 M PBS, pH 7.4) for 60 min at specified time points and permeabilized. Sham-injured control wells (placed in the injury apparatus without injury) were fixed 20 min after sham injury (n = 3 wells/group). Injured cultures were fixed at 24 hr post-single injury (n = 3 wells/group), the time point prior to a second injury. Cultures were then incubated overnight at 4°C with polyclonal rabbit anti-pan NaCh antibody (06-811; 1:200; Upstate Biotechnology, Lake Placid, NY) specific for the intracellular III-IV loop of the NaCh α-subunit, rinsed with PBS, and incubated for 60 min with Alexa 488-conjugated anti-rabbit IgG (Invitrogen). Although quantitative examinations cannot be performed with this technique, qualitative assessment was performed by determining the relative fluorescence of 10 randomly selected fascicles in each (minus back-ground fluorescence) in ImageJ software and then averaged.
NaCh Western Blot Analysis
The gap regions of isolated axons in the wells were examined by Western blot analysis of either control axons (no injury) or axons 24 hr postinjury (the time point prior to a second injury). The wells were placed on dry ice, and the gap region (2 × 16 mm area of injury) was excised with a scalpel. Notably, there was an anticipated very low yield of protein from this region, necessitating the pooling of material from eight wells for each examination. The dissected tissue was placed in RIPA buffer with 3× Halt Protease Inhibitor Cocktail (Pierce Biotechnology, Rockford, IL). The samples were sonicated and centrifuged at 10,000g at 4°C for 10 min. The supernatant from each was run on a 7% Tris-acetate gel and transferred to an Immobilon-FL PVDF membrane (Millipore Corporation, Billerica, MA) for fluorescent evaluation or an Immobilon-P PVDF membrane for chemiluminescent evaluation. For fluorescent evaluation, anti-PAN monoclonal antibody (Sigma) specific to the III-IV linker domain of the a-subunit of the NaCh was applied at 1:500 overnight at 4°C. Anti-mouse secondary (IRDye 680; LI-COR Biosciences, Lincoln, NE) was applied at 1:5,000 for 1 hr. The blot was analyzed on the LI-COR Odyssey Infrared Imaging System. For chemiluminescent evaluation, anti-PAN polyclonal antibody (Alomone Labs, Jerusalem, Israel) was applied at 1:500 overnight at 4°C. Goat anti-rabbit HRP-conjugated antibody was then applied at 1:125,000 for 1 hr, followed by SuperSignal West Dura Extended Substrate (Pierce Biotechnology). For both blots, optical density was measured in ImageJ. These data were analyzed by using an unpaired t-test.
RESULTS
Threshold for [Ca2+]i Increase and Morphological Changes After Single Injury
Fluo-4 analysis of injured axons indicated that a 5% strain was the minimum level of injury necessary to induce calcium influx. At 3% strain (“mild” subthreshold injury), Fluo-4 fluorescence remained at baseline level (F/F0 = 0.77). At strains ≥5% (suprathreshold injury), Fluo-4 fluorescence increased significantly over baseline levels (P < 0.001). Undulations typical of axon injury were not observed in subthreshold injuries but were present in all suprathreshold populations (Fig. 1).
Changes in [Ca2+]i After Repetitive Mild Injury
When axonal injury below the threshold of 5% strain was repeated 24 hr later, we found a significant increase in [Ca2+]i (F/F0 = 1.92, P < 0.001) as well as morphological changes such as minor undulations and swellings of the axons (Fig. 2). The level of [Ca2+]i following repetitive subthreshold injury was similar to that of a strain level of single injury over four times greater (Fig. 2). TTX treatment prior to the second injury prevented changes in [Ca2+]i, which remained the same as in noninjured axons (F/F0 = 0.93, ns; Fig. 2). This finding indicates that the posttraumatic increase in intraaxonal calcium was dependent on sodium entering through the TTX-sensitive NaChs.
Fig. 2.
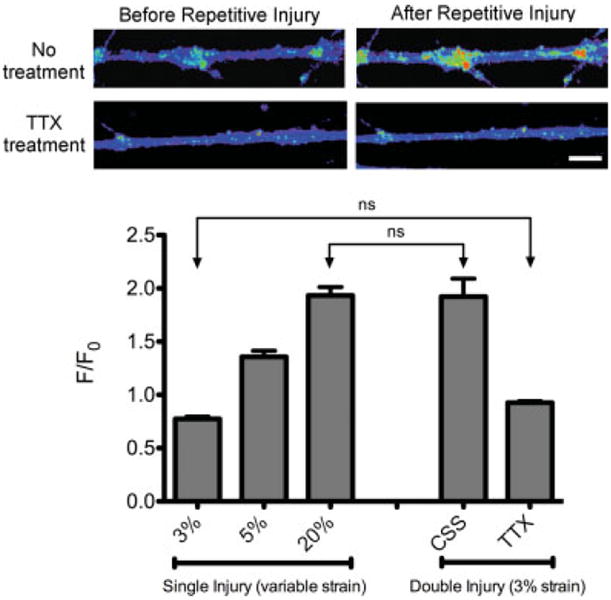
Changes in calcium influx after stretch injury. Top: A second mild injury 24 hr after the first insult resulted in an increase in intraaxonal calcium. TTX treatment prior to the second injury completely abolished postinjury increases of [Ca2+]i. The intensity of the fluorescent calcium dye was translated into a color spectrum. Bottom: Quantification of changes in intraaxonal Ca2+ fluorescence for single injury at 3% strain (subthreshold), 5% strain (above threshold), and 20% strain, along with double injuries at 3% strain with and without TTX treatment. F/F0 = change in Ca2+ fluorescence over initial fluorescence. Values reported are mean ± SEM. A one-way ANOVA was performed to compare the means of all groups. All comparisons were statistically significant (P < 0.001) except for those labeled “ns” (P > 0.05). Scale bar = 20 μm.
Sodium Channel Immunocytochemistry
Qualitative examination showed a substantial increase in immunoreactivity to NaChs for axon fascicles at 24 hr following a subthreshold injury (Fig. 3). These overt changes were consistent in all wells, with no overlap in the extent of fluorescence between wells with no injury and those examined 24 hr after injury.
Fig. 3.

Representative qualitative increases in NaCh immunoreactivity 24 hr after mild axon stretch injury were observed on axon fascicles (A,B). These changes were confirmed by quantitative Western blot examination (C), with which a 28% change was found. Values reported as mean ± SEM, *P < 0.03. Scale bar = 20 μm.
Sodium Channel Western Blot Analysis
Western blot analysis with two additional antibodies on separate blots corroborated the immunocytochemical findings (Fig. 3). The blots revealed a band of protein at 220 kD, the molecular weight of NaChs. The average optical density measurement for the injured axons increased 28% over that of noninjured axons.
DISCUSSION
In the present study, we have identified a mechanism whereby a single mTBI triggers a substantially worsened response following a repeated mild injury 24 hr later. After an initial mild injury, otherwise normal-appearing axons were found to display an increased expression of NaChs by 24 hr. After a second, identical mild injury, pathologic increases in [Ca2+]i were observed, leading to axon degeneration. The central role of NaChs in this response was demonstrated by treatment with TTX prior to the second injury, which completely abolished postinjury increases in [Ca2+]i. These in vitro data suggest that mild axonal trauma induces a form of sodium channelopathy on axons that can greatly exacerbate the pathophysiologic response to subsequent injuries. These observations may have important implications for repetitive mTBI, in which axonal injury and dysfunction are thought to be important neuropathological features.
Although it is often assumed that one mTBI exacerbates outcome from a second mTBI, there has been scant evidence. For TBI patients, it is difficult to gauge the actual levels of successive injuries in order to conclude whether there was an exacerbated response. However, there have been controversial claims of a “second-impact syndrome” for individuals displaying devastating brain swelling after a second TBI occurring days or weeks after the first injury. In addition, terms such as “sports concussion” have been used in anecdotal accounts of a “period of vulnerability,” when one mild injury is thought to predispose an individual to greatly exaggerated responses, such as a loss of consciousness and cognitive dysfunction. However, an actual vulnerability after mTBI and potential mechanisms involved have not been elucidated.
Repetitive TBI has been explored in animal models, in which the injury levels can be measured and controlled. In a mouse model of TBI, a potential “vulnerable state” was found when a second injury was induced between 3 and 5 days following the first TBI, leading to worsened cognitive outcome and axonal pathology (Laurer et al., 2001). An increase in axonal pathology after repetitive TBI has also been observed in a pig model (Raghupathi et al., 2004), and enhanced calpain activation in axons has been found in a rat model (Huh et al., 2007). Additionally, in a transgenic “Alzheimer’s disease” mouse, repetitive TBI was found to accelerate deposition of Aβ peptides, cerebral atrophy, and cognitive deficits, potentially through formation of Aβ in damaged axons (Smith et al., 1999a; Uryu et al., 2002; Yoshiyama et al., 2005). However, as with clinical observations, these experimental data could not distinguish whether the worsened outcomes after repeat exposure to TBI were a cumulative effect or reflected a mechanism that actually exacerbated outcome. Nonetheless, it is notable that a common finding with the animal models was enhanced axonal pathology.
The classical histopathologic definition of DAI is the formation of axonal swellings throughout the white matter resulting from posttraumatic interruption of axonal transport. However, it is widely believed that swollen axons represent only a small fraction of the overall pathologic changes and that many otherwise “normal” appearing axons are rendered dysfunctional. DAI has been shown to be responsible for the immediate loss of consciousness after injury and has been implicated in the classic postconcussive syndrome. This functional link between mTBI and DAI suggests a potential mechanism that leads to vulnerability with a repeat injury. Specifically, phenotypic or physiologic changes of the injured axons would likely influence outcome following repetitive mTBI. Our present in vitro results reveal how such a mechanism can lower the injury threshold for mildly injured axons.
We found that dynamic stretch of isolated axons below 5% strain at strain rates of 20 sec−1 did not induce changes in axon morphology (e.g., undulations), increases in [Ca2+]i, swelling, or degeneration. Notably, these parameters are also below the boundary predicted for injury resulting from rapid brain deformation in TBI based on computational and physical models (Zhang et al., 2004). Although no overt changes were observed, immunocytochemical and Western blot analyses revealed an increase in NaCh immunoreactivity. One day after the first stretch injury, a second, identical mild axon stretch injury was applied. In this case, however, substantial increases in [Ca2+]i were observed. This increase was found to be equivalent to [Ca2+]i increases induced from a relatively high level of a single stretch injury at 20% strain, fully five- to six-fold the strain that was actually applied. Despite the mild strain used for the second injury, the increases in [Ca2+]i induced axon degeneration within hours. These data indicate an induced vulnerability, whereby a single mild injury can condition axons to display a substantially exaggerated response after subsequent mild injuries.
The connection between a change in axonal NaChs after one mild injury and an exacerbated response with a second mild injury was demonstrated by NaCh blockade with TTX. Specifically, TTX treatment prior to the second injury completely abolished postinjury increases of [Ca2+]i, in turn blocking degeneration. The mechanism linking NaCh dysregulation with increases in [Ca2+]i has previously been elucidated examining single axon stretch injury at high levels of strain (Wolf et al., 2001). Dynamic mechanical deformation of axons was shown to induce Na+ influx through NaChs, triggering pathologic Ca2+ influx via the activation of voltage-sensitive calcium channels as well as the reversal of sodium-calcium exchangers. The resulting progressive increases in [Ca2+]i were found to fuel further pathologic changes, including proteolysis and degeneration (Iwata et al., 2004). Thus, traumatic axonal injury can induce a unique “feed-forward,” deleterious process of NaCh dysregulation and Ca2+ influx that leads to degeneration.
Our current findings present evidence that the posttraumatic increase in intraaxonal calcium was dependent on sodium entering through the TTX-sensitive NaChs. However, in addition to an indirect effect of NaCh dysregulation on Ca2+ influx following the second mild trauma, it is also possible that Ca2+ entered directly through NaChs, as has been shown following heightened NaCh stimulation (Lev-Ram et al., 1992; Rosenbluth et al., 2009).
Although an initial mild axon stretch injury in the present study did not elicit detectable increases in [Ca2+]i, it appears to have been sufficient to induce a form of sodium channelopathy. This channelopathy, characterized by an abnormally increased expression of NaChs, may provide a mechanistic basis of induced vulnerability after a mild injury. Potentially, an abnormally increased density of NaChs is a liability to axons undergoing dynamic deformation and results in an exaggerated influx of cations. This phenotypic change may essentially lower the injury strain threshold where axons are pushed into the deleterious feed-forward process. As such, even though a second injury may be mild in terms of strain, it could nonetheless induce NaCh dysregulation, leading to increases in [Ca2+]i and degeneration.
It is unclear why sodium channelopathy would develop on axons after a single mild insult. It may be that axonal NaChs are sensitive to even mild mechanical deformations, which could change their functional capacity and reduce conduction. For example, NaChs on mildly injured axons may become inactivated. If so, these axons may undergo compensatory changes akin to processes proposed for the developing nervous system and axon regeneration. Indeed, remodeling of growing axons during development is accompanied by hyperexpression of NaChs, which is thought to play a compensatory role in the maintenance of normal NaCh conduction (Lupa et al., 1993; Pfister et al., 2006). We found an increase in NaCh expression on axons at 24 hr posttrauma, demonstrating that this phenotypic change was relatively rapid. However, this potentially compensatory change appears to have come at a price, by predisposing the axons to a greatly exacerbated response following subsequent mild injuries.
Sodium channelopathy of various forms has previously been implicated in other nervous system disorders. Changes in the expression pattern of NaCh isoforms have been found in association with swollen and degenerating axons in multiple sclerosis lesions (Craner et al., 2004; Waxman, 2006). Many conditions such as epilepsy, chronic pain, neurodegenerative diseases, and spasticity are also linked with an abnormally increased activity or noninactivation of sodium channels (Baker and Wood, 2001; Rhodes et al., 2004; Tarnawa et al., 2007).
Overall, the present data demonstrate that axons can be conditioned by one mild traumatic insult to respond in an exaggreated fashion to a second mild injury and that this response appears to be accompanied by the development of a form of sodium channelopathy following the first injury. Given that mounting evidence implicates axonal injury as a major anatomical substrate for mTBI, the results of this study provide an important first step in identifying a mechanism responsible for the exacerbated outcome seen in mTBI.
Acknowledgments
The authors thank Catherine von Reyn, Theresa Lusardi, and Min Tang-Schomer for their excellent technical assistance and insight.
Contract grant sponsor: National Institutes of Health; Contract grant numbers: NS38104 and NS38104 (to Douglas H. Smith).
References
- Adams JH, Graham DI, Murray LS, Scott G. Diffuse axonal injury due to nonmissile head injury in humans: an analysis of 45 cases. Ann Neurol. 1982;12:557–563. doi: 10.1002/ana.410120610. [DOI] [PubMed] [Google Scholar]
- Adams JH, Doyle D, Ford I, Gennarelli TA, Graham DI, McLellan DR. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology. 1989;15:49–59. doi: 10.1111/j.1365-2559.1989.tb03040.x. [DOI] [PubMed] [Google Scholar]
- Baker MD, Wood JN. Involvement of Na+ channels in pain pathways. Trends Pharmacol Sci. 2001;22:27–31. doi: 10.1016/s0165-6147(00)01585-6. [DOI] [PubMed] [Google Scholar]
- Bazarian JJ, Zhong J, Blyth B, Zhu T, Kavcic V, Peterson D. Diffusion tensor imaging detects clinically important axonal damage after mild traumatic brain injury: a pilot study. J Neurotrauma. 2007;24:1447–1459. doi: 10.1089/neu.2007.0241. [DOI] [PubMed] [Google Scholar]
- Collins MW, Grindel SH, Lovell MR, Dede DE, Moser DJ, Phalin BR, Nogle S, Wasik M, Cordry D, Daugherty KM, Sears SF, Nicolette G, Indelicato P, McKeag DB. Relationship between concussion and neuropsychological performance in college football players. JAMA. 1999;282:964–970. doi: 10.1001/jama.282.10.964. [DOI] [PubMed] [Google Scholar]
- Covassin T, Stearne D, Elbin R. Concussion history and postconcussion neurocognitive performance and symptoms in collegiate athletes. J Athl Train. 2008;43:119–124. doi: 10.4085/1062-6050-43.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craner MJ, Newcombe J, Black JA, Hartle C, Cuzner ML, Waxman SG. Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc Natl Acad Sci USA. 2004;101:8168–8173. doi: 10.1073/pnas.0402765101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham DI, Adams JH, Gennarelli TA. Mechanisms of non-penetrating head injury. Prog Clin Biol Res. 1988;264:159–168. [PubMed] [Google Scholar]
- Huh JW, Widing AG, Raghupathi R. Basic science; repetitive mild non-contusive brain trauma in immature rats exacerbates traumatic axonal injury and axonal calpain activation: a preliminary report. J Neurotrauma. 2007;24:15–27. doi: 10.1089/neu.2006.0072. [DOI] [PubMed] [Google Scholar]
- Inglese M, Makani S, Johnson G, Cohen BA, Silver JA, Gonen O, Grossman RI. Diffuse axonal injury in mild traumatic brain injury: a diffusion tensor imaging study. J Neurosurg. 2005;103:298–303. doi: 10.3171/jns.2005.103.2.0298. [DOI] [PubMed] [Google Scholar]
- Iverson GL, Gaetz M, Lovell MR, Collins MW. Cumulative effects of concussion in amateur athletes. Brain Inj. 2004;18:433–443. doi: 10.1080/02699050310001617352. [DOI] [PubMed] [Google Scholar]
- Iwata A, Stys PK, Wolf JA, Chen XH, Taylor AG, Meaney DF, Smith DH. Traumatic axonal injury induces proteolytic cleavage of the voltage-gated sodium channels modulated by tetrodotoxin and protease inhibitors. J Neurosci. 2004;24:4605–4613. doi: 10.1523/JNEUROSCI.0515-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurer HL, Bareyre FM, Lee VM, Trojanowski JQ, Longhi L, Hoover R, Saatman KE, Raghupathi R, Hoshino S, Grady MS, McIntosh TK. Mild head injury increasing the brain’s vulnerability to a second concussive impact. J Neurosurg. 2001;95:859–870. doi: 10.3171/jns.2001.95.5.0859. [DOI] [PubMed] [Google Scholar]
- Lev-Ram V, Miyakawa H, Lasser-Ross N, Ross WN. Calcium transients in cerebellar Purkinje neurons evoked by intracellular stimulation. J Neurophysiol. 1992;68:1167–1177. doi: 10.1152/jn.1992.68.4.1167. [DOI] [PubMed] [Google Scholar]
- Lupa MT, Krzemien DM, Schaller KL, Caldwell JH. Aggregation of sodium channels during development and maturation of the neuromuscular junction. J Neurosci. 1993;13:1326–1336. doi: 10.1523/JNEUROSCI.13-03-01326.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser RS, Schatz P, Jordan BD. Prolonged effects of concussion in high school athletes. Neurosurgery. 2005;57:300–306. doi: 10.1227/01.neu.0000166663.98616.e4. discussion 300–306. [DOI] [PubMed] [Google Scholar]
- National Center for Injury Prevention Control. Report to Congress on Mild Traumatic Brain Injury in the United States: Steps to Prevent a Serious Public Health Problem. Atlanta: Centers for Disease Control and Prevention; 2003. [Google Scholar]
- Pfister BJ, Bonislawski DP, Smith DH, Cohen AS. Stretch-grown axons retain the ability to transmit active electrical signals. FEBS Lett. 2006;580:3525–3531. doi: 10.1016/j.febslet.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povlishock JT. Traumatically induced axonal injury: pathogenesis and pathobiological implications. Brain Pathol. 1992;2:1–12. [PubMed] [Google Scholar]
- Raghupathi R, Mehr MF, Helfaer MA, Margulies SS. Traumatic axonal injury is exacerbated following repetitive closed head injury in the neonatal pig. J Neurotrauma. 2004;21:307–316. doi: 10.1089/089771504322972095. [DOI] [PubMed] [Google Scholar]
- Rhodes TH, Lossin C, Vanoye CG, Wang DW, George AL., Jr Noninactivating voltage-gated sodium channels in severe myoclonic epilepsy of infancy. Proc Natl Acad Sci USA. 2004;101:11147–11152. doi: 10.1073/pnas.0402482101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluth J, Schiff R, Lam P. Effects of osmolality on PLP-null myelin structure: implications re axon damage. Brain Res. 2009;1253:191–197. doi: 10.1016/j.brainres.2008.11.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DH, Meaney DF. Axonal damage in traumatic brain injury. Neuroscientist. 2000;6:483–495. [Google Scholar]
- Smith DH, Chen XH, Nonaka M, Trojanowski JQ, Lee VM, Saatman KE, Leoni MJ, Xu BN, Wolf JA, Meaney DF. Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J Neuropathol Exp Neurol. 1999a;58:982–992. doi: 10.1097/00005072-199909000-00008. [DOI] [PubMed] [Google Scholar]
- Smith DH, Wolf JA, Lusardi TA, Lee VM, Meaney DF. High tolerance and delayed elastic response of cultured axons to dynamic stretch injury. J Neurosci. 1999b;19:4263–4269. doi: 10.1523/JNEUROSCI.19-11-04263.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi A, Camacho P, Lechleiter JD, Herman B. Measurement of intracellular calcium. Physiol Rev. 1999;79:1089–1125. doi: 10.1152/physrev.1999.79.4.1089. [DOI] [PubMed] [Google Scholar]
- Tarnawa I, Bolcskei H, Kocsis P. Blockers of voltage-gated sodium channels for the treatment of central nervous system diseases. Rec Patents CNS Drug Discov. 2007;2:57–78. doi: 10.2174/157488907779561754. [DOI] [PubMed] [Google Scholar]
- Uryu K, Laurer H, McIntosh T, Pratico D, Martinez D, Leight S, Lee VM, Trojanowski JQ. Repetitive mild brain trauma accelerates Abeta deposition, lipid peroxidation, and cognitive impairment in a transgenic mouse model of Alzheimer amyloidosis. J Neurosci. 2002;22:446–454. doi: 10.1523/JNEUROSCI.22-02-00446.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall SE, Williams WH, Cartwright-Hatton S, Kelly TP, Murray J, Murray M, Owen A, Turner M. Neuropsychological dysfunction following repeat concussions in jockeys. J Neurol Neurosurg Psychiatry. 2006;77:518–520. doi: 10.1136/jnnp.2004.061044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG. Axonal conduction and injury in multiple sclerosis: the role of sodium channels. Nat Rev Neurosci. 2006;7:932–941. doi: 10.1038/nrn2023. [DOI] [PubMed] [Google Scholar]
- Wilde EA, McCauley SR, Hunter JV, Bigler ED, Chu Z, Wang ZJ, Hanten GR, Troyanskaya M, Yallampalli R, Li X, Chia J, Levin HS. Diffusion tensor imaging of acute mild traumatic brain injury in adolescents. Neurology. 2008;70:948–955. doi: 10.1212/01.wnl.0000305961.68029.54. [DOI] [PubMed] [Google Scholar]
- Wolf JA, Stys PK, Lusardi T, Meaney D, Smith DH. Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J Neurosci. 2001;21:1923–1930. doi: 10.1523/JNEUROSCI.21-06-01923.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Uryu K, Higuchi M, Longhi L, Hoover R, Fujimoto S, McIntosh T, Lee VM, Trojanowski JQ. Enhanced neurofibrillary tangle formation, cerebral atrophy, and cognitive deficits induced by repetitive mild brain injury in a transgenic tauopathy mouse model. J Neurotrauma. 2005;22:1134–1141. doi: 10.1089/neu.2005.22.1134. [DOI] [PubMed] [Google Scholar]
- Zhang L, Yang KH, King AI. A proposed injury threshold for mild traumatic brain injury. J Biomech Eng. 2004;126:226–236. doi: 10.1115/1.1691446. [DOI] [PubMed] [Google Scholar]