

Abstract
Traumatic brain injury (TBI) is a risk factor for developing Alzheimer's disease (AD). Additionally, TBI induces AD‐like amyloid β (Aβ) plaque pathology within days of injury potentially resulting from massive accumulation of amyloid precursor protein (APP) in damaged axons. Here, progression of Aβ accumulation was examined using brain tissue from 23 cases with post‐TBI survival of up to 3 years. Even years after injury, widespread axonal pathology was consistently observed and was accompanied by intra‐axonal co‐accumulations of APP with its cleavage enzymes, beta‐site APP cleaving enzyme and presenilin‐1 and their product, Aβ. However, in marked contrast to the plaque pathology noted in short‐term cases post TBI, virtually no Aβ plaques were found in long‐term survivors. A potential mechanism for Aβ plaque regression was suggested by the post‐injury accumulation of an Aβ degrading enzyme, neprilysin. These findings fail to support the premise that progressive plaque pathology after TBI ultimately results in AD.
Keywords: amyloid‐precursor protein, BACE, beta‐amyloid, diffuse axonal injury, dystrophic neurites, human, kinesin, neprilysin, PS‐1, traumatic brain injury
INTRODUCTION
In humans, traumatic brain injury (TBI) has been shown to induce neuropathological changes similar to those found in Alzheimer's disease (AD). In particular, plaques composed of amyloid beta (Aβ), a hallmark pathology of AD, have been found deposited throughout the brain in about 30% of fatal TBI cases 20, 37, 44. However, in contrast to AD, extensive plaque formation after TBI can occur within days, even in young individuals (37). In addition, several studies have shown that a history of TBI can increase the risk of developing AD 18, 28, 29, 31, 33, 36, 41. Thus, it has been suggested that progressive Aβ plaque pathology after TBI may play a role in the epidemiological link with AD.
Axonal injury, a very common feature of TBI 3, 43, may provide a mechanism for rapid Aβ plaque formation. Disruption of axonal transport after TBI results in the rapid and massive accumulation of amyloid precursor protein (APP), potentially providing ample substrate for Aβ production (14). Indeed, in a model of diffuse axonal injury (DAI) in the pig and subsequently in humans, Aβ plaques were found in association with extensive axonal accumulations of Aβ throughout the white matter within days after injury 44, 45. Aβ has also been found accumulating in damaged axons in rat models of TBI, although in the absence of Aβ plaques 21, 49. More recently in the pig model, APP was consistently found to co‐accumulate in axonal swellings with the enzymes required for Aβ formation, including β‐site APP cleaving enzyme (BACE) and presenilin‐1 (PS‐1) (9). Thus, after TBI there may be a unique mechanism of Aβ formation within the membrane compartment of damaged axons. Lysis or leakage of injured axons would release Aβ into the tissue, potentially contributing to plaque formation.
Intra‐axonal production of Aβ is not without precedent. Cleavage of APP by BACE and PS‐1 recently observed in human peripheral nerve axons 25, 26. It was further noted that APP, PS‐1 and BACE are actually transported together down the axon by kinesin (26). However, these findings are controversial and contradicted by another report (27). Nonetheless, a more recent study also suggests that interrupted axon transport may stimulate the proteolysis of APP, leading to the development of Aβ plaques 47, 48.
It remains unknown how axonal pathology and associated Aβ accumulation evolves over time following TBI. In particular, it is unclear whether Aβ accumulation is a self‐limiting, acute‐phase response or whether it persistently accumulates over a more protracted time course, possibly leading to AD. Furthermore, potential catabolic mechanisms that might clear Aβ after TBI have not been explored. For example, a major endogenous Aβ degrading enzyme, neprilysin, which has been implicated in the pathogenesis of AD (40), has not been examined in TBI.
Here we used immunohistochemistry (IHC) to evaluate axonal pathology and accumulation of APP, PS‐1, BACE, Aβ, kinesin and neprilysin following TBI in humans. We examined tissue from cases who died acutely following injury as well as cases who survived long‐term, up to 3 years, post trauma.
MATERIALS AND METHODS
Brain tissue
Brain tissue from 23 cases (22 male and 1 female) was obtained at autopsy from fatal head injury cases treated at The Southern General Hospital, Glasgow, UK. Cases were aged between 16–79 years (mean 47 years). For comparisons between short‐ and long‐term changes, five cases were selected with survival times ranging from 19 to 120 h (mean 64 h) post injury, and 18 cases were selected with post‐injury survival times ranging from 27 days to 3 years (mean 245 days). Causes of injury included automobile crashes, falls and physical assault. Post‐mortem delays ranged from 12 h to 8 days (mean 64 h) (Table 1). Controls consisted of three cases (2 male, 1 female), aged 15–33 years (mean 23 years) who died from non‐neurological causes and with no known history of TBI. DAI was graded according to Adams et al (1). Hypoxic brain damage was assessed by Graham et al (16). Evidence of there having been raised intracranial pressure was determined according to Adams and Graham(2). Clinical and neuropathological information for each subject is listed in Table 1.
Table 1.
Clinical Characteristics of Cases
| Age (yrs) | Sex | Surv. | Cause of Injury | PM delay | Skull # | ICH | Contusions | DAI | HBD | Swelling | Herniation | Cause Death |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Acute Cases | ||||||||||||
| 16 | M | 48 h | RTA | 43 h | + | RSDH | + | 1 | Severe | B | + | Raised ICP |
| 59 | M | 96 h | Fall | 48 h | + | LEDH (e) | + | − | − | − | + | Raised ICP |
| 59 | F | 120 h | Fall | 144 h | + | LSDH | + | − | Mod | L | + | Raised ICP |
| 71 | M | 19 h | Fall | 27 h | + | BSDH | + | − | Mild | − | + | Raised ICP |
| 17 | M | 37 h | RTA | 72 h | − | − | + | 3 | Severe | B | + | Raised ICP |
| Long Term Cases | ||||||||||||
| 31 | M | 27 d | Fall | 4 d | + | LSDH (e) | + | 1 | − | − | Pneumonia | |
| 76 | M | 5 w | Fall | 47 h | + | RP | + | 1 | − | − | − | Pneumonia |
| 76 | M | 6 w | Assault | 32 h | + | RSDH (e) | + | 1 | Mod | − | + | Pneumonia |
| 50 | M | 7 w | NK | 90 h | − | RSDH (e) | + | 1 | Mod | − | + | Pneumonia |
| 18 | M | 4 w | Assault | 1 day | + | − | + | 2 | Mild | − | − | Pneumonia |
| 23 | M | 4 w | Assault. | 81 h | − | − | − | 2 | − | − | − | Pneumonia |
| 18 | M | 3 m | RTA | 64 h | + | − | + | 3 | − | − | + | Pneumonia |
| 79 | M | 4 m | RTA | 75 h | − | LSDH (e) | + | 1 | Mod | − | − | Cardiac Failure |
| 65 | M | 5 m | NK | 5 d | + | − | − | − | − | − | − | Pneumonia |
| 56 | M | 5 m | NK | 5 d | − | − | + | − | Mild | − | − | Pneumonia |
| 31 | M | 6 m | RTA | 3 d | − | − | + | 3 | − | − | − | Pneumonia |
| 39 | M | 6 m | RTA | 12 h | − | − | + | 3 | − | − | − | Pneumonia |
| 67 | M | 8 m | Fall | 3 d | + | RSDH (e) | + | 1 | Mod | − | + | Pneumonia |
| 28 | M | 9 m | RTA | 8 d | − | − | − | 3 | Mild | − | − | Pneumonia |
| 58 | M | 9 m | RTA | NK | − | − | − | 3 | − | − | − | Pneumonia |
| 64 | M | 18 m | Assault | 24 h | − | − | − | 3 | Mild | − | − | Pneumonia |
| 48 | M | 2 y | Fall | 5 d | + | LSDH (e) | + | − | Mild | − | − | Alcohol Abuse |
| 56 | M | 3 y | Assault | 20 h | − | − | + | 3 | − | − | − | Pneumonia |
| Controls | ||||||||||||
| 21 | F | N/A | N/A | 63 h | − | − | − | − | − | − | − | Septic Shock |
| 33 | M | N/A | N/A | 36 h | − | − | − | − | − | − | − | Acute Tracheobronchitis |
| 15 | M | N/A | N/A | 5 d | − | − | − | − | − | − | − | Aspiration pneumonia |
F = female; M = male; PMD = post‐mortem delay; # = fracture; Survl = survival time post‐trauma; R = right, L = left, B = bilateral; RTA = road traffic accident; SDH = subdural hematoma; HBD = hypoxic brain damage; DAI = traumatic axonal injury as graded by Adams et al (1989); ICH = intracerebral hematoma; EDH = Extradural Hematoma; (e) = evacuated; RICP = raised intracranial pressure; + = present; − = absent; NK = not known; N/A = not applicable.
All brain sections that were examined contained white matter tracts from the subcortex, deep white matter and corpus callosum.
Use of tissue for this study was approved by the Ethics Committee of the Southern General Hospital, South Glasgow University Hospitals NHS Trust.
Immunohistochemistry
IHC staining was performed on serial paraffin‐embedded sections. Single, double or triple labeled IHC was performed using a well‐characterized panel of primary antibodies (Table 2).
Table 2.
Summary of antibodies used for immunohistochemistry. Abbreviations: M = monoclonal; P = polyclonal; APP = amyloid precursor protein; Aβ = amyloid beta; NF = neurofilament.
| Antibody | Epitope protein/Amino acids | Type | Dilution | Company |
|---|---|---|---|---|
| 22C11 | APP/60‐100 | M | 1:40 | Chemicon |
| Karen | APP/N terminal | P | 1:800 | * |
| 6F/3D | Aβ/8‐17 | M | 1:50 | Dako |
| 4G8 | Aβ/17‐24 | M | 1:1000 | * |
| 13335 | Aβ/1‐42 | P | 1:1000 | * |
| AMY33 | Aβ/1‐28 | M | 1:500 | Zymed |
| BCO5 | Aβ‐42 C terminal | M | 1:10000 | * |
| BACE | β‐secretase Asp1 | P | 1:500 | Alpha |
| PS‐1 | C‐terminal presenilin | P | 1:200 | Zymed |
| L1 | Kinesin‐L | M | 1:200 | Chemicon |
| N52 | NF‐H | M | 1:400 | Sigma |
| CD10 | Neprilysin | M | 1:50 | Novocastra |
| NEP | Neprilysin | P | 1:500 | Chemicon |
| OX42 | Macrophage | M | 1:100 | Serotec |
| Macrophage | macrophage inflammatory protein | P | 1:100 | Abcam |
| SMI‐94 | Mylin basic protein | M | 1:1000 | Covance |
Antibody obtained courtesy of Dr V.M.‐Y. Lee
Neurofilament (NF) was detected using monoclonal antibody N52 at a dilution of 1:400 (Sigma, St Louis, MO, USA). APP was detected using monoclonal antibody 22C11 at a dilution of 1:40 (Chemicon, Temocular, CA, USA) and polyclonal antibody Karen (courtesy of Dr V.M.‐Y. Lee) at a dilution of 1:800. Amyloid beta peptide (Aβ) was detected using antibodies 4G8 (54) (courtesy of Dr V.M.‐Y. Lee), 13335 (30) (courtesy of Dr V.M.‐Y. Lee), 6F/3D (6) (Dako Corporation, Carpentaria, CA, USA), Amy33 (7) (Zymed, San Francisco, CA, USA) and BC05 (50) (courtesy of Dr V.M.‐Y. Lee) at dilutions of 1:1000, 1:1000, 1:50, 1:500, 1:10,000, respectively. Because of the potential cross‐reactivity of antibody 4G8 with both Aβ and APP, this antibody was used for analysis of plaques only, not Aβ in axons.
Beta‐site APP cleaving enzyme (BACE) was detected using polyclonal antibody BACE (Alpha Laboratories, Hampshire, UK) at dilution 1:500. Presenilin‐1 (PS‐1) was detected using polyclonal antibody PS‐1 (Zymed) at a dilution of 1:200. Kinesin was detected using monoclonal antibody L1 (Chemicon) at a dilution of 1:200. Neprilysin was detected using monoclonal antibody CD10 (Novocastra Laboratories, Newcastle Upon Tyne, UK) and polyclonal antibody NEP (Chemicon) at dilutions of 1:50 and 1:500, respectively. These anti‐neprilysin antibody dilutions were chosen so that no background staining could be detected in the control cases, that is, permitting interpretation of significant staining in trauma cases.
Macrophages/microglia were detected using antibody CD11b, at dilution1:100 (Serotec Inc., Raleigh, NC, USA) and rabbit polyclonal antibody to macrophage inflammatory protein 1 alpha at dilution 1:100 (Abcam Inc., Cambridge, MA, USA). Myelin basic protein was stained for using; SMI 94, at dilution 1:1000 (Covance: Berkeley, CA, USA).
Single‐label IHC examination
The sections were deparaffinized and endogenous peroxidase activity was quenched using 20% H2O2 (Sigma) in methanol (Fisher, Pittsburgh, PA, USA) for 30 minutes. Antigen retrieval was achieved by immersion in 88% formic acid (Sigma) for 5 minutes followed by boiling (using a microwave) for 5 minutes. Sections were then blocked by 2% normal horse serum (Sigma) in 0.1 M Tris, pH 7.4 for 30 minutes. The slides were incubated overnight with a primary monoclonal antibody or polyclonal antibody at 4°C and then incubated with the appropriate biotinylated secondary antibodies at room temperature for 1 h (ABC kit 1:1000, Vector Laboratories Inc., Burlingame, CA, USA). Visualization was achieved using the DAB peroxidase substrate kit (Vector). Antibodies were diluted in 0.1 M/L Tris buffer with 5% normal horse serum and 0.1% Triton X‐100, and tissue sections were washed with 0.1 M/L Tris buffer. Sections were counterstained with hematoxylin (1:20), dehydrated and coverslipped. In order to obtain a negative control, the above process was performed with omission of primary antibody.
Double or triple label examination
To examine the co‐accumulation of proteins within the tissue, sections were incubated using various combinations of primary antibodies (details on request).
Processing of each primary antibody on the sections was as described above. Incubations of fluorescent secondary antibodies were performed in a darkroom. The secondary antibodies were: fluorescein conjugated goat anti‐rabbit IgG (1:200, Molecular probes Inc., Eugene, OR, USA); Texas red conjugated horse anti‐mouse IgG (1:200, Vector laboratories Burlingame); fluorescein conjugated goat Fab fragment anti‐rabbit IgG (1:200, Jackson ImmunoReseach Laboratories Inc., West Grove, PA, USA), and AMCA donkey anti‐goat IgG (1:200, Jackson ImmunoReseach Laboratories). In general, IHC staining was performed sequentially; that is, one primary antibody was applied followed by its respective secondary antibody and the procedure repeated using secondary and tertiary staining sequences. As a negative control, each pair of primary antibodies was processed separately to confirm the lack of cross‐reactivity between the two or three secondary and primary antibodies.
All sections were examined with fluorescence microscopy using a Nikon Microphoto SA with a UFX‐DX camera system (Optical Apparatus, Ardmore, PA, USA) and appropriate filters. The images were captured using a Nikon Eclipse E600 with spot RT digital camera (Diagnostic Instruments Inc., Sterling Heights, MI, USA).
Luxol fast blue staining
Tissue was deparaffinized and hydrated to 95% alcohol. Luxol fast blue solution (Sigma) was applied overnight at 60°C. Following rinsing using distilled water, lithium carbonate solution was applied (5 minutes) followed by emersion in 70% alcohol (2 × 10s). Steps beyond deparaffinization and hydration were repeated until a sharp contrast between the blue of the white matter and colorless gray matter is evident. Following this, slides were rinsed in 70% alcohol and counterstained with eosin and cresyl violet.
Semi‐quantitative analysis
Observations were conducted blind to the demographic and clinical information for all cases. For each stain, two sections from each block of tissue were examined. Only immunoreactive profiles that could be clearly identified based on morphological characteristics (axonal bulbs and plaques) were counted in each section. Profiles were counted manually using light microscopy at ×200 magnifications over five fields (each field measuring 1.2 mm2) for each brain section. The average number of each profile type was then determined. The average number of profiles was categorized by applying the following ranked scale; minimal—fewer than 5 profiles (+); moderate—6 to 15 profiles (+ +) or extensive—greater than 15 profiles (+ + +).
RESULTS
A summary of all immunohistochemical findings are shown in Table 3.
Table 3.
Summary of Immunohistochemical Data
| Case No. | Survl. | PMD | Age | Axonal Bulbs | Soma | Aβ Plaques | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Aβ | APP | NF | BACE | PS1 | Kinesin | NEP | NEP | |||||
| Acute Cases | ||||||||||||
| 1 | 48 h | 43 h | 16 | ++ | +++ | +++ | ++ | ++ | + | ++ | + | ++ |
| 2 | 96 h | 48 h | 59 | + | + | ++ | + | + | + | + | − | ++ |
| 3 | 120 h | 144 h | 59 | − | + | + | + | + | + | − | − | − |
| 4 | 19 h | 27 h | 71 | − | + | + | + | + | + | − | − | − |
| 5 | 37 h | 72 h | 17 | ++ | +++ | +++ | ++ | + | ++ | ++ | + | ++ |
| Long‐Term cases | ||||||||||||
| 1 | 27 d | 96 h | 31 | + | + | + | + | − | + | ++ | +++ | − |
| 2 | 5 w | 47 h | 76 | + | + | + | − | − | + | + | + | − |
| 3 | 6 w | 32 h | 76 | + | + | + | − | − | + | − | + | − |
| 4 | 7 w | 90 h | 50 | ++ | ++ | ++ | + | + | + | ++ | +++ | − |
| 5 | 4 w | 24 h | 18 | ++ | ++ | ++ | + | + | + | ++ | − | − |
| 6 | 4 w | 81 h | 23 | ++ | ++ | ++ | + | + | + | ++ | +++ | − |
| 7 | 12 w | 64 h | 18 | + | + | + | + | − | + | + | ++ | − |
| 8 | 16 w | 75 h | 79 | + | + | + | + | + | + | − | − | − |
| 9 | 5 m | 120 h | 65 | + | ++ | + | + | + | + | ++ | +++ | + |
| 10 | 5 m | 120 h | 56 | + | + | + | + | − | + | − | − | − |
| 11 | 6 m | 72 h | 31 | ++ | + | + | + | + | + | ++ | +++ | − |
| 12 | 6 m | 12 h | 39 | + | + | ++ | + | + | + | − | − | − |
| 13 | 8 m | 72 h | 67 | + | + | ++ | + | + | + | + | +++ | − |
| 14 | 9 m | 192 h | 28 | + | + | + | + | + | + | − | − | − |
| 15 | 9 m | NK | 58 | + | + | + | + | + | + | − | − | − |
| 16 | 18 m | 24 h | 64 | + | + | + | − | − | − | − | − | − |
| 17 | 2 yr | 120 h | 48 | + | + | + | + | + | + | − | ++ | − |
| 18 | 3 yr | 20 h | 56 | + | ++ | + | − | + | − | − | ++ | − |
| Control Cases | ||||||||||||
| C1 | n/a | NK | 21 | − | − | − | − | − | − | − | − | − |
| C2 | n/a | NK | 33 | − | − | − | − | − | − | − | − | − |
| C3 | n/a | 4 d | 15 | − | − | − | − | − | − | − | − | − |
+ = fewer than 5 profiles; ++ = 6 to 15 profiles; +++ = greater than 15 profiles; PMD = post‐mortem delay; Survl = survival time post‐trauma; PS1 = presenilin‐1; BACE = beta‐site APP cleaving enzyme; APP = amyloid precursor protein; Aβ = amyloid beta; NF = neurofilament; NEP = neprilysin; NK = Not known.
Axonal pathology
Axonal pathology was identified immunohistochemically using antibodies targeting NF (N52) and APP proteins (22C11 and Karen (9)), in accordance with previously published criteria for the detection of axonal pathology in long‐term survival cases 4, 5, 24.
Within the TBI group, axonal bulbs and axonal swellings were detected in all cases, both with acute and long‐term survival groups, using antibodies specific for both APP and NF (Figure 1). Most axonal bulbs had the classic appearance of discrete spherical profiles surrounded by a halo where the bulb has pulled away from the tissue. These bulb profiles were up to 40 µm in diameter, connected to the proximal portion of the axon which was comparatively normal in diameter. The morphology of the axonal bulbs was distinct from the elongated, varicose swellings often seen effecting non‐disconnected axon shafts. These varicosities at times spanned up to several hundred micrometers.
Figure 1.
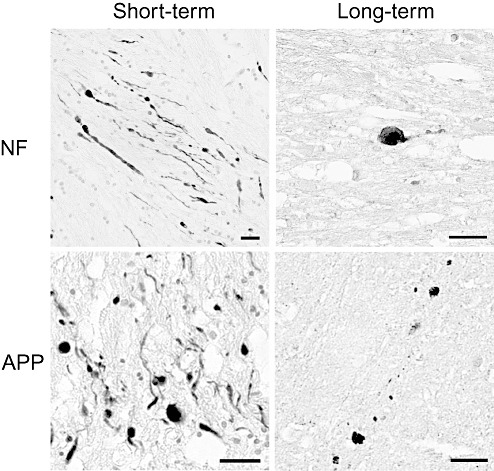
Axonal pathology. Representative immunoreactivity for neurofilament (NF) and amyloid precursor protein (APP) demonstrating axonal bulbs and varicosities in both short‐ and long‐term survival cases of traumatic brain injury. Scale bar = 50 µm.
Cases with a short‐term post‐injury survival time generally appeared to have more profiles indicative of axonal pathology than long‐term survivors (27 days to 3 years).
As was noted previously (46), no axonal injury, protein accumulation or Aβ plaque‐like profiles were detected by stains in any control cases.
BACE, PS‐1, kinesin and APP accumulation
Immunoreactivity demonstrating accumulation of BACE in swollen axonal profiles was established by specific antibody BACE (Table 2). BACE immunoreactivity was found in the axonal bulbs in virtually all head injury cases. Within the short‐term survival group, BACE staining was noted to be extensive. Cases with long‐term survival tended to have less abundant immunoreactivity although it was still detectable in axons in all but four cases (2, 3).
Figure 2.
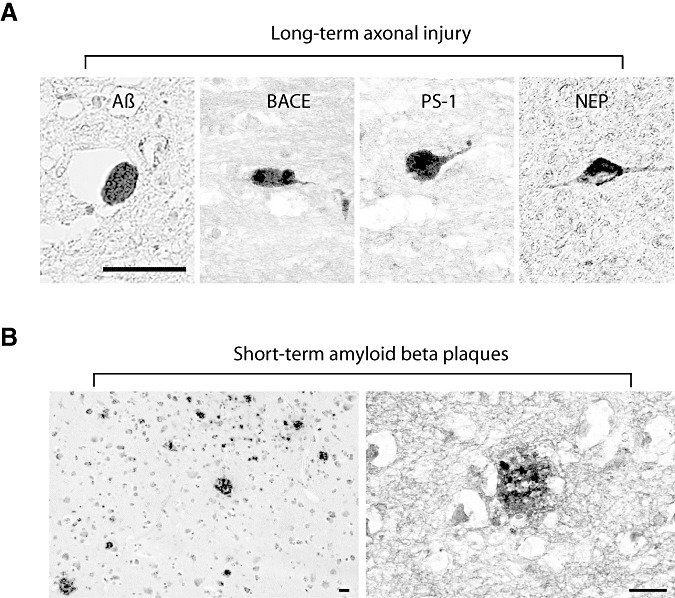
Long‐term intra‐axonal pathology and short‐term amyloid plaques. (A) Axonal bulb immunoreactivity for amyloid β (Aβ) (6F/3D) antibody, β‐site APP cleaving enzyme (BACE), presenilin‐1 (PS‐1) and neprilysin in long‐term survival cases. (B) Aβ plaques found in short‐term survival cases (4G8 antibody). Scale bar = 50 µm.
Figure 3.
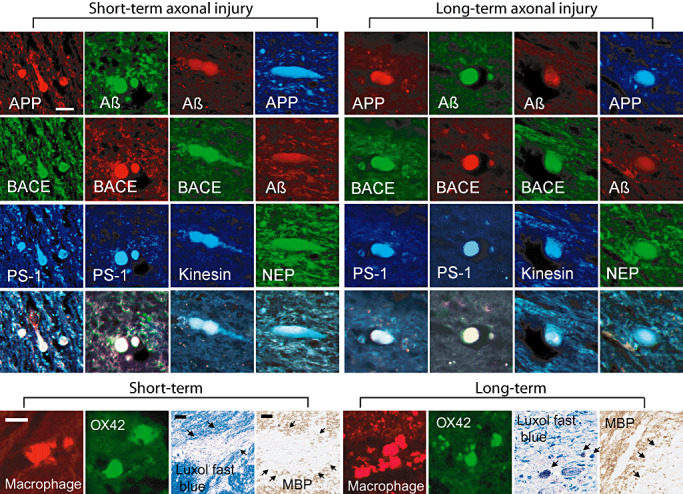
Axonal bulb accumulations, macrophages/microglia and myelin. Top panels: representative triple labeling of amyloid precursor protein (APP), amyloid β (Aβ), β‐site APP cleaving enzyme (BACE), presenilin‐1 (PS‐1), kinesin, neprilysin (NEP), and APP within both short‐ and long‐term survival cases. Immunoreactivity gives the typical appearance of these proteins accumulating within axonal bulbs in the subcortical white matter. 13335 and Amy33 antibodies demonstrate Aβ within axonal bulbs. Neprilysin can be found co‐localizing with APP and Aβ. Scale bar = 50 µm. Bottom Panels: Immunofluorescence demonstrated macrophages/ microglia in the brain tissue in short‐ and long‐term survival cases detected by antibodies OX42 and macrophage inflammatory protein. Luxol fast blue and immunostaining for myelin basic protein (SMI 94) revealed patchy loss of immunoreactivity in the subcortical white matter in both short‐ and long‐term survival patients. Scale bar = 50 µm.
Immunoreactivity for PS‐1 protein in swollen axonal profiles was detected using the specific antibody PS‐1. PS‐1 immunoreactivity was less prevalent than that of BACE with predominantly minimal immunoreactivity being detected in both groups. However, it was clear that PS‐1 immunoreactivity was elevated in cases surviving up to 3 years post TBI (2, 3).
Immunoreactivity for kinesin protein in swollen axonal profiles was identified by antibody specific to kinesin (L1). Kinesin immunoreactivity was demonstrated in almost all cases in axonal bulbs. In terms of the extent of immunoreactivity, both the long‐ and short‐term survival groups had moderate to extensive staining (Figure 3).
Using double or triple‐fluorescence IHC, we consistently found co‐immunoreactivity of combinations of APP or Aβ with BACE and PS‐1 in axonal profiles by using specific antibodies (Figure 3). This co‐immunoreactivity was seen in cases who died shortly after trauma as well as in those who survived long‐term (Figure 3).
Aβ accumulation
In total, a panel of five antibodies were used to detect various Aβ epitopes. Initially, for detection of plaque‐like profiles, 13335 was used in all sections from all cases. 4G8 was used on 11 sections from 11 cases to confirm the presence or absence of plaque pathology as identified by 13335. Staining for Aβ plaque pathology also revealed the presence of Aβ within axon bulbs. Thus, confirmatory detection of Aβ accumulation in damaged axons was accomplished using BC05, 6F/3D and Amy33; all of which are specific for Aβ and do not cross‐react with full‐length APP. BC05 was used in all cases, 6F/3D in 16 cases, and Amy33 in 11 cases. When compared, these antibodies revealed consistent immunoreactivity to Aβ.
Axonal swellings stained for Aβ had the typical appearance of axonal bulbs at the terminal ends of disconnected axons following TBI. Double immunostaining demonstrated co‐localization of Aβ with markers of axonal pathology (APP and NF) within some of the axonal bulbs. Although APP and Aβ co‐localization could be seen, this was not the case in all axon bulbs, with some bulbs staining with APP alone. Classically observed varicose swellings were demonstrated within non‐disconnected axons, yet Aβ accumulation was not detected within these sites despite the accumulation of other proteins including APP and NF. Aβ accumulation appeared to be restricted to the axonal bulbs. Notably, the multiple antibodies used, with their different epitope recognition, may suggest that multiple forms of Aβ accumulate in axon bulbs following trauma.
Within the short‐term survival group, Aβ accumulation was consistently observed within axonal bulbs in three of the five cases, as detected by anti‐Aβ antibodies. In addition to the intra‐axonal accumulation, three of five cases in the short‐term survival group also demonstrated plaque‐like profiles in the extracellular space, as shown by antibodies 13335 and 4G8 (Figure 2). Aβ containing plaque‐like profiles were found in both gray and white matter, often, but not exclusively, found close to immunoreactive axons.
The long‐term survival group also demonstrated accumulation of Aβ within axonal bulbs in virtually all cases, detected by anti‐Aβ antibodies (2, 3). However, within this group, only one case was found to have a minimal number of extracellular plaques.
Macrophage/microglia and mylein staining
Immunofluorescence and distinct morphologic characteristics demonstrated macrophages/microglia in the brain tissue in short‐ and long‐term survival patients detected by antibodies OX42 and macrophage inflammatory protein (Figure 3). Luxol fast blue and immunostaining for myelin basic protein (SMI 94) revealed patchy loss of immunoreactivity in the subcortical white matter in both short‐ and long‐term survival patients (Figure 3; scale bar = 50 µm).
Distribution of immunoreactivity for neprilysin
Within the short‐term survival TBI group, specific antibodies (CD10 and NEP used in all cases with consistent immunostaining) demonstrated immunoreactivity for neprilysin protein in axonal bulbs in the subcortical white matter in two of five cases. (2, 4). Notably, this staining demonstrated the classic appearance of axonal bulbs or varicosities surrounded by a halo caused by tissue dehydration.
Figure 4.
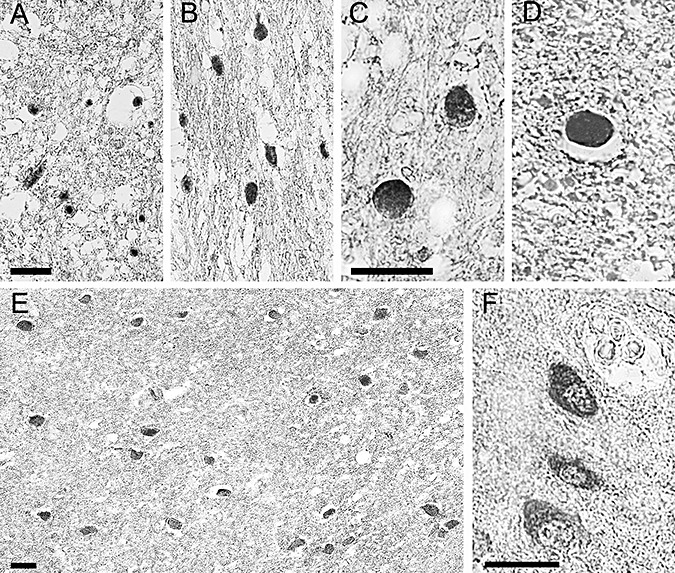
Neprilysin staining. Photomicrographs demonstrating immunoreactivity to neprilysin representative of positive cases found up to 8 months post injury. (A and B) Clusters of positively stained axons are seen within the subcortical white matter. Note the classic appearance of halos surrounding the swollen axon terminals. This phenomenon occurs secondary to dehydration of the tissue during processing. (C and D) Swollen axon bulbs seen at higher magnification. (E) Clusters of soma within the subcortical white matter demonstrating positive neprilysin staining. (F) Somal neprilysin staining seen at a higher magnification. Note the staining has a granulated quality. All scale bars = 50 µm.
Cases with a long‐term survival were positive for neprilysin immunoreactivity in axons in approximately half of the cases examined. However, it was not observed past 8 months survival (2, 4). Within the soma of these cases, neprilysin immunoreactivity was detected in the majority of cases (11 of 18), up to 3 years post TBI (Figure 4E and F). Staining of soma appeared almost granular in nature.
Following injury, neprilysin immunoreactivity was also observed in cells displaying the morphological features of macrophages/microglia, particularly within the short‐term survival group.
In control cases, no immunoreactivity for neprilysin protein was demonstrated in axons or soma as was predicted by setting dilutions for detection of a significant staining threshold beyond control cases.
DISCUSSION
This study provides the first evidence that axonal swelling and degeneration persists for years after TBI in humans. These axonal swellings were consistently found to contain accumulations of Aβ and the proteins necessary for its generation, including APP, BACE and PS‐1. Although Aβ plaques were found shortly after TBI as has been reported earlier 20, 37, 44, almost no Aβ plaques were found months to years after injury, despite apparent Aβ production in axons. This finding was a surprise considering that progressive and escalating plaque pathology after injury was anticipated. To the contrary, these data suggest that plaques may actually regress over time after TBI. If so, this process may be mediated via proteolysis of Aβ by neprilysin, for which immunoreactivity was also increased in injured brains. These collective data provide the first evidence that TBI in humans triggers progressive axonal degeneration in association with the anabolism and possible catabolism of Aβ for years after injury. However, the absence of Aβ plaques in long‐term survivors of TBI fails to provide a pathologic mechanism that might explain the epidemiological association between TBI and AD.
Axonal injury is one of the most frequently found, and arguably most important, pathological findings following TBI 3, 43. It is characterized by the presence of axonal swellings, either as discrete bulb formations or as elongated varicosities. As the axon undergoes cytoskeletal disorganization and progressive protein accumulation, secondary axotomy will eventually result with swollen axonal bulbs seen at the axon terminal 34, 43. The heterogeneous nature of TBI is such that axonal pathology may be attributed to not only the direct traumatic mechanical disruption of axons, but also potentially to secondary acute changes such as raised intracranial pressure or compromised vasculature and resultant hypoxia (12). However, a far greater number of cases than those evaluated in the present study would be necessary to determine the relative contribution of primary and secondary pathologic processes on the extent of axonal pathology and protein accumulation.
APP and NF (transported by fast and slow axonal transport, respectively) are useful markers of protein accumulation associated with axonal pathology 8, 15, 55. Using these markers, swollen and disconnected axons were found in virtually all the TBI cases examined. It is generally believed that this traumatic axonal pathology is a relatively acute feature of brain trauma, being completely cleared after a few months following injury (1). Here it is shown that axonal pathology actually persists for at least 3 years after injury. However, the degree of axonal pathology found within the long‐term survival group was generally less extensive than those cases who survived for a shorter duration, as has previously been shown in a pig model of DAI (9). Why axons should continue to swell and disconnect over such a protracted time course is not clear. The long duration and persistent nature of this pathology suggests that TBI can induce a progressive neurodegenerative process. Thus, there may be two phases of axon degeneration after TBI; (i) an immediate and reactive process of axons undergoing acute degeneration subsequent either to devastating mechanical disruption or secondary pressure/vascular complications; and (ii) a delayed process in axons that, despite remaining relatively intact shortly after injury, are primed to undergo degeneration even years later through unknown mechanisms. It may be that this is a form of wallerian degeneration that has not been described previously.
This chronic axon degeneration is also associated with a patchy loss of myelin staining, potentially related to short‐ and long‐term death of oligodendrocytes that has previously been observed after TBI in humans 42, 53. Additionally, there appears to a be long‐term inflammatory response after TBI, with the identification of macrophages/microglia in brain tissue even years after injury, as has previously been observed (13). However, it remains unknown whether these phagocytic cells are simply clearing debris resulting from ongoing axonal degeneration or are actively involved in the degenerative process (10).
A final step towards axon degeneration in TBI is failed axonal transport, a process that has recently gained momentum as a mechanism for several neurodegenerative diseases, including AD 39, 48, 51. The current data demonstrate that for TBI, the pathologic accumulation of Aβ caused by impaired axonal transport continues for at least 3 years following injury in humans. As previously shown in swine (9), long‐term axonal accumulation of Aβ in humans was accompanied by the co‐accumulation of proteins necessary for its generation, including APP, BACE and PS‐1. This is strongly suggestive that the generation of Aβ is intra‐axonal. Under normal conditions, APP, BACE and PS‐1 are transported as distinct cargoes in axons. Disruption of axonal transport may provide a unique environment whereby pathologic accumulations of these proteins interact resulting in the production of Aβ. Indeed, recent studies have described Aβ production mediated by BACE and PS‐1 in the axonal membrane compartment of peripheral nerves 25, 26. Although these observations are in debate (27), a more recent study has shown that interruption of axonal transport may stimulate the proteolysis of APP, leading to the development of Aβ plaques (48).
A potential intra‐axonal location of Aβ metabolism is unclear when considering that Aβ is conventionally thought to be processed via the cell membrane. However, there is accumulating evidence implicating lipid rafts as important sites of Aβ processing within neuron cell bodies 10, 19. Considering that axons are rich with lipid rafts, this may provide a mechanism for Aβ production completely within the axonal membrane compartment after TBI.
If damaged axons provide an environment conducive to Aβ formation and accumulation, subsequent lysis or degeneration of these axons could result in the expulsion of Aβ into the parenchymal tissue. Indeed, Aβ containing plaque‐like structures have been identified in brain injured tissue just hours after trauma, often close to damaged axons 17, 20, 37, 44. Additionally, elevated Aβ peptide has been identified in the cerebrospinal fluid of TBI cases 11, 35. Our data suggest that the anabolism of Aβ remains elevated in axons both acutely and up to 3 years after trauma. However, plaque formation appears to be minimal in those cases surviving long term.
Potential long‐term regression of Aβ plaques formed acutely after TBI may reflect a change in the balance between Aβ anabolism and catabolism. In terms of catabolism, neprilysin has emerged as a primary endogenous Aβ degrading enzyme (23) and has increasingly been implicated in the pathogenesis of AD (for review, see (22)). Neprilysin is a transmembrane glycoprotein whose C‐terminal catalytic site lies on the extracellular surface 38, 52. Here extensive neprilysin immunoreactivity was found in the soma of the majority of long‐term survival cases as well as being identifiable in swollen axonal bulbs. Interestingly, these cases comprise the same group that have a virtual absence of Aβ plaques. These findings may reflect a long‐term process involving an upregulation of neprilysin that continually clears both intracellular and of extracellular Aβ, thereby promoting plaque regression.
What stimulates neprilysin production is not well understood, although a recent study demonstrated that amyloid intracellular domain, a cytoplasmic fragment generated from PS‐1 dependant cleavage of Aβ, acts as a mediator of neprilysin expression at a transcriptional level (32). Thus the persistent generation of Aβ may trigger a feedback loop that results in its own clearance. However, it is unclear whether this process can occur in response to Aβ production within the axonal compartment.
If neprilysin is playing a role in Aβ clearance after trauma, variations in this mechanism may explain why some TBI patients form plaques while others do not. Further work is required to clarify the exact role neprilysin is playing under post‐traumatic conditions and how neprilysin effects Aβ accumulation, not only in relation to TBI, but also how it may contribute to the development of AD. It may be such that aberrations of neprilysin mediated Aβ catabolism combined with TBI as an environmental trigger produces a unique subset of TBI patients who go on to develop AD.
Overall, our data suggest a long‐term process of Aβ metabolism initiated by TBI. Ongoing axonal pathology appears to supply all of the essential elements for both the anabolism and catabolism of Aβ. However, the absence of Aβ plaques in the brains of long‐term survivors for up to 3 years after TBI does not support the premise that plaque pathology progressively fulminates over time, ultimately resulting in the clinical manifestations of AD. To the contrary, based on the present data, Aβ plaques formed in the initial weeks after injury may actually regress with time. Here, a continuously renewed supply of Aβ in degenerating axons may be kept in check through degradation by endogenous mediators such as neprilysin. Nonetheless, our data do not rule out the possibility that in some individuals with TBI, the balance between Aβ anabolism and catabolism eventually shifts during aging, accounting for the epidemiologic evidence of a link between TBI and AD.
ACKNOWLEDGMENTS
We thank the families of patients whose generosity made this research possible. We would also like to thank Dr David I. Graham (Academic Unit of Neuropathology, Division of Clinical Neuroscience, Institute of Neurological Sciences, Southern General Hospital NHS Trust, Glasgow, UK) for his generous efforts in providing access to the Glasgow Brain Archive, and Dr V.M.‐Y. Lee (The Center for Neurodegenerative Disease Research, University of Pennsylvania School of Medicine) for her generous contribution of antibodies. This work was supported by grants from the National Institutes of Health, including the National Institute of Neurological Disorders and Stroke (NS38104 and NS056202 to D.H.S.) and National Institute of Aging (AG10124 and AF11542 to J.Q.T.). J.Q.T. is the William Maul Measey‐Truman G Schnabel, Jr, Professor of Geriatric Medicine and Gerontology.
REFERENCES
- 1. Adams JH, Doyle D, Ford I, Gennarelli TA, Graham DI, McLellan DR (1989) Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology 15:49–59. [DOI] [PubMed] [Google Scholar]
- 2. Adams JH, Graham DI (1976) The relationship between ventricular fluid pressure and the neuropathology of raised intracranial pressure. Neuropathol Appl Neurobiol 2:323–332. [Google Scholar]
- 3. Adams JH, Graham DI, Gennarelli TA, Maxwell WL (1991) Diffuse axonal injury in non‐missile head injury. J Neurol Neurosurg Psychiatry 54:481–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Adams JH, Graham DI, Jennett B (2000) The neuropathology of the vegetative state after an acute brain insult. Brain 123:1327–1338. [DOI] [PubMed] [Google Scholar]
- 5. Adams JH, Graham DI, Jennett B (2001) The structural basis of moderate disability after traumatic brain damage. J Neurol Neurosurg Psychiatry 71:521–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Akiyama H, Schwab C, Kondo H, Mori H, Kametani F, Ikeda K, McGeer PL (1996) Granules in glial cells of patients with Alzheimer's disease are immunopositive for C‐terminal sequences of beta‐amyloid protein. Neurosci Lett 206:169–172. [DOI] [PubMed] [Google Scholar]
- 7. Bickel U, Lee VM, Trojanowski JQ, Pardridge WM (1994) Development and in vitro characterization of a cationized monoclonal antibody against beta A4 protein: a potential probe for Alzheimer's disease. Bioconjug Chem 5:119–125. [DOI] [PubMed] [Google Scholar]
- 8. Blumbergs PC, Scott G, Manavis J, Wainwright H, Simpson DA, McLean AJ (1995) Topography of axonal injury as defined by amyloid precursor protein and the sector scoring method in mild and severe closed head injury. J Neurotrauma 12:565–572. [DOI] [PubMed] [Google Scholar]
- 9. Chen XH, Siman R, Iwata A, Meaney DF, Trojanowski JQ, Smith DH (2004) Long‐term accumulation of amyloid‐beta, beta‐secretase, presenilin‐1, and caspase‐3 in damaged axons following brain trauma. Am J Pathol 165:357–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cordy JM, Hussain I, Dingwall C, Hooper NM, Turner AJ (2003) Exclusively targeting beta‐secretase to lipid rafts by GPI‐anchor addition up‐regulates beta‐site processing of the amyloid precursor protein. Proc Natl Acad Sci USA 100:11735–11740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Emmerling MR, Morganti‐Kossmann MC, Kossmann T, Stahel PF, Watson MD, Evans LM et al (2000) Traumatic brain injury elevates the Alzheimer's amyloid peptide a beta 42 in human CSF. A possible role for nerve cell injury. Ann N Y Acad Sci 903:118–122. [DOI] [PubMed] [Google Scholar]
- 12. Geddes JF, Whitwell HL, Graham DI (2000) Traumatic axonal injury: practical issues for diagnosis in medicolegal cases. Neuropathol Appl Neurobiol 26:105–116. [DOI] [PubMed] [Google Scholar]
- 13. Gentleman SM, Leclercq PD, Moyes L, Graham DI, Smith C, Griffin WS, Nicoll JA (2004) Long‐term intracerebral inflammatory response after traumatic brain injury. Forensic Sci Int 146:97–104. [DOI] [PubMed] [Google Scholar]
- 14. Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW (1993) Beta‐amyloid precursor protein (beta APP) as a marker for axonal injury after head injury. Neurosci Lett 160:139–144. [DOI] [PubMed] [Google Scholar]
- 15. Grady MS, McLaughlin MR, Christman CW, Valadka AB, Fligner CL, Povlishock JT (1993) The use of antibodies targeted against the neurofilament subunits for the detection of diffuse axonal injury in humans. J Neuropathol Exp Neurol 52:143–152. [DOI] [PubMed] [Google Scholar]
- 16. Graham DI, Ford I, Adams JH, Doyle D, Teasdale GM, Lawrence AE, McLellan DR (1989) Ischaemic brain damage is still common in fatal non‐missile head injury. J Neurol Neurosurg Psychiatry 52:346–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Graham DI, Gentleman SM, Lynch A, Roberts GW (1995) Distribution of beta‐amyloid protein in the brain following severe head injury. Neuropathol Appl Neurobiol 21:27–34. [DOI] [PubMed] [Google Scholar]
- 18. Guo Z, Cupples LA, Kurz A, Auerbach SH, Volicer L, Chui H et al (2000) Head injury and the risk of AD in the MIRAGE study. Neurology 54:1316–1323. [DOI] [PubMed] [Google Scholar]
- 19. Hama E, Shirotani K, Iwata N, Saido TC (2004) Effects of neprilysin chimeric proteins targeted to subcellular compartments on amyloid beta peptide clearance in primary neurons. J Biol Chem 279:30259–30264. [DOI] [PubMed] [Google Scholar]
- 20. Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VM et al (2004) Alzheimer's pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol 190:192–203. [DOI] [PubMed] [Google Scholar]
- 21. Iwata A, Chen XH, McIntosh TK, Browne KD, Smith DH (2002) Long‐term accumulation of amyloid‐beta in axons following brain trauma without persistent upregulation of amyloid precursor protein genes. J Neuropathol Exp Neurol 61:1056–1068. [DOI] [PubMed] [Google Scholar]
- 22. Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP et al (2001) Metabolic regulation of brain Abeta by neprilysin. Science 292:1550–1552. [DOI] [PubMed] [Google Scholar]
- 23. Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E et al (2000) Identification of the major Abeta1‐42‐degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med 6:143–150. [DOI] [PubMed] [Google Scholar]
- 24. Jennett B, Adams JH, Murray LS, Graham DI (2001) Neuropathology in vegetative and severely disabled patients after head injury. Neurology 56:486–490. [DOI] [PubMed] [Google Scholar]
- 25. Kamal A, Almenar‐Queralt A, LeBlanc JF, Roberts EA, Goldstein LS (2001) Kinesin‐mediated axonal transport of a membrane compartment containing beta‐secretase and presenilin‐1 requires APP. Nature 414:643–648. [DOI] [PubMed] [Google Scholar]
- 26. Kamal A, Stokin GB, Yang Z, Xia CH, Goldstein LS (2000) Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin‐I. Neuron 28:449–459. [DOI] [PubMed] [Google Scholar]
- 27. Lazarov O, Morfini GA, Lee EB, Farah MH, Szodorai A, DeBoer SR et al (2005) Axonal transport, amyloid precursor protein, kinesin‐1, and the processing apparatus: revisited. J Neurosci 25:2386–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lye TC, Shores EA (2000) Traumatic brain injury as a risk factor for Alzheimer's disease: a review. Neuropsychol Rev 10:115–129. [DOI] [PubMed] [Google Scholar]
- 29. Mortimer JA, French LR, Hutton JT, Schuman LM (1985) Head injury as a risk factor for Alzheimer's disease. Neurology 35:264–267. [DOI] [PubMed] [Google Scholar]
- 30. Murai H, Pierce JE, Raghupathi R, Smith DH, Saatman KE, Trojanowski JQ et al (1998) Twofold overexpression of human beta‐amyloid precursor proteins in transgenic mice does not affect the neuromotor, cognitive, or neurodegenerative sequelae following experimental brain injury. J Comp Neurol 392:428–438. [DOI] [PubMed] [Google Scholar]
- 31. Nemetz PN, Leibson C, Naessens JM, Beard M, Kokmen E, Annegers JF, Kurland LT (1999) Traumatic brain injury and time to onset of Alzheimer's disease: a population‐based study. Am J Epidemiol 149:32–40. [DOI] [PubMed] [Google Scholar]
- 32. Pardossi‐Piquard R, Petit A, Kawarai T, Sunyach C, Alves da Costa C, Vincent B et al (2005) Presenilin‐dependent transcriptional control of the Abeta‐degrading enzyme neprilysin by intracellular domains of betaAPP and APLP. Neuron 46:541–554. [DOI] [PubMed] [Google Scholar]
- 33. Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D et al (2000) Documented head injury in early adulthood and risk of Alzheimer's disease and other dementias. Neurology 55:1158–1166. [DOI] [PubMed] [Google Scholar]
- 34. Povlishock JT, Becker DP (1985) Fate of reactive axonal swellings induced by head injury. Lab Invest 52:540–552. [PubMed] [Google Scholar]
- 35. Raby CA, Morganti‐Kossmann MC, Kossmann T, Stahel PF, Watson MD, Evans LM et al (1998) Traumatic brain injury increases beta‐amyloid peptide 1‐42 in cerebrospinal fluid. J Neurochem 71:2505–2509. [DOI] [PubMed] [Google Scholar]
- 36. Rasmusson DX, Brandt J, Martin DB, Folstein MF (1995) Head injury as a risk factor in Alzheimer's disease. Brain Inj 9:213–219. [DOI] [PubMed] [Google Scholar]
- 37. Roberts GW, Gentleman SM, Lynch A, Murray L, Landon M, Graham DI (1994) Beta amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer's disease. J Neurol Neurosurg Psychiatry 57:419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roques BP, Noble F, Dauge V, Fournie‐Zaluski MC, Beaumont A (1993) Neutral endopeptidase 24.11: structure, inhibition, and experimental and clinical pharmacology. Pharmacol Rev 45:87–146. [PubMed] [Google Scholar]
- 39. Roy S, Zhang B, Lee VM, Trojanowski JQ (2005) Axonal transport defects: a common theme in neurodegenerative diseases. Acta Neuropathol (Berl) 109:5–13. [DOI] [PubMed] [Google Scholar]
- 40. Saido TC, Iwata N (2006) Metabolism of amyloid beta peptide and pathogenesis of Alzheimer's disease towards presymptomatic diagnosis, prevention and therapy. Neurosci Res 54:235–253. [DOI] [PubMed] [Google Scholar]
- 41. Schofield PW, Tang M, Marder K, Bell K, Dooneief G, Chun M et al (1997) Alzheimer's disease after remote head injury: an incidence study. J Neurol Neurosurg Psychiatry 62:119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shaw K, MacKinnon MA, Raghupathi R, Saatman KE, McLntosh TK, Graham DI (2001) TUNEL‐positive staining in white and grey matter after fatal head injury in man. Clin Neuropathol 20:106–112. [PubMed] [Google Scholar]
- 43. Smith DH (2000) Axonal damage in traumatic brain injury. Neuroscientist 6:483–495. [Google Scholar]
- 44. Smith DH, Chen XH, Iwata A, Graham DI (2003) Amyloid beta accumulation in axons after traumatic brain injury in humans. J Neurosurg 98:1072–1077. [DOI] [PubMed] [Google Scholar]
- 45. Smith DH, Chen XH, Nonaka M, Trojanowski JQ, Lee VM, Saatman KE et al (1999) Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J Neuropathol Exp Neurol 58:982–992. [DOI] [PubMed] [Google Scholar]
- 46. Smith DH, Uryu K, Saatman KE, Trojanowski JQ, McIntosh TK (2003) Protein accumulation in traumatic brain injury. Neuromolecular Med 4:59–72. [DOI] [PubMed] [Google Scholar]
- 47. Stokin GB, Goldstein LS (2006) Axonal transport and Alzheimer's disease. Annu Rev Biochem 75:607–627. [DOI] [PubMed] [Google Scholar]
- 48. Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL et al (2005) Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science 307:1282–1288. [DOI] [PubMed] [Google Scholar]
- 49. Stone JR, Okonkwo DO, Singleton RH, Mutlu LK, Helm GA, Povlishock JT (2002) Caspase‐3‐mediated cleavage of amyloid precursor protein and formation of amyloid Beta peptide in traumatic axonal injury. J Neurotrauma 19:601–614. [DOI] [PubMed] [Google Scholar]
- 50. Suzuki N, Cheung TT, Cai XD, Odaka A, Otvos L Jr, Eckman C et al (1994) An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science 264:1336–1340. [DOI] [PubMed] [Google Scholar]
- 51. Terwel D, Dewachter I, Van Leuven F (2002) Axonal transport, tau protein, and neurodegeneration in Alzheimer's disease. Neuromolecular Med 2:151–165. [DOI] [PubMed] [Google Scholar]
- 52. Turner AJ, Isaac RE, Coates D (2001) The neprilysin (NEP) family of zinc metalloendopeptidases: genomics and function. Bioessays 23:261–269. [DOI] [PubMed] [Google Scholar]
- 53. Williams S, Raghupathi R, MacKinnon MA, McIntosh TK, Saatman KE, Graham DI (2001) In situ DNA fragmentation occurs in white matter up to 12 months after head injury in man. Acta Neuropathol (Berl) 102:581–590. [DOI] [PubMed] [Google Scholar]
- 54. Wisniewski HM, Wen GY, Kim KS (1989) Comparison of four staining methods on the detection of neuritic plaques. Acta Neuropathol (Berl) 78:22–27. [DOI] [PubMed] [Google Scholar]
- 55. Yaghmai A, Povlishock J (1992) Traumatically induced reactive change as visualized through the use of monoclonal antibodies targeted to neurofilament subunits. J Neuropathol Exp Neurol 51:158–176. [DOI] [PubMed] [Google Scholar]