

Abstract
Nutlin-3a is an MDM2 inhibitor that is under investigation in preclinical models for a variety of pediatric malignancies, including retinoblastoma, rhabdomyosarcoma, neuroblastoma, and leukemia. We used physiologically based pharmacokinetic (PBPK) modeling to characterize the disposition of nutlin-3a in the mouse. Plasma protein binding and blood partitioning were assessed by in vitro studies. After intravenous (10 and 20 mg/kg) and oral (50, 100, and 200 mg/kg) dosing, tissue concentrations of nutlin-3a were determined in plasma, liver, spleen, intestine, muscle, lung, adipose, bone marrow, adrenal gland, brain, retina, and vitreous fluid. The PBPK model was simultaneously fit to all pharmacokinetic data using NONMEM. Nutlin-3a exhibited nonlinear binding to murine plasma proteins, with the unbound fraction ranging from 0.7 to 11.8%. Nutlin-3a disposition was characterized by rapid absorption with peak plasma concentrations at approximately 2 h and biphasic elimination consistent with a saturable clearance process. The final PBPK model successfully described the plasma and tissue disposition of nutlin-3a. Simulations suggested high bioavailability, rapid attainment of steady state, and little accumulation when administered once or twice daily at dosages up to 400 mg/kg. The final model was used to perform simulations of unbound tissue concentrations to determine which dosing regimens are appropriate for preclinical models of several pediatric malignancies.
Introduction
Nutlin-3a (2-piperazinone, 4-[[(4S,5R)-4,5-bis(4-chlorophenyl)-4,5-dihydro-2-[4 methoxy-2-(1-methylethoxy)phenyl]-1H-imidazol-1-yl]carbonyl]-) is currently undergoing preclinical investigation as a p53 reactivation agent. Although many cancers and tumor types express mutated forms of p53 (Hollstein et al., 1991), a subset of cancers, and particularly pediatric tumors, retain wild-type p53 (Tweddle et al., 2003). In these cases, cancer cells frequently use other mechanisms to abrogate p53 function. One such mechanism is overexpression or amplification of the murine double minute (MDM2) protein. This molecule binds directly to p53 to accelerate its turnover and inhibits transcription of downstream targets, including cell cycle and apoptotic genes (Momand et al., 1992, 2000). Disruption of the MDM2-p53 interaction is proposed as a novel strategy for treatment of cancers that do not have p53 alterations (Shangary and Wang, 2008a,b).
Nutlins are a class of small molecules that target the p53-binding pocket of MDM2 (Klein and Vassilev, 2004; Vassilev et al., 2004). Treatment of multiple types of cancer cells, including leukemias (Kojima et al., 2005; Gu et al., 2008), neuroblastoma (Barbieri et al., 2006b), rhabdomyosarcoma (Miyachi et al., 2009), and retinoblastoma (Elison et al., 2006), with nutlin-3a induces p53-dependent cell cycle arrest and cell death, whereas in normal cells, nutlin-3a exposure leads to cell cycle arrest without cell death (Vassilev, 2005). Nutlin-3a has antitumor activity in a preclinical xenograft model of neuroblastoma (Van Maerken et al., 2009) and was tested in several other preclinical models of malignancies (Vassilev et al., 2004; Sarek and Ojala, 2007).
To date, the pharmacokinetics of nutlin-3a have not been reported. An understanding of the systemic disposition of nutlin-3a, as well as the distribution to target tissue or tumor sites, will provide a rational basis for the selection of dosage regimens for preclinical models. In addition, because the in vitro tumor cell line sensitivities to nutlin-3a have been determined, pharmacokinetic modeling can be used to determine the dose and schedule necessary to achieve appropriate unbound nutlin-3a concentrations at the tumor site. One approach to analyze these data is the use of whole-body physiologically based pharmacokinetic model (PBPK) models, which are based on anatomical compartments and blood flow.
Thus, we performed pharmacokinetic studies to develop a PBPK model describing the disposition of nutlin-3a in plasma and tissues, including adipose, adrenal gland, bone marrow, brain, liver, lung, intestine, muscle, retina, spleen, and vitreous fluid. The PBPK model was used to perform simulations, which in combination with in vitro cell sensitivity data provided rationale for choosing dosing regimens for mouse models of common childhood cancers, including retinoblastoma, neuroblastoma, rhabdomyosarcoma, and acute lymphoblastic leukemia (ALL).
Materials and Methods
Animals.
Adult C57BL/6 mice were purchased from Charles River (Bar Harbor, ME). Mice were housed in a temperature-controlled room on a normal 12-h light/dark cycle, with free access to water and standard laboratory food. All procedures were approved by the St. Jude Institutional Animal Care and Use Committee and conducted in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals (Institute of Laboratory Animal Resources, 1996). The animal facility is accredited by the American Association for Accreditation of Laboratory Animal Care.
Chemicals.
cis-Nutlin-3a (98% purity, lot no. 08252008) was synthesized and supplied by the Department of Chemical Biology and Therapeutics at St. Jude Children's Research Hospital (Memphis, TN). The oral formulation (Vassilev et al., 2004) used in the pharmacokinetic studies was nutlin-3a suspended in 2% Klucel (Conservation Resources International, LLC, Springfield, VA) and 0.5% Tween 80 (Sigma-Aldrich, St. Louis, MO), and the intravenous formulation used was nutlin-3a in 4% ethanol, 35% propylene glycol (Fisher Scientific, Pittsburgh, PA), 10% PEG-400 (Sigma-Aldrich), and 51% phosphate-buffered saline (PBS) (Mediatech Inc., Herndon, VA). The internal standard ketoconazole was purchased from Sigma-Aldrich. Blank murine plasma was obtained from Hilltop Laboratory Animals, Inc. (Scottsdale, PA). All other reagents were of analytical grade or higher.
Blood/Plasma Ratio.
Pooled whole blood from healthy male C57BL/6 mice was collected into heparin tubes. Nutlin-3a was spiked into aliquots of whole blood at final concentrations of 0.1, 1, 10, and 100 μM in triplicate. Samples were mixed and incubated at 37°C for 30 min with additional mixing every 5 min. After incubation, 50 μl of whole blood was removed and immediately placed on dry ice. The remainder of the sample was centrifuged at 16,000 rpm for 2 min, and a 50-μl plasma sample was removed and immediately placed on dry ice. All samples were stored at −80°C until analysis. The blood/plasma concentration ratio was calculated using the following equation:
where CP is the concentration in plasma and CWB is the concentration in whole blood.
Nutlin-3a Protein Binding Studies.
Equilibrium dialysis was performed in a 96-well dialysis plate with a 5-kDa cutoff membrane (Harvard Apparatus, Holliston, MA). For mouse plasma and cell culture media protein binding, 200 μl of PBS buffer was added into the wells on one side of the membrane and an equivalent volume of male C57BL/6 plasma or cell culture media (RPMI 1640 medium with 10% fetal bovine serum and 2 mM l-glutamine) containing varying concentrations of nutlin-3a was added into the wells on the opposite side. The plate was sealed and fixed onto a dual plate rotator (Harvard Apparatus) at 37°C in a humidified incubator containing 5% CO2. Equilibrium dialysis was performed at 0.2, 20, and 100 μM nutlin-3a in triplicate for 24 h. For vitreous protein binding, 150 μl of rodent vitreous containing 0.5 μM nutlin-3a was added to the sample side and equal volume of PBS buffer was added to the buffer side. Equilibrium dialysis was performed in triplicate for 24 h. The samples were analyzed using the analytical method described below. The bound concentration was considered equal to the plasma side and the free concentration equal to the PBS side (Kariv et al., 2001; Wan and Rehngren, 2006). Binding parameters were estimated with nonlinear regression using the Langmuir equation (Xu et al., 1993):
where CP,b is the bound plasma concentration, Bmax is the quantity of plasma protein binding sites, KA is the binding association constant, and CP,f is the unbound plasma concentration.
Drug Administration and Sample Collection.
Two pharmacokinetic studies were conducted. For the first pharmacokinetic study, 145 adult C57BL/6 mice (128 male and 17 female) were divided into three groups: two oral dosage groups (100 and 200 mg/kg) and one intravenous dosage group (10 mg/kg). Nutlin-3a was administered as a single bolus dose by oral gavage or by intravenous tail vein injection. Each dosing group (n = 5 mice) and vehicle controls had nine collection time points (0.5, 1, 2, 4, 8, 12, 24, 36, and 48 h). At each time point, blood was collected under isoflurane anesthesia via cardiac puncture. Whole-blood samples were centrifuged immediately at 12,000g for 5 min at 4°C to separate plasma. Tissue samples, including brain, vitreous, retina, liver, spleen, and bone marrow, were dissected simultaneously. Each sample was put on dry ice immediately after collection and stored at −80°C until analysis.
In the second pharmacokinetic study, 210 adult male C57BL/6 mice were used. Two oral dosages (50 and 100 mg/kg) and two intravenous dosages (10 and 20 mg/kg) were administered. Each dosing group (n = 5 for 10 mg/kg intravenous and 100 mg/kg oral dosages; n = 10 for 20 mg/kg intravenous and 50 mg/kg oral dosages) and vehicle control had seven collection time points (0.5, 1, 2, 4, 8, 12, and 24 h for the intravenous dosing; 0.5, 1, 2, 4, 8, 12, and 16 h for oral dosing). Serial plasma samples were collected from all mice. Tissue samples, including brain, lung, liver, spleen, kidney, adrenal gland, muscle, fat, and intestine from three mice per time point from the 20 mg/kg intravenous group, were collected. Each sample was put on dry ice immediately after the sample collection and stored at −80°C until analysis.
Quantitative Analysis of Nutlin-3a in Mouse Tissues and PBS.
Nutlin-3a mouse plasma samples were analyzed based on our previously published liquid chromatography electrospray ionization tandem mass spectrometry analytical method (Bai et al., 2009). For each sample type (cerebellum, brain, vitreous, retina, lung, heart, liver, gall bladder, spleen, kidney, adrenal gland, muscle, fat, bone marrow, intestine, whole blood, and PBS), standard curves and controls were generated using the corresponding untreated tissue or PBS to eliminate any matrix effect. For larger tissues, sections were cut, weighed, and stored on ice for further processing. Ten microliters of ice-cold homogenization buffer (5 mM HCOONH4, pH = 7) was added per milligram of tissue. For smaller tissue samples, including vitreous, retina, adrenal gland, and gall bladder, the amount of homogenization buffer used was increased to a minimum volume of 70∼100 μl. Tissue samples were then sonicated on ice for 15 s, with 5-s intervals. The number of total sonications varied depending on the tissue types. Homogenated tissues and whole-blood samples were extracted and analyzed using protein precipitation, and the PBS samples were extracted using the liquid-liquid extraction method as described previously (Bai et al., 2009).
Whole-Body PBPK Model Development.
We developed a whole-body PBPK model for nutlin-3a based on in vitro blood cell partitioning, plasma protein binding, and pooled concentration-time data from all plasma and tissue samples collected from both pharmacokinetic studies. This PBPK model consisted of a series of mass balance differential equations describing the concentration of nutlin-3a in various tissues, which were connected by blood flow. Physiological values for mouse organ size and blood flow are presented in Table 1. A schematic representation of the model is shown in Fig. 1.
TABLE 1.
List of physiological parameters
| Tissue | Symbol | Mass | Q B | Reference |
|---|---|---|---|---|
| %b.wt. | ml/h | |||
| Blood | BLO | 4.9 | 839 | Brown et al., 1997 |
| Adipose | ADI | 6.8 | 58.7 | Brown et al., 1997 |
| Adrenal gland | ADR | 0.048 | 2.52 | Brown et al., 1997 |
| Bone marrow | MRW | 5.8 | 92.3 | Brown et al., 1997 |
| Brain | BRA | 1.65 | 27.7 | Brown et al., 1997 |
| Intestines | INT | 3.62 | 109 | Brown et al., 1997 |
| Liver | LIV | 5.49 | 16.8 | Brown et al., 1997 |
| Lung | LUN | 0.73 | 839 | Brown et al., 1997 |
| Muscle | MUS | 38.4 | 133 | Brown et al., 1997 |
| Retina | RET | 0.04 | 3.16 | Experimental; Wright et al., 2009 |
| Spleen | SPL | 0.35 | 9.48 | Brown et al., 1997 |
| Vitreous fluid | VIT | 0.035 | 0 a | Experimental |
| Remainder | RES | 29.9 | 256.9 |
Vitreous assumed to have no direct blood flow.
Fig. 1.

Schematic diagram of PBPK model for nutlin-3a in mice. C,b, bound drug concentration; C,f, free drug concentration. Arrows connecting compartments represent blood flows from literature values.
Plasma concentrations were converted to whole-blood concentrations based on the in vitro blood partitioning experiment. Unbound plasma concentrations were described by the following equation (Xu et al., 1993):
where CP,u is the unbound plasma concentration, CP is the total plasma concentration, and Bmax and KA were determined from in vitro plasma protein binding studies. The unbound fraction (fub) was calculated by
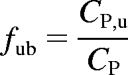 |
Most organs fit well to a perfusion-limited model, and thus were described by the following equation:
where Vi is the volume of organ, Ai is the amount of drug in the organ, Ci is the concentration in the organ, Ki is the partition coefficient, and CART is the concentration of arterial plasma.
Liver blood flow (QLIV) was the sum of the blood flow to the hepatic artery, spleen, and liver, and the concentration of blood entering the liver (CBLO,LIV) was based on the arterial concentration and the venous outflow of the portal circulation. The liver contained an elimination term (ke) for metabolism, based on experiments showing that nutlin-3a is metabolized by mouse liver microsomes (K. Guy, unpublished data):
The intestine was modeled with a separate lumen and tissue compartment. Absorption from the lumen was assumed to be linear based on an absorption rate constant (ka):
The eye was fit to a two-compartment model consisting of the retina and vitreous. Input into the vitreous was by diffusion from the retina. The following equations were used for the retina and vitreous:
where PAVIT is the permeability-surface area product.
All tissues that were not sampled were lumped together in a residual compartment. Modeling this compartment as perfusion-limited did not adequately describe the multiexponential profile of nutlin-3a. Therefore, the residual compartment was modeled as diffusion-limited, with the vascular space assumed to be 5% of the residual volume. The equations for the residual vascular space and tissue are as follows:
The input into the venous pool of blood was modeled as the sum of the output from all tissues except the lung. The volume of the venous pool was fixed to 75% of the total blood volume. The lungs received all venous input, and the arterial input was the output from the lungs. The equation for the lungs was as follows:
The arterial concentrations were based on the output from the lungs and contained an additional saturable elimination term:
Elimination terms in both the blood compartment and liver compartment were necessary for a good model fit to the data from both oral and intravenous administration.
Simulations.
After development of the PBPK model, tissue concentrations were simulated with NONMEM after multiple oral and intravenous doses at 50, 100, 200, and 400 mg/kg given both once daily and twice daily. The AUC0–24 at steady state was calculated with the log-linear trapezoidal method applied to the simulated data. Bioavailability was estimated using the ratio of AUC0-τ, IV/AUC0-τ, PO with the simulated steady-state data.
Results
Blood-to-Plasma Partitioning and Plasma Protein Binding of Nutlin-3a.
Blood-to-plasma partitioning showed an average blood/plasma concentration ratio of 0.70, indicating that 30% of nutlin-3a partitions to blood cells (Fig. 2A). Binding of nutlin-3a to mouse plasma proteins was nonlinear, with fub ranging from 0.007 at 0.1 μM to 0.118 at 300 μM (Fig. 2B). Nonlinear regression of unbound versus bound plasma concentrations using the Langmuir equations resulted in a Bmax of 286 and a KA of 0.085 (Supplemental Fig. S1).
Fig. 2.

Analysis of nutlin-3a characteristics in murine blood. A, nutlin-3a blood cell partitioning. Nutlin-3a was spiked into murine whole blood at various concentrations and incubated for 30 min at 37°C. In one aliquot, nutlin-3a was measured in whole blood, and in another aliquot nutlin-3a was measured in the plasma. B, nutlin-3a plasma protein binding. Nutlin-3a was spiked into murine plasma at various concentrations and incubated for 30 min at 37°C. Plasma protein binding was determined by equilibrium microdialysis and is expressed as the percentage of the total nutlin-3a plasma concentration that is unbound. Bars represent the mean, and error bars represent the S.D.
Nutlin-3a Pharmacokinetics in Mice.
Plasma and tissue concentrations of nutlin-3a were measured from 0 to 48 h in mice after a single intravenous dose of 10 or 20 mg/kg or a single oral dose of 50, 100, or 200 mg/kg. After oral administration, nutlin-3a tissue concentrations rose rapidly to reach a maximum value at approximately 2 h. Nutlin-3a concentrations in the intestine, liver, and spleen were higher than those in the plasma; concentrations in adipose, adrenal gland, lung, muscle, and retina were similar to plasma concentrations; and concentrations in the brain, bone marrow, and vitreous were significantly lower than in the plasma (Supplemental Fig. S2).
Rapid elimination was observed in the 10 mg/kg intravenous dosage group. At higher dosages, slower elimination was observed at higher concentrations, indicating saturable elimination of nutlin-3a. After 24 h, all data were below the limit of quantitation of the assay. Models with linear elimination, Michaelis-Menten elimination, and combined linear and Michaelis-Menten elimination were fit to the data. Ultimately, the combination of both linear and Michaelis-Menten elimination had the best fit to the data. The concentration-time data of nutlin-3a in all modeled tissues are plotted against the model-predicted concentrations in Fig. 3 (data not shown for oral 50 mg/kg dosage group, because only plasma was collected). The estimated pharmacokinetic parameters are listed in Table 2.
Fig. 3.

Concentration-time plots of nutlin-3a in tissues. Symbols are data points from individual mice, and the lines represent the model-predicted concentrations. Data from the 50 mg/kg oral group are not shown. Data below the lower limit of quantitation are not shown.
TABLE 2.
Estimated PBPK model parameters
| Parameter | Value |
|---|---|
| k a | 0.409 |
| K e | 0.0160 |
| V max | 0.0287 |
| K m | 0.050 |
| K ADI | 1.61 |
| K ADR | 2.05 |
| K BRA | 0.055 |
| K INT | 12.2 |
| K LIV | 7.54 |
| K LUN | 1.78 |
| K MUS | 2.08 |
| K RET | 4.01 |
| K SPL | 2.72 |
| K VIT | 0.012 |
| PAVIT | 0.0036 |
| K RES | 4.8 |
| PARES | 0.0048 |
| IIV ka | 31.2% |
| IIV ke | 6.4% |
| IIV Vmax | 40.6% |
| Residual error | 35.6% |
Using the final model, we simulated nutlin-3a plasma concentrations after multiple doses on a once-daily and twice-daily schedule (Fig. 4). Little to no accumulation was predicted to occur on a once daily schedule with intravenous or oral dosages up to 400 mg/kg. For twice daily dosing, steady state was predicted to occur within three doses, but accumulation would remain minimal. The predicted accumulation (Cmin single dose/Cmin at steady state) was dose-dependent and at 200 mg/kg is 1.5-fold for intravenous administration and 1.3-fold for oral administration. The AUC0–24 at steady state increased in an approximately dose-proportional manner, and the AUC0–24 was approximately twice as high with twice-daily dosing versus once-daily dosing (Supplemental Fig. S3).
Fig. 4.

Simulated concentration-time plot of plasma nutlin-3a after multiple oral doses with once-daily (QD) and twice-daily (BID) dosing. Simulations were based on the final PBPK model.
Predicted bioavailability was dose- and schedule-dependent and ranged from 75% at 25 mg/kg once-daily to 91% at 400 mg/kg once-daily. Bioavailability was predicted to be near 100% when given twice daily at dosages of 50 mg/kg or higher.
Application of PBPK Model to the Design of Nutlin-3a Dosing Regimens in Mice.
The nutlin-3a PBPK model was used to choose dosing regimens of nutlin-3a to target 1) the retina and vitreous for models of retinoblastoma, 2) the adrenal gland for models of neuroblastoma, 3) the muscle for models of rhabdomyosarcoma, and 4) the plasma, spleen, and bone marrow for models of leukemia. The fraction of unbound nutlin-3a in tissues was assumed to be the same as the plasma unbound fraction, except for vitreous fluid which had a measured unbound fraction of 14.4%. Nutlin-3a binding to cell culture media was measured and shown to be nonlinear over the range of nutlin-3a concentrations used in in vitro cell cytotoxicity assays (Supplemental Fig. S4). The media protein binding value was used to convert the published nutlin-3a IC50 values of various cell lines (Barbieri et al., 2006a; Gu et al., 2008; Miyachi et al., 2009) to the unbound IC50 (Supplemental Table 1).
The simulated unbound retina and vitreous nutlin-3a concentrations were compared to the in vitro unbound IC50 for the Weri1 retinoblastoma cell line to choose optimal dosing regimens for mouse models of retinoblastoma (Fig. 5). Oral dosing of nutlin-3a twice daily was predicted to achieve unbound concentrations in the retina that were consistently above the unbound IC50 at dosages of 200 or 400 mg/kg (Supplemental Table 2). However, even at 400 mg/kg twice daily, unbound concentrations in the vitreous were predicted to be above the unbound IC50 for only 17% of the time, and at lower dosages the concentration of unbound nutlin-3a never reached the unbound IC50 level. Simulated concentration-time plots of unbound nutlin-3a after various dosing regimens are also shown in the adrenal gland for neuroblastoma (Supplemental Fig. S5), muscle for rhabdomyosarcoma (Supplemental Fig. S6), and plasma, bone marrow, and spleen for leukemia (Supplemental Fig. S7). The percent times above the unbound IC50 are also listed in Supplemental Table 2.
Fig. 5.

Simulated concentration-time plot of unbound nutlin-3a in the retina and vitreous after multiple oral doses given once daily (QD) or twice daily (BID). The horizontal lines represent the unbound IC50 of nutlin-3a for Weri1 cells.
Discussion
Nutlin-3a is undergoing preclinical studies examining its potential efficacy for the treatment of several childhood malignancies. Nutlin-3a interrupts the p53-MDM2 protein-protein interaction, which may lead to apoptosis or cell cycle arrest. Treatment of cells with nutlin-3a leads to reversal of multidrug resistance (Michaelis et al., 2009), reduced cell migration (Secchiero et al., 2007), reduced angiogenesis (LaRusch et al., 2007; Secchiero et al., 2007), radiosensitization of hypoxic cancer cells (Supiot et al., 2008), and inhibition of tumor adaptation to hypoxia (Lee et al., 2009). In this study, we developed a mouse PBPK model of nutlin-3a in plasma and multiple tissues of therapeutic interest. This is the first study to provide comprehensive pharmacokinetic data of nutlin-3a in any species. The design of our study included both oral and intravenous dosing at several dosage levels. This method permitted the development of a robust model that accurately describes the disposition of nutlin-3a over a wide range of concentrations. The PBPK model was used to design rational dosing regimens for preclinical models of several malignancies based upon achieving adequate cytotoxic nutlin-3a concentrations within the target organ. This approach is superior to optimizing dosing based solely on plasma concentrations because drug penetration to different organs can vary widely.
Our model describes the key pharmacokinetic properties of nutlin-3a in plasma: rapid absorption, high bioavailability, and saturable elimination that is very rapid at concentrations below 10 μM. Standard noncompartmental calculations of bioavailability of nutlin-3a were greater than 100% due to saturable elimination and different ranges of intravenous (10–20 mg/kg) and oral (50–100 mg/kg) dosages. Using the model to simulate the concentration-time profiles after the same dosage administered both intravenous and oral, we were able to estimate nutlin-3a oral bioavailability. Although we performed simulations after multiple doses, the model was based on data from only single doses of nutlin-3a and, therefore, should be interpreted with caution.
Nutlin-3a is a substrate for the ATP binding cassette transporter P-glycoprotein (P-gp), but at higher concentrations it can also inhibit P-gp efflux activity (Michaelis et al., 2009). The inhibition of P-gp function may explain why nutlin-3a is capable of rapid absorption and high bioavailability despite being a P-gp substrate. It is also possible that inhibition of P-gp function underlies saturable nutlin-3a elimination, because P-gp can excrete drugs into both the bile and urine (International Transporter Consortium, 2010). It is unknown whether nutlin-3a may also inhibit its own metabolism at higher concentrations.
The partition coefficients showed a greater than 1000-fold difference between the tissues with the lowest and highest penetration. The liver and intestine showed the highest penetration. High penetration to the liver could be due to uptake transporters expressed at the hepatocyte membranes, which cause intracellular accumulation of nutlin-3a. The intestine had an atypical profile, possibly due to biliary excretion of nutlin-3a. The blood flow-limited model did not fit well to the intestinal concentration data, limiting the ability to accurately estimate the partition coefficient for this organ.
Retinoblastoma is a tumor of the eye for which a number of orthotopic xenograft and genetic murine models have been developed (Laurie et al., 2005). Daily subconjunctival administration of nutlin-3a for 5 days was effective in a model of retinoblastoma, and when combined with topotecan, an 82-fold reduction in tumor burden with no systemic or ocular side-effects was observed (Laurie et al., 2006). Our PBPK model shows that with the nutlin-3a regimen most commonly used in preclinical studies (200 mg/kg administered orally twice daily), unbound concentrations of nutlin-3a in the retina continuously exceeded the unbound IC50. However, due to poor penetration, the unbound IC50 was never achieved in the vitreous with this regimen and was achieved only transiently at 400 mg/kg twice daily, suggesting that subconjunctival dosing would be more appropriate for targeting retinoblastoma vitreous seeds (Amemiya et al., 1979; Shields, 2008). Although the PBPK model cannot predict nutlin-3a pharmacokinetics after subconjunctival administration because the ocular absorption is not known, data from a limited experiment could be combined with the PBPK model to predict exposures in various tissues after ocular administration.
At diagnosis, 98% of neuroblastoma tumors contain wild-type p53 (Vogan et al., 1993; Tweddle et al., 2001, 2003), and thus these patients are likely to benefit from reactivation of this pathway. Sensitivity to nutlin-3a has been shown in multiple p53 wild-type neuroblastoma cell lines (Barbieri et al., 2006b; Michaelis et al., 2009). Treatment of a subcutaneous UKF-NB-3rDOX20 xenograft model with twice-daily oral nutlin-3a (200 mg/kg) only partially inhibited tumor growth (Van Maerken et al., 2009), despite good sensitivity (IC50 of 5.56 μM) of these cells in vitro (Michaelis et al., 2009). Because the racemic nutlin-3 mixture was used in this experiment, the assumed equivalent dose would be 100 mg/kg nutlin-3a twice daily. If unbound plasma concentrations are evaluated (because this was not an orthotopic xenograft), unbound nutlin-3a concentrations are predicted to be continually below the established IC50 with this regimen. However, the pharmacokinetic properties of the racemic mixture are unknown, and it is possible that nutlin-3b influences the saturable elimination or plasma protein binding of nutlin-3a. For a subcutaneous or orthotopic xenograft with similar cell sensitivity, 400 mg/kg oral nutlin-3a twice daily may result in better activity, because concentrations would continuously be above the IC50. Although the literature and our unpublished observations suggest that twice-daily, 200 mg/kg oral dosing is well tolerated in mice (Van Maerken et al., 2009), further toxicity studies will need to be performed to determine whether higher dosages are tolerable.
Nutlin-3a has also demonstrated cytotoxicity in rhabdomyosarcoma cell lines (Michaelis et al., 2009; Miyachi et al., 2009), although it has not yet been tested in a preclinical model of rhabdomyosarcoma. Our model predictions show that the standard twice-daily oral 200 mg/kg nutlin-3a regime is sufficient to achieve unbound muscle nutlin-3a concentrations that were continuously above the IC50 for the RMS-YM cell line.
Nutlin-3a concentrations above the IC50 for primary T-ALL cells (Kojima et al., 2005; Gu et al., 2008) was achieved in the plasma and spleen with a twice-daily regimen of 100 mg/kg oral nutlin-3a. However, penetration to the bone marrow was poor, and to target this compartment, 400 mg/kg twice daily is recommended, which is predicted to result in unbound nutlin-3a concentrations that are above the IC50 77% of the time.
Although the model has a number of applications, it also has limitations. First, we did not perform experiments to determine the route of elimination of nutlin-3a. Preliminary unpublished observations (K. Guy, unpublished data) indicate that nutlin-3a is metabolized by mouse liver microsomes, but a model with elimination only from the liver did not adequately fit the nutlin-3a plasma concentration-time data. Not modeling the elimination mechanistically could limit the ability to extrapolate the PBPK model to other species. Second, we performed all experiments in nontumor-bearing mice. Compared to normal tissues, the altered environment in tumors (e.g., vasculature, pH, interstitial fluid pressure) may lead to different local disposition. Although it was not feasible to perform pharmacokinetic studies in multiple preclinical models, data obtained from studies in tumor-bearing mice could be easily incorporated into this PBPK model. Another limitation is the assumption that the unbound fraction of nutlin-3a in tissues was equivalent to the unbound fraction in plasma. We did directly measure nutlin-3a binding in vitreous fluid, which is mostly water but has a variety of proteins (Shires et al., 1993). We also performed simulations at dosages beyond those used to develop the model (i.e., 400 mg/kg). Although we were able to characterize the nonlinear elimination at higher plasma concentrations, it is possible that there are unknown nonlinear absorption or elimination processes occurring at this higher dosage, which would make these model predictions inaccurate.
In summary, we performed extensive mouse pharmacokinetic studies of nutlin-3a and developed a PBPK model, which was used to design nutlin-3a dosing regimens for preclinical models of pediatric malignancies. Although there are limitations to extrapolating in vitro cytotoxicity data, this analysis provides a starting point for further pharmacokinetic/pharmacodynamic studies in tumor-bearing mice. For models of retinoblastoma, the disposition of nutlin-3a after subconjunctival administration should be explored.
Supplementary Material
Acknowledgments
We thank Zaifang Huang, Mohamad Elmeliegy, Lei Yang, Shelly Wilkerson, and Frederique Zindy for help with dosing animals and collecting tissue samples. We thank Daniel Groepper for helping with the protein binding study in mouse plasma.
This work was supported in part by the National Institutes of Health National Cancer Institute [Grant N01-CM-42216]; and by the American Lebanese Syrian Associated Charities.
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.110.035915.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
- MDM2
- murine double minute 2
- PBPK
- physiologically based pharmacokinetic
- ALL
- acute lymphoblastic leukemia
- PBS
- phosphate-buffered saline
- AUC
- area under the curve
- P-gp
- P-glycoprotein.
Authorship Contributions
Participated in research design: F. Zhang, Mallari, Miller, Guy, Dyer, Williams, Nemeth, Boulos, Panetta, and Stewart.
Conducted experiments: F. Zhang, Mallari, Miller, Dyer, Roussel, Nemeth, Zhu, Boulos, and J. Zhang.
Contributed new reagents or analytic tools: Lu.
Performed data analysis: F. Zhang, Tagen, Panetta, and Stewart.
Wrote or contributed to the writing of the manuscript: F. Zhang, Tagen, Throm, Mallari, Guy, Dyer, Williams, Roussel, Boulos, Panetta, and Stewart.
References
- Amemiya T, Yoshida H, Ishigooka H. (1979) Vitreous seeds in retinoblastoma, clinical significance and ultrastructure. Albrecht Von Graefes Arch Klin Exp Ophthalmol 211:205–213 [DOI] [PubMed] [Google Scholar]
- Bai F, Zhu F, Tagen M, Miller L, Owens TS, Mallari J, Derrick E, Zhang F, Stewart CF. (2010) Determination of nutlin-3a in murine plasma by liquid chromatography electrospray ionization tandem mass spectrometry (LC-ESI-MS/MS). J Pharm Biomed Anal 51:915–920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri A, Sabatini L, Indiveri P, Bonfiglioli R, Lodi V, Violante FS. (2006a) Simultaneous determination of low levels of methotrexate and cyclophosphamide in human urine by micro liquid chromatography/electrospray ionization tandem mass spectrometry. Rapid Commun Mass Spectrom 20:1889–1893 [DOI] [PubMed] [Google Scholar]
- Barbieri E, Mehta P, Chen Z, Zhang L, Slack A, Berg S, Shohet JM. (2006b) MDM2 inhibition sensitizes neuroblastoma to chemotherapy-induced apoptotic cell death. Mol Cancer Ther 5:2358–2365 [DOI] [PubMed] [Google Scholar]
- Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, Beliles RP. (1997) Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health 13:407–484 [DOI] [PubMed] [Google Scholar]
- Elison JR, Cobrinik D, Claros N, Abramson DH, Lee TC. (2006) Small molecule inhibition of HDM2 leads to p53-mediated cell death in retinoblastoma cells. Arch Ophthalmol 124:1269–1275 [DOI] [PubMed] [Google Scholar]
- International Transporter Consortium, Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, et al. (2010) Membrane transporters in drug development. Nat Rev Drug Discov 9:215–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu L, Zhu N, Findley HW, Zhou M. (2008) MDM2 antagonist nutlin-3 is a potent inducer of apoptosis in pediatric acute lymphoblastic leukemia cells with wild-type p53 and overexpression of MDM2. Leukemia 22:730–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollstein M, Sidransky D, Vogelstein B, Harris CC. (1991) p53 mutations in human cancers. Science 253:49–53 [DOI] [PubMed] [Google Scholar]
- Institute of Laboratory Animal Resources (1996) Guide for the Care and Use of Laboratory Animals 7th ed Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council, Washington DC [Google Scholar]
- Kariv I, Cao H, Oldenburg KR. (2001) Development of a high throughput equilibrium dialysis method. J Pharm Sci 90:580–587 [DOI] [PubMed] [Google Scholar]
- Klein C, Vassilev LT. (2004) Targeting the p53-MDM2 interaction to treat cancer. Br J Cancer 91:1415–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima K, Konopleva M, Samudio IJ, Shikami M, Cabreira-Hansen M, McQueen T, Ruvolo V, Tsao T, Zeng Z, Vassilev LT, et al. (2005) MDM2 antagonists induce p53-dependent apoptosis in AML: implications for leukemia therapy. Blood 106:3150–3159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRusch GA, Jackson MW, Dunbar JD, Warren RS, Donner DB, Mayo LD. (2007) Nutlin3 blocks vascular endothelial growth factor induction by preventing the interaction between hypoxia inducible factor 1alpha and Hdm2. Cancer Res 67:450–454 [DOI] [PubMed] [Google Scholar]
- Laurie NA, Donovan SL, Shih CS, Zhang J, Mills N, Fuller C, Teunisse A, Lam S, Ramos Y, Mohan A, et al. (2006) Inactivation of the p53 pathway in retinoblastoma. Nature 444:61–66 [DOI] [PubMed] [Google Scholar]
- Laurie NA, Gray JK, Zhang J, Leggas M, Relling M, Egorin M, Stewart C, Dyer MA. (2005) Topotecan combination chemotherapy in two new rodent models of retinoblastoma. Clin Cancer Res 11:7569–7578 [DOI] [PubMed] [Google Scholar]
- Lee YM, Lim JH, Chun YS, Moon HE, Lee MK, Huang LE, Park JW. (2009) Nutlin-3, a Hdm2 antagonist, inhibits tumor adaptation to hypoxia by stimulating the FIH-mediated inactivation of HIF-1alpha. Carcinogenesis 30:1768–1775 [DOI] [PubMed] [Google Scholar]
- Michaelis M, Rothweiler F, Klassert D, von Deimling A, Weber K, Fehse B, Kammerer B, Doerr HW, Cinatl J., Jr (2009) Reversal of P-glycoprotein-mediated multidrug resistance by the murine double minute 2 antagonist nutlin-3. Cancer Res 69:416–421 [DOI] [PubMed] [Google Scholar]
- Miyachi M, Kakazu N, Yagyu S, Katsumi Y, Tsubai-Shimizu S, Kikuchi K, Tsuchiya K, Iehara T, Hosoi H. (2009) Restoration of p53 pathway by nutlin-3 induces cell cycle arrest and apoptosis in human rhabdomyosarcoma cells. Clin Cancer Res 15:4077–4084 [DOI] [PubMed] [Google Scholar]
- Momand J, Wu HH, Dasgupta G. (2000) MDM2–master regulator of the p53 tumor suppressor protein. Gene 242:15–29 [DOI] [PubMed] [Google Scholar]
- Momand J, Zambetti GP, Olson DC, George D, Levine AJ. (1992) The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69:1237–1245 [DOI] [PubMed] [Google Scholar]
- Sarek G, Ojala PM. (2007) p53 reactivation kills KSHV lymphomas efficiently in vitro and in vivo: new hope for treating aggressive viral lymphomas. Cell Cycle 6:2205–2209 [DOI] [PubMed] [Google Scholar]
- Secchiero P, Corallini F, Gonelli A, Dell'Eva R, Vitale M, Capitani S, Albini A, Zauli G. (2007) Antiangiogenic activity of the MDM2 antagonist nutlin-3. Circ Res 100:61–69 [DOI] [PubMed] [Google Scholar]
- Shangary S, Wang S. (2008a) Small-molecule inhibitors of the MDM2–p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annu Rev Pharmacol Toxicol 49:223–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shangary S, Wang S. (2008b) Targeting the MDM2–p53 interaction for cancer therapy. Clin Cancer Res 14:5318–5324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields CL. (2008) Forget-me-nots in the care of children with retinoblastoma. Semin Ophthalmol 23:324–334 [DOI] [PubMed] [Google Scholar]
- Shires TK, Faeth JA, Pulido JS. (1993) Protein levels in the vitreous of rats with streptozotocin-induced diabetes mellitus. Brain Res Bull 30:85–90 [DOI] [PubMed] [Google Scholar]
- Supiot S, Hill RP, Bristow RG. (2008) Nutlin-3 radiosensitizes hypoxic prostate cancer cells independent of p53. Mol Cancer Ther 7:993–999 [DOI] [PubMed] [Google Scholar]
- Tweddle DA, Malcolm AJ, Bown N, Pearson AD, Lunec J. (2001) Evidence for the development of p53 mutations after cytotoxic therapy in a neuroblastoma cell line. Cancer Res 61:8–13 [PubMed] [Google Scholar]
- Tweddle DA, Pearson AD, Haber M, Norris MD, Xue C, Flemming C, Lunec J. (2003) The p53 pathway and its inactivation in neuroblastoma. Cancer Lett 197:93–98 [DOI] [PubMed] [Google Scholar]
- Van Maerken T, Ferdinande L, Taildeman J, Lambertz I, Yigit N, Vercruysse L, Rihani A, Michaelis M, Cinatl J, Jr, Cuvelier CA, et al. (2009) Antitumor activity of the selective MDM2 antagonist nutlin-3 against chemoresistant neuroblastoma with wild-type p53. J Natl Cancer Inst 101:1562–1574 [DOI] [PubMed] [Google Scholar]
- Vassilev LT. (2005) p53 activation by small molecules: application in oncology. J Med Chem 48:4491–4499 [DOI] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303:844–848 [DOI] [PubMed] [Google Scholar]
- Vogan K, Bernstein M, Leclerc JM, Brisson L, Brossard J, Brodeur GM, Pelletier J, Gros P. (1993) Absence of p53 gene mutations in primary neuroblastomas. Cancer Res 53:5269–5273 [PubMed] [Google Scholar]
- Wan H, Rehngren M. (2006) High-throughput screening of protein binding by equilibrium dialysis combined with liquid chromatography and mass spectrometry. J Chromatogr A 1102:125–134 [DOI] [PubMed] [Google Scholar]
- Wright WS, Messina JE, Harris NR. (2009) Attenuation of diabetes-induced retinal vasoconstriction by a thromboxane receptor antagonist. Exp Eye Res 88:106–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Selick P, Pang KS. (1993) Nonlinear protein binding and enzyme heterogeneity: effects on hepatic drug removal. J Pharmacokinet Biopharm 21:43–74 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.