

Abstract
The endocannabinoid system regulates a wide range of physiological processes including pain, inflammation, and cognitive/emotional states. URB597 is one of the best characterized covalent inhibitors of the endocannabinoid-degrading enzyme fatty acid amide hydrolase (FAAH). Here, we report the structure of the FAAH-URB597 complex at 2.3 Å resolution. The structure provides insights into mechanistic details of enzyme inactivation and experimental evidence of a previously uncharacterized active site water molecule that likely is involved in substrate deacylation. This water molecule is part of an extensive hydrogen-bonding network, and is coordinated indirectly to residues lining the cytosolic port of the enzyme. In order to corroborate our hypothesis concerning the role of this water molecule in FAAH’s catalytic mechanism, we determined the structure of FAAH conjugated to a urea-based inhibitor, PF-3845, to a higher resolution (2.4 Å) than previously reported. The higher resolution structure confirms the presence of the water molecule in a virtually identical location in the active site. Examination of the structures of serine hydrolases that are non-homologous to FAAH, such as elastase, trypsin, or chymotrypsin, shows a similarly positioned hydrolytic water molecule and suggest a functional convergence between the amidase signature enzymes and serine proteases.
Keywords: Fatty Acid Amide Hydrolase, FAAH; URB597; crystal structure; deacylating water; catalytic mechanism
INTRODUCTION
Fatty acid amide hydrolase (FAAH) degrades lipid signaling molecules such as the endogenous cannabinoid (endocannabinoid), anandamide (N-arachidonyl ethanolamine) and the sleep-inducing substance oleamide (cis-9-octadecenamide) (1-3). Pharmacological studies in rodents and experiments with knock-out mice have demonstrated that blockade of FAAH activity raises endogenous levels of fatty acid amides (2-6). Such studies have also linked elevations in fatty acid amides to therapeutically desirable analgesic, anti-inflammatory, and neuropsychiatric effects (3-18). These effects occur without many of the untoward side effects observed with direct cannabinoid receptor-1 (CB1) agonists, such as hypothermia and cognitive and motor dysfunction. Finding selective and potent inhibitors of FAAH with drug-like properties has thus been the subject of intense medicinal chemistry efforts (17, 18). Many of the most promising inhibitors identified through these efforts are mechanism-based covalent modifiers of the enzyme.
FAAH is a member of the amidase signature family (1, 19-20). Enzymes in this family are found in bacteria, archaea and eukaryotes, and hydrolyze diverse substrates ranging from glutamine (21) to peptide amides (22) to simple hydrophilic amides, such as acetamide or malonamide (23). As might be expected, these enzymes have correspondingly diverse substrate binding pockets (20, 22-23). In FAAH, a pocket adjacent to the catalytic site branches into an acyl-chain binding pocket and a tunnel leading to the membrane (membrane access channel) (20) (Figure 1A). Consistent with their proposed roles in binding the fatty acid chains of substrates, these portions of FAAH have hydrophobic surfaces (20). A second channel, the cytosolic port, connects the active site to the cytoplasm. This hydrophilic cavity interacts with the head groups of FAAH substrates (20, 24, 25). It provides a route through which hydrophilic products (ethanolamine in the case of anandamide) and the leaving groups of certain covalent inhibitors are released into the cytoplasm (20, 24, 25). FAAH and its homolog FAAH-2 (26) are unusual members of the amidase signature family in that they are membrane associated. Nevertheless, FAAH shares a core set of residues, including a catalytic Ser-Ser-Lys triad, with other members of the family (20, 22-23).
Figure 1.



(A) The FAAH dimer bound to URB597. A partially transparent surface in the monomer shown on the right illustrates the location of the membrane access channel (MAC), the acyl-chain binding pocket (ABP) and the cytosolic port (CP). (B) Proposed reaction mechanism of FAAH, comparing irreversible acyl intermediates and substrates. The catalytic intermediate states are indicated (S0 to S4). We propose that the water molecule shown in blue corresponds with W1 in Figure 2. The states corresponding to S3 must differ between substrates and irreversible inhibitors such that the latter cannot proceed to S4. (C) Structures of substrates and inhibitors of FAAH.
The catalytic mechanism of FAAH has attracted considerable interest (2, 27-29). In broad outline, it involves four steps: (1) nucleophilic attack of the substrate by an activated serine to generate an enzyme-linked tetrahedral intermediate, (2) subsequent elimination of a substrate leaving group to form an acyl-enzyme intermediate, (3) attack of an activated water to create a second tetrahedral intermediate and (4) resolution into the acid product and regenerated enzyme. (Figure 1B). Ser241 was identified as the nucleophile in the active site of FAAH through mutational and affinity labeling studies (27). Mutational studies also showed the importance of Lys142, which acts as both a base to activate Ser241 and as an acid that helps protonate the substrate leaving group (28). Structural studies established that both of these activities of Lys142 are indirect and mediated through the hydroxyl side chain of Ser217 (20). FAAH is an atypical serine hydrolase; it catalyzes the hydrolysis of both amide and ester substrates at comparable rates, with an acylation rate limiting step (28). It has been proposed that this unusual enzymatic profile is critical for FAAH activity in vivo, where lipid esters are far more abundant than lipid amides (28). Despite these differences, some classes of FAAH inhibitors, such as carbamates, fluorophosphonates, and α-ketoheterocycles, also inhibit non-homologous serine hydrolases, suggesting the enzymes share some degree of mechanistic similarity (2, 17).
URB597 is a widely used carbamate inhibitor of FAAH with a relatively simple chemical structure, consisting of a cyclohexyl moiety, a carbamate reactive group and a meta-biphenylamide leaving group (Figure 1C; 30-31). It displays excellent selectivity for FAAH in the nervous system, although the inhibitor does inactivate additional peripheral hydrolases (32). The carbamate inhibitors were initially derived from an acetylcholine esterase inhibitor with very poor FAAH potency (compound 19, Figure 1C), but were rapidly optimized into compounds such as URB524, which has an apparent potency of 63 nM (Figure 1C, 30). Addition of an amide group to the meta-biphenyl moiety yielded URB597, an inhibitor with an IC50 of 4.6 nM (31), making it one of the earliest drug like inhibitors of FAAH. Initial modeling studies suggested that the biphenyl moiety of the URB compounds might bind in the hydrophobic pocket of FAAH, with the cyclohexyl group extending into the cytosolic port (30, 31). Subsequent experiments indicated that the original modeling was incorrect, instead invoking a flipped orientation of the compounds in the active site of FAAH (33).
Here we describe the URB597-carbamoylated structure of humanized rat FAAH (h/rFAAH), determined at 2.3 Å resolution. The h/rFAAH protein used in these and past crystallographic studies contains six active site mutations that replace non-conserved residues in the rat sequence with their human counterparts (25). These mutations allow us to take advantage of the superior expression and biophysical properties of rat FAAH while mimicking the inhibitor binding profile of the human enzyme. Because it contains a plateau composed of two helices with exposed hydrophobic residues, h/rFAAH remains fully membrane-associated even though it lacks the hydrophobic N-terminal sequence of the wild-type enzyme (20). The structure of FAAH–URB597 establishes the orientation of the cyclohexyl-carbamate moiety within the acyl chain-binding tunnel and is consistent with the results of structure activity relationship experiments. The structure also shows the location of a water molecule in the active site that is close to the carbonyl carbon of the inhibitor. The water is held in place through hydrogen bonds to both protein and a second water molecule. Re-determination of the structure of the urea-based inhibitor, PF-3845 (18), at a higher resolution reveals similarly placed waters. The water molecules close to the inhibitor carbonyl carbon atom in the active site of FAAH superimpose on those seen in high resolution structures of non-homologous serine hydrolases, indicating that some of the mechanistic insights derived from decades of analysis of serine proteases may be applicable to FAAH.
RESULTS
We have determined the structure of h/rFAAH conjugated with the carbamate inhibitor URB597 at a resolution of 2.30 Å and determined the structure of h/rFAAH conjugated with the urea inhibitor PF-3845 at a higher resolution (2.42 Å) than previously reported (Table 1). Both h/rFAAH conjugates crystallized in space group P212121, with a biological dimer in the asymmetric unit with 1,550 Å2 (7.5 %) of the monomer surface buried upon dimer formation. The structures have been refined to crystallographic R values of 17.6 and 16.0% respectively (Rfree: 21.4 and 20.0%), with good geometry. All residues of the crystallographic construct have been built into the electron density maps, except for some disordered residues at the extreme N- (model starts at residue Thr32) and C- (model ends at Pro578 in the FAAH–URB597 structure) termini of the protein. The two monomers within each asymmetric unit are very similar in structure and, together, constitute what is likely the physiological dimeric form of the enzyme. Although the unit cell dimensions of the FAAH–PF-3845 structure differ substantially from those of our earlier data set (PDB ID 2WAP), the structures show no global conformational changes. The root mean squared deviations (RMSDs) calculated over all pairs of ordered Cα atoms in the dimer between the current two structures of FAAH–URB597 and FAAH–PF-3845 and the reference model PDB ID 2WAP are 0.32 for and 0.38Å, respectively.
Table 1.
Data Collection and Refinement Statistics of FAAH Crystals.
| FAAH–URB597 | FAAH–PF-3845 | |
|---|---|---|
| X-ray source | APS-GM/CA-CAT | APS-GM/CA-CAT |
| Crystal data | ||
| Space group | P212121 | P212121 |
| Cell dimensions | ||
| a, b, c (Å) | 102.73, 105.08, 147.80 | 71.59, 105.32, 221.56 |
| α=β=γ (°) | 90.0 | 90.0 |
| Data collection | ||
| Wavelength (Å) | 1.03310 | 0.97934 |
| Resolution (Å) | 30.0-2.30(2.40-2.30) | 40.0-2.42(2.50-2.42) |
| Rsym (%) | 7.0(52.0) | 13.7(48.5) |
| I / σI | 10.6(2.2) | 11.3(3.0) |
| Completeness (%) | 97.6(98.5) | 99.5(95.5) |
| No. of unique reflections | 70007 | 64295 |
| Redundancy | 3.3(3.3) | 5.9(4.1) |
| Refinement | ||
| Resolution (Å) | 2.30(2.33-2.30) | 2.42(2.46-2.42) |
| Rwork / Rfree (%) | 17.6(27.1)/21.4(30.3) | 16.0(24.3)/20.0(30.3) |
| No. atoms | 8951 | 9142 |
| Protein | 8450 | 8408 |
| Ligand/ion | 20 | 54 |
| Water | 481 | 680 |
| Average B overall (Å2) | 45 | 29 |
| R.m.s.d. bond length (Å) | 0.012 | 0.012 |
| R.m.s.d. bond angle (°) | 1.279 | 1.293 |
| Ramachandran Plot | ||
| Core (%) | 89.5 | 89.8 |
| Allowed (%) | 10.4 | 10.2 |
| Gener. Allowed (%) | 0.1 | 0.0 |
Unbiased electron density maps show the locations and orientations of the inhibitors in the active sites and reveal the positions of ordered water molecules that we suggest are mechanistically important (Figure 2). All atoms of the carbamoylated portion of the inhibitors are ordered and visible in the electron density maps. As expected, the inhibitor carbonyl carbon atoms are covalently bonded to the catalytic Ser241 (Figures 2-4).
Figure 2.

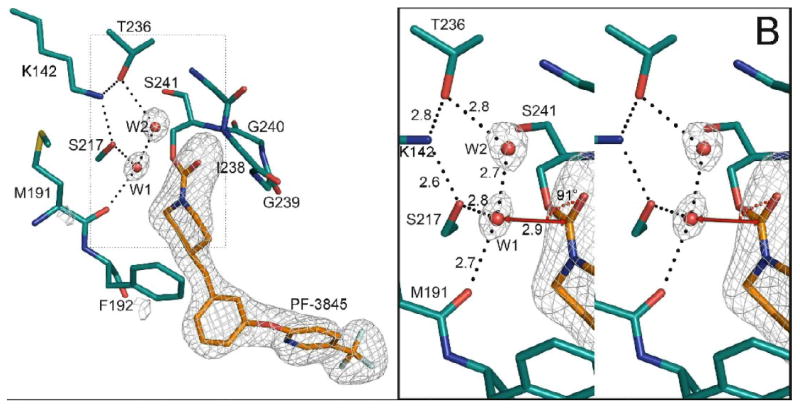
Structure of the catalytic core of FAAH bound to URB597 (panel A) and PF3845 (panel B). Unbiased electron density maps were calculated with the inhibitors and two water molecules omitted from the model. The Fo-Fc omit electron density maps have been contoured at 4.0 σ. The geometry around water molecule 1 is illustrated in the right hand panels. The vector connecting water 1 (W1) to the carbonyl carbon is shown as a red line. The W1⋯C=O angles range from 88 to 92° when measured in the four crystallographically independent monomers in our FAAH structures.
Figure 4.

Superposition of the FAAH–URB597 and FAAH–PF-3845 structures. The FAAH-URB597 structure is shown with inhibitor carbon atoms colored yellow, and protein carbon atoms colored green. The corresponding carbon atoms in the FAAH-PF-3845 structure are colored orange and cyan. A shift is apparent when comparing residues 192-195.
FAAH–inhibitor interactions can be conveniently divided into three regions: those that occur in the hydrophobic pocket (including the acyl-chain binding pocket and membrane access channels), those in the cytosolic port, and those localized to the catalytic core. The URB597 and PF-3845 inhibitors are trapped in a stable carbamoylated intermediate form, a species that is formed concomitant with the loss of inhibitor leaving groups. The interactions of these inhibitors with the cytosolic ports are thus not observed in the current structures. Protein-inhibitor interactions are therefore presented below in the context of the hydrophobic pocket and the catalytic core.
Protein-inhibitor interactions in the hydrophobic pocket of FAAH
The cyclohexyl moiety of URB597 binds in a cavity that connects the active site residues to the membrane surface of FAAH (Figure 3). This cavity is lined with hydrophobic residues that can assume multiple conformations depending on the structures of bound inhibitors (24). Three residues in particular, Phe432, Met436, and Met495, possess distinct conformations that cause profound alterations of the relative sizes of the membrane access channel and acyl-chain binding pocket (24, 25).
Figure 3.


Interactions of URB597 with the active site of FAAH. (A) URB597 covalently bound in the active site of h/rFAAH. Hydrogen bonds are indicated with dashed lines. The van der Waal’s surface of the inhibitor is shown as a dotted surface. Protein residues that contact URB597 are shown as sticks within transparent white spheres. (B) A schematic representation of URB597: FAAH interactions. Hydrogen bonds and CH-π interactions are shown in dashed and dotted lines, respectively.
Phe432 is located at the junction of the membrane access channel and acyl-binding pocket, and two distinct rotamers of this residue have been observed in previous structures. In the FAAH–URB597 structure, Phe432 has an orientation seen previously in the PF-3845 and MAFP containing structures, with a well defined acyl-chain binding pocket and a constricted membrane access channel. In this context, it contrasts with the OL-135 and PF-750 inhibitor bound structures. The FAAH–URB597 structure is unique among currently available structures in that no inhibitor atoms are in direct contact with Phe432. However, difference electron density maps contain unaccounted for electron density in the acyl-chain binding pocket near Phe432, suggesting that components from the crystallization buffer are present there at a low occupancy. It is therefore unclear whether the pose of Phe432 is a consequence of a lack of inhibitor contact (and potentially reflective of its pose in apo FAAH) or is influenced by the crystallization conditions.
Closer to the catalytic core, shifts of the main chain atoms of residues 192-195 occur in the context of various inhibitors (18, 24, 25). The hydrophobicity and flexibility of this region of FAAH is consistent with its role in binding the diverse acyl-chains of its various endogenous lipid substrates. Structural, mutational, and chemical approaches have been used to better characterize this portion of the enzyme with the twin goals of understanding substrate specificity and the generation of improved inhibitors (2, 17-18, 20, 24, 25, 30-33).
The region of the active site pocket occupied by the cyclohexyl group of URB597 overlaps that occupied by the piperidyl moieties of PF-750 and PF-3845 and the alkyl chains of inhibitors such as OL-135 or MAFP (18, 20, 24, 25). Although both URB597 and the PF compounds place six membered rings in this part of the active site, the poses of these rings differ (Figure 4). In contrast to the piperidyl rings of the PF inhibitors which are separated from the Oγ of Ser241 by one atom, the cyclohexyl of URB597 is separated by two atoms from the Oγ. The two classes of inhibitors thus have different inhibitor trajectories. As a consequence of these differences and to alleviate a protein–inhibitor steric clash, PF-750 and PF-3845 induce a shift in the position of the main chain atoms of residues 192-195 that is not observed in the URB597, OL-135 or FAAH–MAFP structures (18, 20, 24, 25). Indeed, the carbamate nitrogen of URB597 is 3.0-3.1 Å from the carbonyl oxygen of Ser193 with which it forms a hydrogen bond with good geometry. This unpredicted hydrogen bond, with the secondary carbamate nitrogen of the URB597 inhibitor, has not been observed previously in the urea structures, which are instead characterized by tertiary carbamate nitrogens (18, 25).
A number of residues appear to make favorable hydrophobic contacts with the cyclohexyl ring of URB597 (Figure 3). Phe192, Ile238, Val491, and Ser193 make van der Waals interactions with this moiety. Overall, approximately 250 Å2 of protein surface is shielded from solvent through interactions with the cyclohexyl ring of URB597. In addition, the inhibitor may form a weak aliphatic CH–π hydrogen bond with aromatic ring of Phe192 (Figure 3).
Protein-inhibitor interactions in the catalytic core of FAAH
As illustrated in Figure 4, the positions of the catalytic triad and oxyanion hole residues in the FAAH–URB597 and FAAH–PF-3845 structures superimpose well. The inhibitor interactions with these residues are well conserved, and have been described previously (20, 24, 25).
The higher resolution of the structures described here leads us to conclude that two unresolved features observed in the active sites of earlier structures and tagged there as dummy atoms (25) are, in fact, water molecules (Figure 2). The height of the W1 peak in the omit electron density map is 10σ in the URB597 and 9σ in the PF-3845 structure. The W2 molecule has corresponding peak heights of 6σ and 9σ. In the URB597 structure, the temperature factor for W1 is 28-30 Å2 and W2 is 32-42 Å2 (the average for all waters is 48 Å2). In the PF-3845 structure, overall temperature factors are slightly lower: W1 has a temperature factor of 16-25 Å2, W2 of 18-22 Å2, and the average for all waters in the structure is 37 Å2. In both inhibitor bound structures, the temperature factors for W1 and W2 are within the range expected for ordered water molecules. Electron density map peaks corresponding to the positions of W1 and W2 have also been observed in the FAAH–PF-750 acyl intermediate structure (atoms UNX1575–1577, PDB code 2VYA), but not in those of FAAH bound to MAFP or OL-135, where the waters are displaced by the methyl or central heterocycle moieties, respectively. W1 and W2 thus appear to be conserved in carbamoylated but not covalent, tetrahedral adducts with FAAH.
W1 is coordinated by FAAH in a fashion that places it close to the carbonyl carbon of the carbamoyl intermediates (Figure 2). It forms hydrogen bonds with the hydroxyl side chain of Ser217 (2.7–2.8 Å), the main chain carbonyl of Met191 (2.7–2.8 Å) and W2 (2.6–2.8 Å), through which it is indirectly coordinated to Thr236 (2.8–3.1 Å) in the cytosolic port and Ile238 in the oxyanion hole. Thr236 in turn also hydrogen bonds with the ε-amino moiety of the catalytically critical residue Lys142. The coordination of W1, places it far out of the FAAH:inhibitor carbamate plane (3.0 Å). A normal vector dropped from W1 down to the plane would come close to the inhibitor carbonyl carbon. To reduce bias, no user-input distance restraints involving W1 were used during the course of refinement with the program REFMAC 5.0. W1 is only 3.0–3.1 Å from the URB597 and 2.9 Å from the PF-3845 carbamate carbons.
DISCUSSION
Structure-activity relationships and the crystal structure of FAAH bound to URB597
Early modeling efforts docked URB597 into the structure of the FAAH–MAP complex in two fundamentally different ways, with the cyclohexyl moiety in the acyl-chain binding pocket or in the cytosolic port. Though initial synthetic studies failed to resolve this ambiguity (30, 31), later work based on mass spectrometry characterization of the carbamoylated-FAAH adduct (34), QM/MM mechanistic modeling efforts (35), and the characterization of a series of carbamic acid biphenyl esters (33) favored, consistent with our current structure, placing the cyclohexyl moiety in the acyl-chain binding pocket.
Piomelli and colleagues have generated extensive structure-activity relationship data replacing the cyclohexyl moiety of URB524 with a variety of alkyl and aromatic groups (33). These data can now be examined in the context of the FAAH–URB597 structure. They observed that substituting the cyclohexyl group with a methyl causes a 220-fold loss in potency, indicating the importance of protein-inhibitor interactions in this region of the active site. Cyclobutyl and cyclopentyl substitutions yielded slightly more favorable IC50s than observed for URB524. A norbornyl-substituted inhibitor can also be docked relatively easily within the active site and showed only a 4-fold drop in potency. However, modeling an adamantyl substitution into the crystal structure shows that it is too large to be easily accommodated within the active site, and indeed resulted in a 65-fold fall in potency.
One of the more dramatic changes in potency is the 60-fold drop observed when the cyclohexyl moiety is replaced by a phenyl group. The FAAH–URB597 structure provides an explanation for this decrease in activity. A search of the Cambridge Structural Database of small molecules yielded 82 high quality phenyl carbamates structures with hydrogens attached to the aniline nitrogen and the ortho positions of the aromatic ring (37, 38). The phenyl and carbamate planes were typically close to coplanar, with a standard deviation of 10.3°. The preferred conformation of a phenyl-substituted inhibitor would therefore place the aromatic plane perpendicular to the orientation of a least–squares fit plane of the six carbon atoms of the cyclohexyl ring in the FAAH–URB597 structure. In this perpendicular orientation, the phenyl moiety would clash with FAAH, indicating that either the phenyl-substituted inhibitor would have to assume a disfavored pose, or it would induce a protein conformational change. Consistent with this explanation, a benzyl rather than phenyl substitution, which would be expected to dramatically alter the plane of the aromatic ring, only decreases potency two-fold.
Replacing the carbamate nitrogen of URB524 with a carbon atom results in a 40-fold drop in potency (33). This effect can be ascribed to two possible causes. First, the substitution might change the intrinsic reactivity of carbonyl carbon. For example, changing the covalently-bound adduct from a carbamate to an ester species increases the electrophilicity of the carbonyl carbon and concomitantly may increase its susceptibility to hydrolysis and the regeneration of active enzyme. Second, the FAAH–URB597 crystal structure shows that the carbamate nitrogen of URB597 forms a hydrogen bond to the carbonyl of Ser193. Loss of this hydrogen bond would presumably directly reduce enzyme–inhibitor affinity and also indirectly further increase the electrophilicity of the carbonyl carbon. Further experiments would be required to determine the relative contributions of these two mechanisms to the loss of potency in the carbon substituted compound. In particular, it would be interesting to know whether the carbon-substituted compound URB524 can form as stable a FAAH adduct as URB597 (the FAAH:URB597 intermediate can survive a 22° C incubation for over 18 hours (39)).
Crystallographic waters in the catalytic core of FAAH
Modeling and analysis of crystallographic waters in the structures described here established the presence of two water molecules close to the carbamate group, which we called W1 and W2. Earlier structures contained unmodeled density peaks (18), or atoms flagged as unknown, at positions similar to those of W1 and W2 (25). However, given the lower resolution of these earlier data sets, we could not at the time unambiguously identify these features as water molecules.
We propose that W1 is the substrate deacylating water of FAAH in the acyl intermediate state conformation and that W2 indirectly coordinates it through Thr236 to Lys142 of the catalytic triad. The W1 to carbamate carbon distances in our FAAH structures range from 2.9 to 3.1 Å and the W1⋯C=O angles span 88–92°.
To confirm that this surprisingly close contact with W1 in these structures was physically reasonable, we searched for similar contacts in higher resolution crystallographic structures. A search of the February 2009 release of the Cambridge Structural Database (37, 38) revealed a number of contacts between carbonyl carbons and water molecules. We restricted our analyses to entries of single, well-ordered crystals of small organic molecules with R factors below 5%, and no annotated errors. The database contains 36 instances with water to carbonyl carbon distances between 2.8 and 3.1 Å, and an additional 45 occurrences between 3.1 and 3.2 Å. For most of these 81 close approaches, the water molecules lie far from the molecular plane containing the carbonyl moiety (further than 2.8 Å for three-quarters of the water molecules). The O··C=O angles have a broad distribution with a mean angle of 93° and a standard deviation of 12.7°. High-resolution small molecule structures containing close oxygen:carbonyl carbon contacts therefore demonstrate that the positions occupied by W1 molecules in our moderate resolution structures are precedented and energetically accessible. Nevertheless, as a class, close W1⋯C=O contacts are rare, and presumably disfavored (the number of such contacts with distances between 2.8 and 3.2 Å is ten times smaller than the number with distances between 3.2 and 3.6 Å)
Remarkably, the position of W1 in the active site of FAAH is similar to that of the deacylating waters in high-resolution structures of acyl-serine protease structures (Figure 5; 40-44). Because the amidase signature and serine protease families are evolutionarily independent and have completely different folds, they cannot be superimposed using Cα coordinates. Instead, a much more limited superposition, using the corresponding pairs of catalytic serine Cβ and inhibitor ester or carbamate atoms is informative. For example, such a superposition brings W1 within 0.4 Å of the catalytic water in a 1.6 Å resolution acyl-intermediate structure of elastase (Figure 5; 43, PDB code: 1HAX). This superposition also demonstrates the overlap of the catalytic triad histidine nitrogen with the hydroxyl of Ser217.
Figure 5.
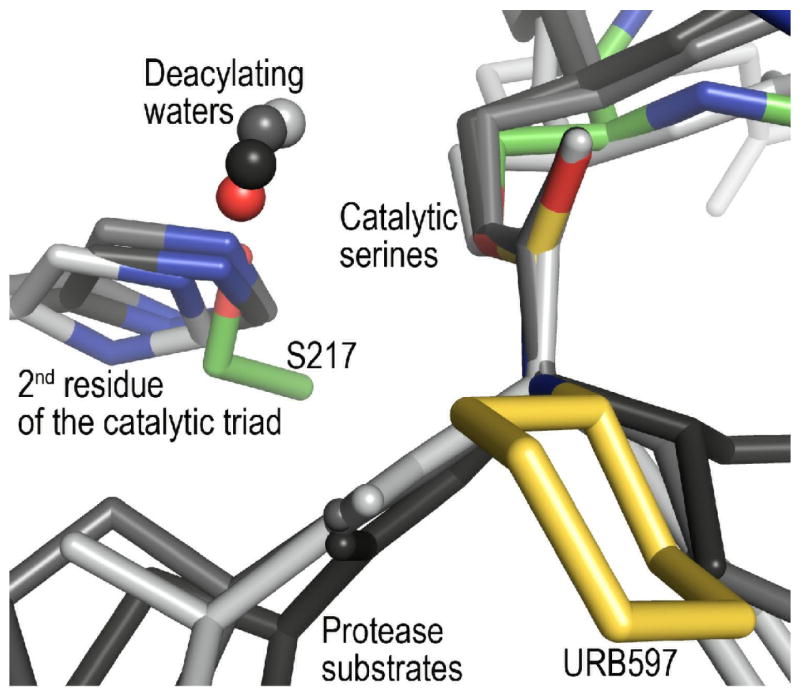
Superposition of FAAH–URB597 with serine proteases elastase (PDB code: 1HAX), trypsin (PDB code: 2AGE), and chymotrypsin (PDB code: 2GCT) with substrate and substrate analogs. The serine protease structures are shown in shades of gray, with their histidine nitrogen atoms colored blue. Atoms proposed to be deacylating waters are indicated by small spheres. Because the amidase signature enzymes are not homologous with the serine proteases, the proteins could not be superimposed using Cα coordinates. Instead, the serine Cβ and inhibitor ester or carbamate atoms were used for the superposition. When the structures are compared, it is apparent that the position of Ser217 in FAAH overlaps with those of the catalytic triad histidines in the serine proteases. The positions of the proposed deacylating water molecules also overlap.
High-resolution structures of acyl-serine protease complexes demonstrate that they have evolved the ability to position water molecules at defined positions close to the carbonyl carbons of acyl-enzyme intermediates (40-44), and our observations indicate that the members of the amidase signature family have also evolved a similar ability. This convergent evolution can be explained using Bürgi-Dunitz theory. From an examination of small molecule crystal structures, Bürgi et al. proposed that the preferred trajectory of nucleophilic attack on a carbonyl would follow a Nu⋯C=O angle of 105 ±5° (45). Subsequent work has defined this “Bürgi-Dunitz angle” as 107° (46). Faced with this fundamental physical property of the carbonyl bond, evolutionarily independent hydrolases appear to have positioned water molecules in active sites so that they can follow the trajectory defined by the Bürgi-Dunitz angle.
Disruption of the water trajectory could also contribute to our ability to obtain complexes that are irreversible, and therefore stable enough for crystallographic studies at stoichiometric ratios of inhibitor to protein. This is clearly the case for methyl arachidonyl phosphonate-labeled FAAH, where the position of the methoxy moiety overlaps that of W1 in the structures presented here, preventing FAAH from effectively coordinating the hydrolytic water.
The disruption of productive water-carbonyl interactions appears to also extend to the more subtle cases such as those presented here, where a hydrolytic water is observed in the crystal structure. The comparison with the serine proteases is again informative. Our FAAH-carbamoyl covalent complex structures show an approximately 15° deviation between their W1⋯C=O angles (88–92°) and the Bürgi-Dunitz angle. In previous crystal structures of trypsin proteases complexed with good substrates, similar deviations are observed with W1⋯C=O angles well below 100° (41). Indeed, even more subtle changes in active site geometry are known to have dramatic effects on reaction rates catalyzed by a wide range of enzymes (47).
On the basis of crystallographically observed atomic motions involved in the acylation (and deacylation) reactions, it has been proposed by Radisky et al. that in the case of the serine proteases the reaction coordinate is characterized by translations of the substrate’s carbonyl carbon and the attacking nucleophile, accompanied by concerted adjustments of the remaining residues of the catalytic triad (41). If similar motions occur in the amide hydrolases, as exemplified by FAAH, then it would be reasonable to expect that an additional source of inhibitor irreversibility be due to the intrinsic geometrical and chemical differences between the α-carbon of substrates (sp3) and the secondary (URB597) or tertiary (PF-3845) nitrogens of the planar carbamic groups. The bulkiness and rigidity of the inhibitors may impede the movements required for the optimal trajectory for the nucleophilic attack to occur. Further mechanistic and structural studies will be required to establish whether, in addition to binding hydrolytic water molecules similarly placed to those in the serine proteases, the amide hydrolases have also convergently evolved similar protein conformational movements.
CONCLUSION
Determining the location, reactivity, and dynamics of active site water molecules is critical for understanding the mechanisms of serine hydrolases, which depend on water for deacylation of acyl-enzyme intermediates and catalytic turnover. Obtaining such information requires the determination of a set of structures reflective of multiple steps in the reaction pathway at resolutions high enough for waters to be identified unambiguously. Despite the difficulties inherent in the iterative determination of crystal structures of a membrane protein, we are now beginning to assemble such a representative set for FAAH. In particular, the structures described here, and their comparison with serine protease structures have led us to identify an active site water that we propose plays a key catalytic role.
MATERIALS AND METHODS
FAAH Expression and Purification
A protocol for FAAH expression and purification has been described in detail previously (25). In brief, the pET28a vector containing the transmembrane-deleted version (ΔTM-FAAH, amino acids 30-579) of the humanized version of the rat FAAH gene (h/rFAAH) was heterologously expressed in the Escherichia coli strain BL21 A.I. (Invitrogen). The h/rFAAH construct replaces six amino acids in the active site of the rat FAAH protein (Leu192Phe, Phe194Tyr, Ala377Thr, Ser435Asn, Ile491Val, Val495Met) in order to recreate the binding profile of the human enzyme (25). Three chromatography steps including metal affinity, cation exchange, and size exclusion chromatography were used to reach a purity of greater than 95% as visualized by Coomassie blue stained SDS PAGE experiments (not shown). Protein concentrations were determined using a reducing-agent compatible BCA protein assay kit (Pierce Biotechnology). The inhibitors were added to the protein sample after the first step of purification and incubated at 4°C overnight. The inhibitor URB597 was purchased from Cayman Chemical and the inhibitor PF-3845 was prepared as previously described (18).
Crystallization and Crystal Structure Determination
The protein sample was concentrated to 25-30 mg/mL in size exclusion chromatography buffer containing 10 mM Hepes (pH 7.0), 500 mM NaCl, 2 mM dithiothreitol, and 0.08% n-undecyl-β-D-maltoside (Anatrace) or 0.1% n-decyl-β-D-maltoside for the crystallization of h/rFAAH–URB597and h/rFAAH–PF-3845 conjugates, respectively. Xylitol (Sigma) was added to the protein samples at a concentration of 12%. The FAAH–URB597 protein sample was supplemented with 1.6% benzyldimethyl(2-dodecyloxyethyl)-ammonium chloride (Aldrich) and mixed at a 1:1 proportion with a crystallization buffer containing (30% PEG400, 100 mM TrisHCl pH 7.5, and 100mM MgCl2). The FAAH–PF-3845 protein sample was supplemented with 1.6% benzyl-dimethyl-dodecyl ammonium bromide (Aldrich) and mixed at a 1:1 ratio with a crystallization buffer containing 30% PEG400, 100 mM 2-(N-morpholino)ethanesulfonate/NaOH pH 5.5, and 400mM LiCl. Crystals were grown by sitting drop vapor diffusion at 14 °C in 96-well plates (Innovaplate SD-2, Innovadyne Technologies) and frozen by plunging into liquid nitrogen directly after harvesting. The data were collected at a temperature of 100 K from a single crystal at the GM/CA-CAT beamline of the Advanced Photon Source (APS, Argonne, IL) using a 10-μm beam collimator. Data were processed using the XDS suite of programs (48). Structures were determined, modified and refined by using Refmac, Coot and other programs contained in the CCP4 package (49-51). The software suite Phenix (52) was used to refine individual atomic displacement parameters. Results from data processing and structure refinement are provided in Table 1. The structures were determined by molecular replacement using the coordinates from the highest resolution FAAH structure to date as a search model (24; PDB code: 2WJ1). Chemical parameters for the inhibitors were calculated by the Dundee PRODRG Web server (53). Figures were made using the program PyMol (54).
Acknowledgments
We gratefully acknowledge Doug Johnson and Scott Lazerwith for providing the inhibitor PF-3845, and the financial support of the National Institutes of Health (DA017259). Use of the Advanced Photon Source at Argonne National Laboratory was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
Footnotes
ACCESSION NUMBERS: Coordinates and structure factors have been deposited in the Protein Data Bank with accession number 3LJ6 (FAAH–PF-3845) and 3LJ7 (FAAH–URB597).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384(6604):83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 2.McKinney MK, Cravatt BF. Structure and function of fatty acid amide hydrolase. Annu Rev Biochem. 2005;74:411–432. doi: 10.1146/annurev.biochem.74.082803.133450. [DOI] [PubMed] [Google Scholar]
- 3.Ahn K, McKinney MK, Cravatt BF. Enzymatic pathways that regulate endocannabinoid signaling in the nervous system. Chem Rev. 2008;108(5):1687–1707. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci USA. 2001;98(16):9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cravatt BF, Saghatelian A, Hawkins EG, Clement AB, Bracey MH, Lichtman AH. Functional disassociation of the central and peripheral fatty acid amide signaling systems. Proc Natl Acad Sci USA. 2004;101(29):10821–10826. doi: 10.1073/pnas.0401292101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kathuria S, et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- 7.Lichtman AH, Leung D, Shelton C, Saghatelian A, Hardouin C, Boger D, Cravatt BF. Reversible inhibitors of fatty acid amide hydrolase that promote analgesia: evidence for an unprecedented combination of potency and selectivity. J Pharmacol Exp Ther. 2004;311(2):441–448. doi: 10.1124/jpet.104.069401. [DOI] [PubMed] [Google Scholar]
- 8.Lichtman AH, Shelton CC, Advani T, Cravatt BF. Mice lacking fatty acid amide hydrolase exhibit a cannabinoid receptor-mediated phenotypic hypoalgesia. Pain. 2004;109(3):319–327. doi: 10.1016/j.pain.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 9.Naidu PS, Varvel SA, Ahn K, Cravatt BF, Martin BR, Lichtman AH. Evaluation of fatty acid amide hydrolase inhibition in murine models of emotionality. Psychopharmacology (Berl) 2007;192(1):61–70. doi: 10.1007/s00213-006-0689-4. [DOI] [PubMed] [Google Scholar]
- 10.Huitron-Resendiz S, Sanchez-Alavez M, Wills DN, Cravatt BF, Henriksen SJ. Characterization of the sleep-wake patterns in mice lacking fatty acid amide hydrolase. Sleep. 2004;27(5):857–865. doi: 10.1093/sleep/27.5.857. [DOI] [PubMed] [Google Scholar]
- 11.Massa F, Marsicano G, Hermann H, Cannich A, Monory K, Cravatt BF, Ferri GL, Sibaev A, Storr M, Lutz B. The endogenous cannabinoid system protects against colonic inflammation. J Clin Invest. 2004;113(8):1202–1209. doi: 10.1172/JCI19465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang L, Luo L, Palmer JA, Sutton S, Wilson SJ, Barbier AJ, Breitenbucher JG, Chaplan SR, Webb M. Inhibition of fatty acid amide hydrolase produces analgesia by multiple mechanisms Brit. J Pharmacol. 2006;148(1):102–113. doi: 10.1038/sj.bjp.0706699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Russo R, Loverme J, La Rana G, Compton TR, Parrott J, Duranti A, Tontini A, Mor M, Tarzia G, Calignano A, Piomelli D. The fatty acid amide hydrolase inhibitor URB597 (cyclohexylcarbamic acid 3’ carbamoylbiphenyl-3-yl ester)reduces neuropathic pain after oral administration in mice. J Pharmacol Exp Ther. 2007;322(1):236–242. doi: 10.1124/jpet.107.119941. [DOI] [PubMed] [Google Scholar]
- 14.Moreira FA, Kaiser N, Lutz B. Reduced anxiety-like behavior induced by genetic and pharmacological inhibition of the endocannabinoid-degrading enzyme fatty acid amide hydrolase (FAAH) is mediated by CB1 receptors. Neuropharmacol. 2008;54(1):141–150. doi: 10.1016/j.neuropharm.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 15.Gobbi G, Bambico FR, Mangieri R, Bortolato M, Campolongo P, Solinas M, Cassano T, Morgese MG, Debonnel G, Duranti A, Tontini A, Tarzia G, Mor M, Trezza V, Goldberg SR, Cuomo V, Piomelli D. Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc Natl Acad Sci USA. 2005;103(7):10821–10826. doi: 10.1073/pnas.0509591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holt S, Comelli F, Fowler CJ. Inhibitors of fatty acid amide hydrolase reduce carrageenan-induced hind paw inflammation in pentobarbital-treated mice: comparison with indomethacin and possible involvement of cannabinoid receptors. Br J Pharmacol. 2005;146(3):467–476. doi: 10.1038/sj.bjp.0706348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seierstad M, Breitenbucher JG. Discovery and development of fatty acid amide hydrolase (FAAH) inhibitors. J Med Chem. 2008;51(23):7327–7343. doi: 10.1021/jm800311k. [DOI] [PubMed] [Google Scholar]
- 18.Ahn K, Johnson DS, Mileni M, Beidler D, Long JZ, McKinney MK, Weerapana E, Sadagopan N, Liimatta M, Smith SE, Lazerwith S, Stiff C, Kamtekar S, Bhattacharya K, Zhang Y, Swaney S, Van Becelaere K, Stevens RC, Cravatt BF. Discovery and characterization of a highly selective FAAH inhibitor that reduces inflammatory pain. Chem Biol. 2009;16(4):411–20. doi: 10.1016/j.chembiol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chebrou H, Bigey F, Arnaud A, Galzy P. Study of the amidase signature group. Biochim Biophys Acta. 1996;1298(2):285–293. doi: 10.1016/s0167-4838(96)00145-8. [DOI] [PubMed] [Google Scholar]
- 20.Bracey MH, Hanson MA, Masuda KR, Stevens RC, Cravatt BF. Structural adaptations in a membrane enzyme that terminates endocannabinoid signaling. Science. 2002;298(5599):1793–1796. doi: 10.1126/science.1076535. [DOI] [PubMed] [Google Scholar]
- 21.Curnow AW, Hong K, Yuan R, Kim S, Martins O, Winkler W, Henkin TM, Söll D. Glu-tRNAGln amidotransferase: a novel heterotrimeric enzyme required for correct decoding of glutamine codons during translation. Proc Natl Acad Sci USA. 1997;94(22):11819–11826. doi: 10.1073/pnas.94.22.11819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Labahn J, Neumann S, Büldt G, Kula MR, Granzin J. An alternative mechanism for amidase signature enzymes. J Mol Biol. 2002;322(5):1053–64. doi: 10.1016/s0022-2836(02)00886-0. [DOI] [PubMed] [Google Scholar]
- 23.Shin S, Lee TH, Ha NC, Koo HM, Kim SY, Lee HS, Kim YS, Oh BH. Structure of malonamidase E2 reveals a novel Ser-cisSer-Lys catalytic triad in a new serine hydrolase fold that is prevalent in nature. EMBO J. 2002;21(11):2509–16. doi: 10.1093/emboj/21.11.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mileni M, Garfunkle J, DeMartino JK, Cravatt BF, Boger DL, Stevens RC. Binding and Inactivation Mechanism of a Humanized Fatty Acid Amide Hydrolase by α-Ketoheterocycle Inhibitors Revealed from Cocrystal Structures. J Am Chem Soc. 2009;131(30):10497–10506. doi: 10.1021/ja902694n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mileni M, Johnson DS, Wang Z, Everdeen DS, Liimatta M, Pabst B, Bhattacharya K, Nugent RA, Kamtekar S, Cravatt BF, Ahn K, Stevens RC. Structure-guided inhibitor design for human FAAH by interspecies active site conversion. Proc Natl Acad Sci USA. 2008;105(35):12820–4. doi: 10.1073/pnas.0806121105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei BQ, Mikkelsen TS, McKinney MK, Lander ES, Cravatt BF. A second fatty acid amide hydrolase with variable distibution among placental mammals. J Biol Chem. 2006;281(48):36569–36578. doi: 10.1074/jbc.M606646200. [DOI] [PubMed] [Google Scholar]
- 27.Patricelli MP, Lovato MA, Cravatt BF. Chemical and mutagenic investigations of fatty acid amide hydrolase: evidence for a family of serine hydrolases with distinct catalytic properties. Biochem. 1999;38(31):9804–12. doi: 10.1021/bi990637z. [DOI] [PubMed] [Google Scholar]
- 28.Patricelli MP, Cravatt BF. Fatty acid amide hydrolase competitively degrades bioactive amides and esters through a nonconventional catalytic mechanism. Biochem. 1999;38(43):14125–39. doi: 10.1021/bi991876p. [DOI] [PubMed] [Google Scholar]
- 29.Tubert-Brohman I, Acevedo O, Jorgensen WL. Elucidation of hydrolysis mechanisms for fatty acid amide hydrolase and its Lys142Ala variant via QM/MM simulations. J Am Chem Soc. 2006;128(51):16904–13. doi: 10.1021/ja065863s. [DOI] [PubMed] [Google Scholar]
- 30.Tarzia G, Duranti A, Tontini A, Piersanti G, Mor M, Rivara S, Plazzi PV, Park C, Kathuria S, Piomelli D. Design, synthesis, and structure-activity relationships of alkylcarbamic acid aryl esters, a new class of fatty acid amide hydrolase inhibitors. J Med Chem. 2003;46(12):2352–60. doi: 10.1021/jm021119g. [DOI] [PubMed] [Google Scholar]
- 31.Mor M, Rivara S, Lodola A, Plazzi PV, Tarzia G, Duranti A, Tontini A, Piersanti G, Kathuria S, Piomelli D. Cyclohexylcarbamic acid 3’- or 4’-substituted biphenyl-3-yl esters as fatty acid amide hydrolase inhibitors: synthesis, quantitative structure-activity relationships, and molecular modeling studies. J Med Chem. 2004;47(21):4998–5008. doi: 10.1021/jm031140x. [DOI] [PubMed] [Google Scholar]
- 32.Ahn K, Johnson DS, Fitzgerald LR, Liimatta M, Arendse A, Stevenson T, Lund ET, Nugent RA, Nomanbhoy TK, Alexander JP, Cravatt BF. Novel mechanistic class of fatty acid amide hydrolase inhibitors with remarkable selectivity. Biochem. 2007;46(45):13019–30. doi: 10.1021/bi701378g. [DOI] [PubMed] [Google Scholar]
- 33.Mor M, Lodola A, Rivara S, Vacondio F, Duranti A, Tontini A, Sanchini S, Piersanti G, Clapper JR, King AR, Tarzia G, Piomelli D. Synthesis and quantitative structure-activity relationship of fatty acid amide hydrolase inhibitors: modulation at the N-portion of biphenyl-3-yl alkylcarbamates. J Med Chem. 2008;51(12):3487–98. doi: 10.1021/jm701631z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alexander JP, Cravatt BF. Mechanism of carbamate inactivation of FAAH: implications for the design of covalent inhibitors and in vivo functional probes for enzymes. Chem Biol. 2005;12(11):1179–87. doi: 10.1016/j.chembiol.2005.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lodola A, Mor M, Rivara S, Christov C, Tarzia G, Piomelli D, Mulholland AJ. Identification of productive inhibitor binding orientation in fatty acid amide hydrolase (FAAH) by QM/MM mechanistic modeling. Chem Commun. 2008;(2):214–216. doi: 10.1039/b714136j. [DOI] [PubMed] [Google Scholar]
- 36.Vacondio F, Silva C, Lodola A, Fioni A, Rivara S, Duranti A, Tontini A, Sanchini S, Clapper JR, Piomelli D, Mor M, Tarzia G. Structure–property relationships of a class of carbamate-based fatty acid amide hydrolase (FAAH) inhibitors: chemical and biological stability. ChemMedChem. 2009;4(9):1495–504. doi: 10.1002/cmdc.200900120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allen FH. The Cambridge Structural Database: a quarter of a million crystal structures and rising. Acta Cryst. 2002;B58:380–8. doi: 10.1107/s0108768102003890. [DOI] [PubMed] [Google Scholar]
- 38.Bruno IJ, Cole JC, Edgington PR, Kessler M, Macrae CR, McCabe P, Pearson J, Taylor R. New software for searching the Cambridge Structural Database and visualising crystal structures. Acta Cryst. 2002;B58:389–97. doi: 10.1107/s0108768102003324. [DOI] [PubMed] [Google Scholar]
- 39.Karbarz MJ, et al. Biochemical and biological properties of 4-(3-phenyl-[1,2,4] thiadiazol-5-yl)-piperazine-1-carboxylic acid phenylamide, a mechanism-based inhibitor of fatty acid amide hydrolase. Anesth Analg. 2009;108(1):330–3. doi: 10.1213/ane.0b013e31818c7cbd. [DOI] [PubMed] [Google Scholar]
- 40.Steinmetz AC, Demuth HU, Ringe D. Inactivation of subtilisin Carlsberg by N-((tert-butoxycarbonyl)alanylprolylphenylalanyl)-O-benzoylhydroxyl- amine: formation of a covalent enzyme-inhibitor linkage in the form of a carbamate derivative. Biochem. 1994;33(34):10535–44. doi: 10.1021/bi00200a040. [DOI] [PubMed] [Google Scholar]
- 41.Radisky ES, Lee JM, Lu CJ, Koshland DE., Jr Insights into the serine protease mechanism from atomic resolution structures of trypsin reaction intermediates. Proc Natl Acad Sci USA. 2006;103(18):6835–40. doi: 10.1073/pnas.0601910103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmidt A, Jelsch C, Ostergaard P, Rypniewski W, Lamzin VS. Trypsin revisited: crystallography at (sub) atomic resolution and quantum chemistry revealing details of catalysis. J Biol Chem. 2003;278(44):43357–62. doi: 10.1074/jbc.M306944200. [DOI] [PubMed] [Google Scholar]
- 43.Wilmouth RC, Clifton IJ, Robinson CV, Roach PL, Aplin RT, Westwood NJ, Hajdu J, Schofield CJ. Structure of a specific acyl-enzyme complex formed between beta-casomorphin-7 and porcine pancreatic elastase. Nat Struct Biol. 1997;4(6):456–62. doi: 10.1038/nsb0697-456. [DOI] [PubMed] [Google Scholar]
- 44.Dixon MM, Brennan RG, Matthews BW. Structure of gamma-chymotrypsin in the range pH 2.0 to pH 10.5 suggests that gamma-chymotrypsin is a covalent acyl-enzyme adduct at low pH. Int J Biol Macromol. 1991;13(2):89–96. doi: 10.1016/0141-8130(91)90054-x. [DOI] [PubMed] [Google Scholar]
- 45.Bürgi HB, Dunitz JD, Shefter E. Chemical reaction paths. IV. Aspects of O…C = O interactions in crystals. Acta Cryst. 1974;B30:1517–27. [Google Scholar]
- 46.Lodge EP, Heathcock CH. On the Origin of Diastereofacial Selectivity in Additions to Chiral Aldehydes and Ketones: Trajectory Analysis. J Am Chem Soc. 1987;109:2819–20. [Google Scholar]
- 47.Mesecar AD, Stoddard BL, Koshland DE., Jr Orbital steering in the catalytic power of enzymes: small structural changes with large catalytic consequences. Science. 1997;277(5323):202–6. doi: 10.1126/science.277.5323.202. [DOI] [PubMed] [Google Scholar]
- 48.Kabsch WJ. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Cryst. 1993;26:795–800. [Google Scholar]
- 49.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. 1997;D53(3):240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 50.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 51.Collaborative Computational Project, Number 4. The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst. 1994;D50:760–63. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 52.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Cryst. 2002;D58:1948–54. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 53.Schüttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Cryst. 2004;D60(8):1355–63. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 54.DeLano WL. The PyMOL Molecular Graphics System. 2002 http://www.pymol.org.