

Abstract
THC2, an autosomal-dominant thrombocytopenia described so far in only two families, has been ascribed to mutations in MASTL or ACBD5. Here, we show that ANKRD26, another gene within the THC2 locus, and neither MASTL nor ACBD5, is mutated in eight unrelated families. ANKRD26 was also found to be mutated in the family previously reported to have an ACBD5 mutation. We identified six different ANKRD26 mutations, which were clustered in a highly conserved 19 bp sequence located in the 5′ untranslated region. Mutations were not detected in 500 controls and are absent from the 1000 Genomes database. Available data from an animal model and Dr. Watson's genome give evidence against haploinsufficiency as the pathogenetic mechanism for ANKRD26-mediated thrombocytopenia. The luciferase reporter assay suggests that these 5′ UTR mutations might enhance ANKRD26 expression. ANKRD26 is the ancestor of a family of primate-specific genes termed POTE, which have been recently identified as a family of proapoptotic proteins. Dysregulation of apoptosis might therefore be the pathogenetic mechanism, as demonstrated for another thrombocytopenia, THC4. Further investigation is needed to provide evidence supporting this hypothesis.
Main Text
Inherited thrombocytopenias are a heterogeneous group of diseases characterized by a reduced number of blood platelets and a bleeding tendency that ranges from very mild to life threatening.1 Fifteen forms of inherited thrombocytopenias are described in OMIM (Online Mendelian Inheritance in Man). For some forms, the genetic defect has been identified in one of the many genes participating in the complex processes of megakaryopoiesis and platelet production, whereas for other forms the gene that is mutated is still unknown.1,2 Moreover, nearly 40% of patients with an inherited form of thrombocytopenia remain without a definite diagnosis because their condition has never been described or was not recognized as pertaining to a known disorder.3
Thrombocytopenia 2 (THC2 [MIM 188000]) is one of the rarest forms of autosomal-dominant thrombocytopenia. It has so far been reported in only two families, one from Italy and the other from North America.4,5 THC2-affected individuals had a degree of thrombocytopenia ranging from mild to severe and suffered from a mild bleeding diathesis without any major bleeding events. Morphological platelet studies did not identify any relevant defect, and in vitro studies did not reveal any functional abnormality. Thrombocytopenia was attributed to defective platelet production because examination of bone marrow found evident dysmegakaryocytopoietic phenomena in both families. The THC2 locus was mapped to chromosome 10p11.1-p12 through linkage analysis in two independent studies.4,5 Two missense changes in different linked genes were found to be causative of the disease: c.501G>C (p.Glu167Asp) (please note that this mutation was incorrectly named as c.565G>C in the original publication)6 of MASTL ([MIM 608221], NM_032844.3) in the North American family6 and c.22C>T (p.His8Tyr) of ACBD5 (NM_001042473.2) in the Italian one.7 Here we report evidence that, at least in the families we studied, THC2 does not derive from defects in either MASTL or ACBD5 but is associated with mutations in a third gene mapping to the same locus.
We studied four pedigrees of Italian ancestry in which 20 individuals showed a clinical phenotype consistent with THC2, in that they had autosomal-dominant, nonsyndromic thrombocytopenia without any morphological or functional platelet defect. Written informed consent was obtained from all study subjects or their parents or legal guardians. This study was approved by the Institutional Review Board of the IRCCS Policlinico San Matteo Foundation and was conducted according to Declaration of Helsinki principles. When all known forms of autosomal-dominant thrombocytopenia were excluded, linkage to the THC2 locus was investigated. Linkage analysis was performed with Merlin version 1.1.28 under a completely penetrant autosomal-dominant model with disease allele frequency of 0.0001. We selected nine microsatellite markers (D10S586, D10S572, D10S1775, D10S197, D10S111, D10S593, CArepeat1, CArepeat2, and D10S174) across the THC2 locus. All these markers were selected from the Marshfield Genetic Map, except for markers CArepeat1 and CArepeat2, which were identified directly from the genome sequence via the on-line tool Tandem Repeat Finder (Table S1). All available family members were genotyped, and the corresponding haplotypes are represented in Figure 1. Marker allele frequencies were inferred from genotyped individuals, and average male/female inter-marker cM distances were drawn from the Marshfield Genetic Map. Best haplotypes were estimated with the haplotyping function implemented in Merlin. Of the two larger pedigrees, pedigree 1 exceeded genome-wide significance for linkage at marker CArepeat1 with a pairwise LOD score of 3.31, whereas pedigree 2 showed consistent linkage to THC2 with a pairwise LOD score of 2.35 (Figure 1 and Table 2). In the two smaller pedigrees 3 and 4, a 10p11.1-p12 haplotype was transmitted consistently with disease segregation (Figure 1), but LOD scores were not significant because of the small size of the families (Table 2). Therefore, we searched for mutations in the coding exons and the respective flanking intronic regions of both MASTL and ACBD5 in probands from the four families. The analysis identified a few SNPs present in dbSNP but did not disclose any unreported variants. Because most of the SNPs were detected in the heterozygous state, we could also exclude large intragenic deletions (data not shown).
Figure 1.
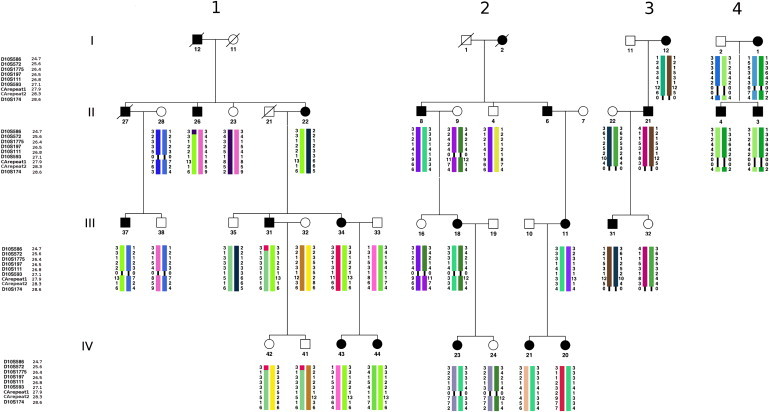
Linkage to Chromosome 10p11.1-p12 in Four THC2 Families
Segregation of microsatellite marker haplotypes in the THC2 locus on chromosome 10p11.1-p12 (with the corresponding Mb positions) in the two large THC2-linked families (Family 1 and 2) and in the two smaller families (families 3 and 4). Black symbols indicate affected individuals, and white symbols indicate healthy ones. Slashed symbols mean that those individuals are deceased. Only individuals for whom the corresponding haplotypes are reported were genotyped. Families 1 and 2, which carry the c.-128G>A and c.-127A>T mutations, respectively (Table 1), are consistent with linkage at the THC2 locus. Families 3 and 4 do not provide significant LOD scores, but their 10p11.1-p12 region segregates consistently with the disease. Family 3 carries the c.-118C>T mutation (Table 1). No mutation in ANKRD26 was found in the affected members of family 4, and therefore segregation of the haplotype is probably not related to the disease in this family. A 0/0 in the haplotype means unsuccessful genotyping for the marker in that individual. Haplotype representation was obtained with Haplopainter version 1.0.17
Table 2.
Single-Point and Multi-Point LOD Scores
| Family | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Marker | Multi-Point LOD Score | |||
| D10S586 | 3.1519 | 2.2887 | 0.6021 | 0.2706 |
| D10S572 | 3.3113 | 2.3979 | 0.6021 | 0.3010 |
| D10S1775 | 3.3113 | 2.3979 | 0.6021 | 0.3010 |
| D10S197 | 3.3113 | 2.3979 | 0.6021 | 0.3010 |
| D10S111 | 3.3113 | 2.4082 | 0.6013 | 0.3010 |
| D10S593 | 3.3113 | 2.4082 | 0.6021 | 0.3010 |
| CArepeat1 | 3.3113 | 2.4082 | 0.6021 | 0.3010 |
| CArepeat2 | 3.3113 | 2.4082 | 0.6021 | 0.3010 |
| D10S174 | 3.3113 | 2.4082 | 0.6021 | 0.3010 |
| Marker | Two-Point LOD Score | |||
| D10S586 | 0.4700 | 0.3010 | 0.6021 | 0.0000 |
| D10S572 | 2.7093 | 1.6428 | 0.6021 | 0.3010 |
| D10S1775 | 1.4313 | 1.4109 | −0.4861 | 0.3010 |
| D10S197 | 1.5065 | −0.2334 | 0.6021 | 0.3010 |
| D10S111 | 3.2325 | 0.1087 | 0.5324 | 0.0000 |
| D10S593 | 0.5523 | 0.5579 | 0.3010 | 0.3010 |
| CArepeat1 | 3.3113 | 2.3502 | 0.6021 | - |
| CArepeat2 | 1.5953 | 2.3087 | 0.6021 | - |
| D10S174 | 2.0967 | 0.4113 | 0.6021 | 0.3 |
A recombination fraction of 0 was used for single-point scores. A dash stands for “LOD score not calculated.”
Recombination events in the pedigrees we analyzed did not refine the THC2 locus, suggesting that a defect in a gene other than MASTL or ACBD5 is responsible for THC2. We therefore analyzed all the other 30 genes in the critical region defined by previous studies. Analysis of the entire coding sequences of all the positional candidate genes detected only known polymorphisms (data not shown). Interestingly, while we were sequencing the 5′ and 3′ untranslated regions (UTRs), we observed different heterozygous single nucleotide substitutions within the 5′ UTR of ANKRD26 (NM_014915.2). Nucleotide changes c.-118C>T, c.-127A>T, and c.-128G>A were described in probands from pedigrees 3, 2, and 1, respectively. We then decided to screen the 5′ UTR of ANKRD26 in 15 additional families, and in the family originally reported to carry a mutation in ACBD54,7. In seven patients from this last family, as well as in another pedigree, the c.-134G>A transition was found, whereas in four other families, c.-127A>T, c.-128G>A, or two further changes, c.-125T>G and c.-116C>T, were detected (Table 1; see also Figure S1). These variants, which always segregated with the linked haplotype along the pedigrees, were not found in 500 controls, nor were they reported in the 1000 Genomes database.
Table 1.
ANKRD26 5′ UTR Mutations Identified in Nine Families with Autosomal-Dominant Thrombocytopenia and Normal Platelet Size
In total, six different ANKRD26 mutations were associated with thrombocytopenia in all the 35 genotyped patients in nine out of 20 independent families, and one of these mutations was found in the original linkage family4. All of them were located in a stretch of 19 nucleotides of the 5′ UTR that is conserved among primates and cattle (Figure 2). Only affected individuals from each family carry the mutation, thus confirming complete penetrance of the trait.
Figure 2.
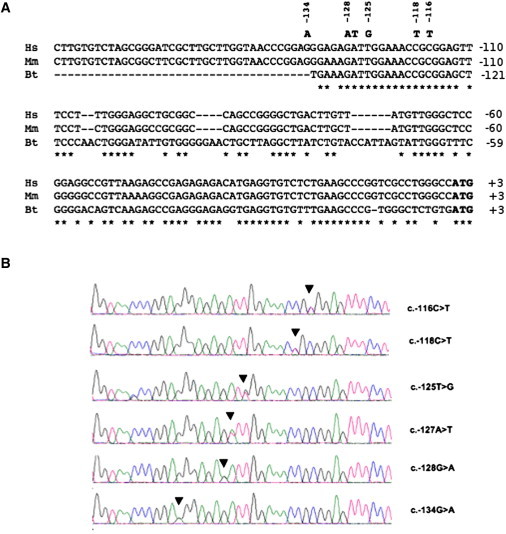
The 5′-UTR-Mutated Sequences of ANKRD26
(A) Alignment of the 5′ UTRs of orthologs from Homo sapiens (Hs: NM_014915.2), Macaca mulatta (Mm: XM_002808496.1), and Bos taurus (Bt: NM_001113767.1). Nucleotide changes are in bold. Stars indicate matching sites.
(B) Electropherograms showing the six different heterozygous mutations identified in THC2 families (Table 1) .
These findings indicate that the ANKRD26 mutations cause thrombocytopenia (Table 1) and support the idea that the p.His8Tyr mutation of ACBD5 segregating within the family is a private rare variant linked to the THC2 locus, rather than being related to the pathogenesis of thrombocytopenia.
ANKRD26 is the ancestor of a family of primate-specific genes termed POTE (Prostate-, Ovary-, Testis-, and placenta-Expressed genes) whose expression is restricted to a few normal tissues and a larger number of pathological tissues, such as breast cancer and many other cancers.9 With regard to human bone marrow cells, Macaulay et al. reported that ANKRD26 is expressed in megakaryocytes, and, to a lesser extent, in erythroid cells.10
The functional role of ANKRD26 is unknown. Mutant mice with partial inactivation of Ankrd26 develop extreme obesity, insulin resistance, and increased body size, whereas their platelet count is normal11 (T.K. Bera, personal communication). The recently released DNA sequence of James D. Watson's genome shows that he carries a heterozygous deletion of about 31.5 Kb involving the last six exons of the gene (Database of Genomic Variants, Variation_39047), but clinical signs of thrombocytopenia are not reported.12 Taken together, these data suggest that THC2 is more likely to be due to a gain of function effect rather than a haploinsufficiency of ANKRD26.
In order to define the pathogenetic effects of the 5′ UTR mutations, we cloned the wild-type and three mutant (c.-127A>T, c.-128G>A, and c.-134G>A) 5′ UTR sequences upstream of a reporter luciferase gene. These are the mutations segregating in three families with conclusive LOD scores, including the family in which the locus was originally mapped. We also included the c.-106T>C variant as a control. The constructs were transiently transfected into two different cell lines: the undifferentiated myeloid K562 cells, which derived from blast crisis of human chronic myelogenous leukemia, and the Dami cells, a human megakaryoblastic cell line whose maturation toward megakaryocytic lineage can be induced by treatment with phorbol 12-myristate 13-acetate (PMA) and thrombopoietin (TPO).13,14
To detect differences in expression, we performed a dual-luciferase reporter assay. We first assayed the K562 cells and noticed an average increase in expression from 2.7 to 4.5 times for the c.-134G>A clone with respect to all the other constructs. We then tested the megakaryoblastic Dami cells, either without or with PMA/TPO stimulation to induce megakaryocytic maturation. We performed a one-factor ANOVA to assess the effect of 5′ UTR ANKRD26 mutations on gene reporter expression levels in the two populations of cells and observed that stimulated Dami cells showed marked differences in expression among mutations (p < 0.001). Then, we carried out a two-tailed Dunnett's test for multiple comparisons against the c.-106T>C as an internal control in Dami – PMA/TPO and Dami + PMA/TPO. In the first group, the c.-128G>A and c.-134G>A but not the c.-127A>T constructs overexpressed the reporter gene with respect to the control. Finally, when we assayed Dami cells in which stimulation with PMA and TPO had induced megakaryocytic maturation (Figure S2), we observed overexpression for all three mutations (p = 0.016 for c.-127A>T and p < 0.001 for c.-128G>A and c.-134G>A; Figure 3).
Figure 3.
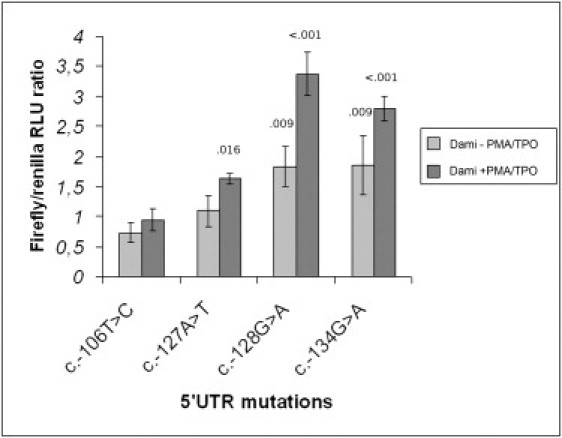
Firefly/Renilla RLU Ratios, Normalized against Wild-Type, for Each of the 5′ UTR Variants in the Functional Study
Scale bars represent means ± standard deviation. Corresponding values of each variant for – PMA/TPO Dami cells (light gray) and + PMA/TPO Dami cells (dark gray) are as follows: c.-106T>C—0.73 ± 0.16, 0.94 ± 0.19; c.-127A>T— 1.09 ± 0.26, 1.63 ± 0.91; c.-128G>A—1.83 ± 0.19, 3.37 ± 0.36; and c.-134G>A—1.84 ± 0.49, 2.79 ± 0.20. The assumption of homogeneity of variances was respected both in −PMA/TPO and in +PMA/TPO Dami (Levene's statistic > .13 and .36, respectively). Normality of the distribution was respected for all the samples (Kolmogorov-Smirnov Z Test). ANOVA rejected the null hypothesis of equality of means in both groups. Significant p values at the 5% level for a Dunnett's test against the c.-106T>C control are reported above the corresponding column of the histogram.
We then estimated the relative contribution of mutations and cell maturation to the variation in expression with a two-factor ANOVA and found that the largest effect was due to the presence of alterations in the 5′ UTR sequence (p < 0.001, partial η2 of 0.9) rather than to PMA-TPO stimulation (p < 0.001, partial η2 of 0.73). This is consistent with a scenario in which the mutation interferes with reporter gene expression, and cell maturation toward a megakaryocytic lineage then amplifies this effect. On the basis of these results, we can speculate that the mutations observed in THC2 patients interfere with the mechanisms controlling the expression of ANKRD26 and affect megakaryopoiesis and platelet production, possibly by induction of apoptosis. Recently, Liu et al. identified POTE as a new family of proapoptotic proteins.15 Morison et al. demonstrated that a different autosomal-dominant thrombocytopenia (THC4, [MIM 612004]) derives from increased apoptotic activity due to a cytochrome c mutation.16 Their observations suggest that platelet formation is particularly sensitive to changes in the intrinsic apoptotic pathway. Bone marrow examination, performed in two patients with different ANKRD26 mutations (families 3 and 12), showed that megakaryocytes were present in normal number and that all their maturation stages were represented. This observation, although preliminary, suggests that thrombocytopenia could derive from a defect of platelet release and/or a reduced platelet life span. The preliminary expression data we present support but do not prove that increased expression and subsequent apoptosis in megakaryocytes is a plausible pathogenic mechanism. These arguments will direct further investigation aimed at clarifying the molecular events leading to THC2.
We conclude that mutations in the 5′ UTR of ANKRD26 are implicated in THC2. Analysis of this gene in the North American family previously described6 will clarify whether THC2 is a genetically heterogeneous disease or the MASTL variant is benign.
Acknowledgments
We thank all patients and their families for participating in the project. We gratefully acknowledge the Comitato Telethon Fondazione Onlus (grant number GGP10089), the IRCCS Burlo Garofolo (Grant Ricerca Corrente 39/09), the Italian ISS (Istituto Superiore di Sanità; Italian/USA Grant on Rare Diseases) and the “Francesco Fede” Department of the Second University of Naples (Grant on Normal and Pathological Hematopoiesis) for financial support of this study. We are grateful to Kerry Rhoden for helpful discussion about the manuscript.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 genomes, http://browser.1000genomes.org/
Database of Genomic Variants, http://projects.tcag.ca/variation/
Marshfield Genetic Map, http://www.bli.uzh.ch/BLI/Projects/genetics/maps/marsh.html
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim
Tandem Repeat Finder, http://tandem.bu.edu/trf/trf.html
References
- 1.Nurden A.T., Nurden P. Inherited thrombocytopenias. Haematologica. 2007;92:1158–1164. doi: 10.3324/haematol.11256. [DOI] [PubMed] [Google Scholar]
- 2.Balduini C.L., Savoia A. Inherited thrombocytopenias: Molecular mechanisms. Semin. Thromb. Hemost. 2004;30:513–523. doi: 10.1055/s-2004-835672. [DOI] [PubMed] [Google Scholar]
- 3.Noris P., Pecci A., Di Bari F., Di Stazio M.T., Di Pumpo M., Ceresa I.F., Arezzi N., Ambaglio C., Savoia A., Balduini C.L. Application of a diagnostic algorithm for inherited thrombocytopenias to 46 consecutive patients. Haematologica. 2004;89:1219–1225. [PubMed] [Google Scholar]
- 4.Savoia A., Del Vecchio M., Totaro A., Perrotta S., Amendola G., Moretti A., Zelante L., Iolascon A. An autosomal dominant thrombocytopenia gene maps to chromosomal region 10p. Am. J. Hum. Genet. 1999;65:1401–1405. doi: 10.1086/302637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Drachman J.G., Jarvik G.P., Mehaffey M.G. Autosomal dominant thrombocytopenia: Incomplete megakaryocyte differentiation and linkage to human chromosome 10. Blood. 2000;96:118–125. [PubMed] [Google Scholar]
- 6.Gandhi M.J., Cummings C.L., Drachman J.G. FLJ14813 missense mutation: A candidate for autosomal dominant thrombocytopenia on human chromosome 10. Hum. Hered. 2003;55:66–70. doi: 10.1159/000071812. [DOI] [PubMed] [Google Scholar]
- 7.Punzo F., Mientjes E.J., Rohe C.F., Scianguetta S., Amendola G., Oostra B.A., Bertoli-Avella A.M., Perrotta S. A mutation in the acyl-coenzyme A binding domain-containing protein 5 gene (ACBD5) identified in autosomal dominant thrombocytopenia. J. Thromb. Haemost. 2010;8:2085–2087. doi: 10.1111/j.1538-7836.2010.03979.x. [DOI] [PubMed] [Google Scholar]
- 8.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin-rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 9.Hahn Y., Bera T.K., Pastan I.H., Lee B. Duplication and extensive remodeling shaped POTE family genes encoding proteins containing ankyrin repeat and coiled coil domains. Gene. 2006;366:238–245. doi: 10.1016/j.gene.2005.07.045. [DOI] [PubMed] [Google Scholar]
- 10.Macaulay I.C., Tijssen M.R., Thijssen-Timmer D.C., Gusnanto A., Steward M., Burns P., Langford C.F., Ellis P.D., Dudbridge F., Zwaginga J.J. Comparative gene expression profiling of in vitro differentiated megakaryocytes and erythroblasts identifies novel activatory and inhibitory platelet membrane proteins. Blood. 2007;109:3260–3269. doi: 10.1182/blood-2006-07-036269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bera T.K., Liu X.F., Yamada M., Gavrilova O., Mezey E., Tessarollo L., Anver M., Hahn Y., Lee B., Pastan I. A model for obesity and gigantism due to disruption of the Ankrd26 gene. Proc. Natl. Acad. Sci. USA. 2008;105:270–275. doi: 10.1073/pnas.0710978105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wheeler D.A., Srinivasan M., Egholm M., Shen Y., Chen L., McGuire A., He W., Chen Y.J., Makhijani V., Roth G.T. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 13.Deutsch V., Bitan M., Friedmann Y., Eldor A., Vlodavsky I. Megakaryocyte maturation is associated with expression of the CXC chemokine connective tissue-activating peptide CTAP III. Br. J. Haematol. 2000;111:1180–1189. doi: 10.1046/j.1365-2141.2000.02476.x. [DOI] [PubMed] [Google Scholar]
- 14.van der Vuurst H., Hendriks M., Lapetina E.G., van Willigen G., Akkerman J.W. Maturation of megakaryoblastic cells is accompanied by upregulation of G(s)alpha-L subtype and increased cAMP accumulation. Thromb. Haemost. 1998;79:1014–1020. [PubMed] [Google Scholar]
- 15.Liu X.F., Bera T.K., Liu L.J., Pastan I. A primate-specific POTE-actin fusion protein plays a role in apoptosis. Apoptosis. 2009;14:1237–1244. doi: 10.1007/s10495-009-0392-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morison I.M., Cramer Bordé E.M., Cheesman E.J., Cheong P.L., Holyoake A.J., Fichelson S., Weeks R.J., Lo A., Davies S.M., Wilbanks S.M. A mutation of human cytochrome c enhances the intrinsic apoptotic pathway but causes only thrombocytopenia. Nat. Genet. 2008;40:387–389. doi: 10.1038/ng.103. [DOI] [PubMed] [Google Scholar]
- 17.Thiele H., Nürnberg P. HaploPainter: A tool for drawing pedigrees with complex haplotypes. Bioinformatics. 2005;21:1730–1732. doi: 10.1093/bioinformatics/bth488. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.