

Abstract
Systemic lupus erythematosus (SLE) is considered to be the prototypic autoimmune disease, with a complex genetic architecture influenced by environmental factors. We sought to replicate a putative association at 11p13 not yet exceeding genome-wide significance (p < 5 × 10−8) identified in a genome-wide association study (GWAS). Our GWA scan identified two intergenic SNPs located between PDHX and CD44 showing suggestive evidence of association with SLE in cases of European descent (rs2732552, p = 0.004, odds ratio [OR] = 0.78; rs387619, p = 0.003, OR = 0.78). The replication cohort consisted of >15,000 subjects, including 3562 SLE cases and 3491 controls of European ancestry, 1527 cases and 1811 controls of African American (AA) descent, and 1265 cases and 1260 controls of Asian origin. We observed robust association at both rs2732552 (p = 9.03 × 10−8, OR = 0.83) and rs387619 (p = 7.7 × 10−7, OR = 0.83) in the European samples with pmeta = 1.82 × 10−9 for rs2732552. The AA and Asian SLE cases also demonstrated association at rs2732552 (p = 5 × 10−3, OR = 0.81 and p = 4.3 × 10−4, OR = 0.80, respectively). A meta-analysis of rs2732552 for all racial and ethnic groups studied produced pmeta = 2.36 × 10−13. This locus contains multiple regulatory sites that could potentially affect expression and functions of CD44, a cell-surface glycoprotein influencing immunologic, inflammatory, and oncologic phenotypes, or PDHX, a subunit of the pyruvate dehydrogenase complex.
Main Text
Systemic lupus erythematosus (SLE [MIM 152700]) is a chronic, heterogeneous autoimmune disorder characterized by inflammation, loss of tolerance to self-antigens, and dysregulated interferon responses. Defining features of the disease are infiltration of lymphocytes into organs such as the kidney and skin, as well as autoantibody production. Both environmental and genetic (sibling risk ratio, λs ≈ 30) factors are important in SLE etiology, though much remains to be learned. Candidate gene studies and, more recently, genome-wide association studies (GWAS), have begun to elucidate the complex genetic architecture of SLE with identification of >30 risk loci.1 These studies have collectively established the importance of several pathways in SLE, including innate immune responses, activation of lymphocytes, and immune complex clearing.1
Although many new loci have been identified as contributing to the pathogenesis of SLE, they collectively do not explain all the risk contributed by heritable factors. For example, recessive effects remain a challenge to detect in the studies conducted to date. Much larger sample sizes (>10,000 cases and 10,000 controls) are needed to detect such effects. Rare or private mutations are also difficult to detect under recently used study designs. The majority of established genetic effects to date have been identified through studies in cohorts of European descent. There are distinct clinical differences between racial groups, including higher risk in African Americans and Asians for developing more severe disease. Previous studies have also suggested that risk haplotypes between groups differ, as illustrated by ITGAM (MIM 120980), in which the differences in haplotype structure were used to identify the causal variant.2
In this study, we sought to replicate a suggestive association at 11p13 not reaching genome-wide significance (p < 5 × 10−8) in our previous GWAS3 that had also been identified in a linkage study of SLE when evaluating multiplex pedigrees with thrombocytopenia.4 This region contains a gene, CD44 (MIM 107269), that has been well studied at the protein level in relation to SLE risk, as well as many other inflammatory conditions. Here we replicate association with two SNPs identified in the GWAS just telomeric to CD44.
The initial GWAS was performed with the Affymetrix Genome-wide Human SNP array 5.0 with a sample size of 431 European SLE cases and 2,155 European controls, as described in Graham et al., 2008.3 The multiethnic replication study consisted of 17,003 total samples (8,922 SLE cases and 8,077 controls), which included self-reported African Americans, Asians, Europeans, Gullahs, Hispanics, and Amerindians (Table 1). The samples were assembled at the Oklahoma Medical Research Foundation (OMRF) after collection through multiple institutions around the world, following ethics committee approval and informed consent. All cases fulfilled American College of Rheumatology criteria for the classification of SLE.5
Table 1.
Summary of Samples Genotyped
| Populations | Number of Samples | Case | Control | Unknown Disease Status | Male | Female | Unknown Gender |
|---|---|---|---|---|---|---|---|
| African Americans | 3462 | 1569 | 1893 | 0 | 723 | 2733 | 6 |
| Asians | 2676 | 1328 | 1348 | 0 | 296 | 2380 | 0 |
| Europeans | 8066 | 4248 | 3818 | 0 | 1642 | 6403 | 21 |
| Gullah | 286 | 155 | 131 | 0 | 34 | 252 | 0 |
| Hispanics | 1383 | 1033 | 350 | 0 | 160 | 1222 | 1 |
| Amerindians | 1126 | 589 | 537 | 0 | 67 | 1,057 | 2 |
| Unknown | 4 | 0 | 0 | 4 | 0 | 4 | 0 |
| Total | 17,003 | 8922 | 8077 | 4 | 2922 | 14,051 | 30 |
The replication data were generated with the Illumina iSelect technology at the OMRF. A total of 119 SNPs encompassing CD44 and within the linkage peak previously observed at 11p13 (2 Mb interval) plus 347 ancestral-informative markers (AIMs) typed throughout the genome were evaluated. SNPs used in the analysis were required to pass stringent quality control criteria that included well-defined cluster scatter plots, >90% call rates across the entire study, Hardy-Weinberg proportions with p > 0.01 in controls and p > 0.0001 in cases, total proportion missing <5%, and p > 0.05 for differential missingness between cases and controls.
Samples with a <90% call rate or increased heterozygosity (>5 standard deviations from the mean) were excluded from the analysis. The remaining samples were then evaluated for duplicates or related individuals, and one individual from each pair was removed if the proportion of alleles shared identity by descent was > 0.4. Samples were assessed for mismatches between reported gender and genetic data. Assigned males were required to have chromosomal X heterozygosity ≤ 10% and be heterozygous at rs2557523, because the G allele for this SNP is only observed on the Y chromosome, and the A allele appears only on the X chromosome. Assigned females were required to have chromosomal X heterozygosity > 10% and be homozygous at rs2557523.
Finally, genetic outliers were removed from further analysis, as determined by principal component analysis (PCA) and admixture estimates (Figure 1).6,7 Price et al. utilized PCA for correcting for population stratification by inferring continuous axes of genetic variation on genotype data that is implemented in EIGENSTRAT software.6 Another method, combining Bayesian and sampling-theory approaches, has been proposed to estimate admixture proportions in multiple populations7–9 and has been implemented in ADMIXMAP software. Both EIGENSTRAT6 and ADMIXMAP8,9 were used to identify population strata within the samples with AIMs, with both yielding similar results. The AIMs were selected to distinguish four continental ancestral populations: Africans, Europeans, American Indians, and East Asians (Figure 1).10,11 We utilized principal components from EIGENSTRAT outputs to identify outliers from each population cluster. After quality control, a total of 1,139 samples was excluded (Table 2). Final numbers of subjects included in analyses were 15,490 for replication and 18,076 with the GWAS samples included (Table 3).
Figure 1.
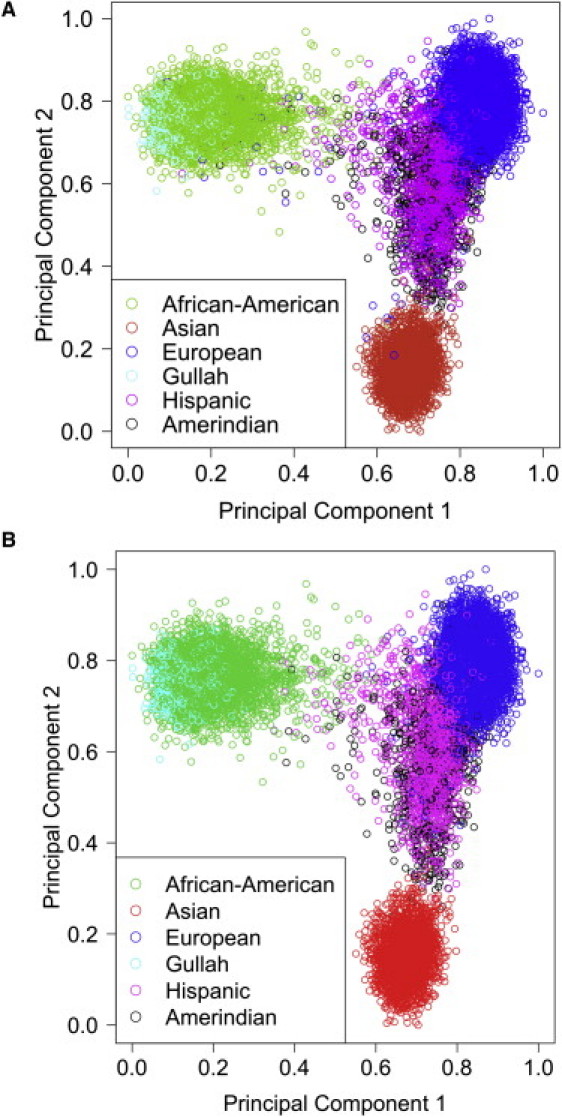
Principal Components Identify Continental Ancestry
Plot of the first two principal components identifying continental population substructure within our sample set before quality control (QC) (A) with 17,003 subjects and after QC (B) with 15,490 subjects. Each circle represents an individual sample, and colors represent the populations based on self-report.
Table 2.
Summary of Samples Remaining after QC
| Populations | Number of Samples | Case | Control | Unknown Disease Status | Male | Female | Unknown Gender |
|---|---|---|---|---|---|---|---|
| African Americans | 3338 | 1527 | 1811 | 0 | 695 | 2643 | 0 |
| Asians | 2525 | 1265 | 1260 | 0 | 253 | 2272 | 0 |
| Europeans | 7053 (7427)a | 3562 (3936)a | 3491 | 0 | 1495 | 5932 | 0 |
| Gullah | 275 | 152 | 123 | 0 | 33 | 242 | 0 |
| Hispanics | 1297 | 961 | 336 | 0 | 149 | 1148 | 0 |
| Amerindians | 1002 | 531 | 471 | 0 | 58 | 944 | 0 |
| Unknown | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Total | 15,490 (15864)a | 7998 (8372)a | 7492 | 0 | 2683 | 13,181 | 0 |
Number of European cases before removing 374 samples to render replication independent from GWAS.
Table 3.
Summary of Sample Data Sets
| Dataset | Cases | Controls | Total |
|---|---|---|---|
| GWAS | 431 | 2155 | 2586 |
| Multiethnic replication | 7998 | 7492 | 15,490 |
| Combined | 8429 | 9647 | 18,076 |
To test for SNP-SLE association in the replication study, we performed logistic regression, as implemented in PLINK.12 The additive, dominant, and recessive models were calculated while adjusting for the first three principle components and gender. Models were also analyzed with the ancestry estimates provided by ADMIXMAP, with no observable difference. Meta-analyses of the SNPs observed in both the GWAS and the multiethnic study were calculated with a weighted Z score (Stouffer's Ztrend), as implemented in METAP.13 Here the weight is the square root of the sample size for each group. This controls for differences in sample size between studies when combining.
Our previously published GWAS identified two SNPs in strong linkage disequilibrium (LD, r2 = 0.94) located ∼74 kb telomeric to CD44, showing suggestive evidence of association with European SLE cases (rs2732552, p = 0.004, odds ratio [OR] = 0.78, 95% confidence interval [CI] = 0.69–0.93; rs387619, p = 0.003, OR = 0.78, 95% CI = 0.68–0.91). In the current independent study, we evaluated this region in a large multiethnic case-control collection of 17,003 subjects (before quality control; Table 1). We observed robust association at both rs2732552 (p = 9.03 × 10−8, OR = 0.83, 95% CI = 0.77–0.88) and rs387619 (p = 7.7 × 10−7, OR = 0.83, 95% CI = 0.77–0.90) in the European samples (Table 4 and Figure 2A). Meta-analyses of these two SNPs between our current European data set and that used in the GWAS, accounting for differences in sample size with a weighted Z score, produced results that surpass genome-wide thresholds for significance and yielded a pmeta = 1.82 × 10−9 (OR = 0.82, 95% CI = 0.76–0.88) for rs2732552 and a pmeta = 1.46 × 10−8 (OR = 0.82, 95% CI = 0.76–0.88) for rs387619 (Table 4 and Figure 2A).
Table 4.
Results of Observed and Imputed SNPs at 11p13 Telomeric of CD44a
| SNP | Position (Mb) |
European |
Asian |
African American |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Allelesb | MAF | GWAS | p | OR (95% CI)c | pmeta (OR, 95% CI) | Allelesb | MAF | p | OR (95% CI)c | Allelesb | MAF | p | OR (95% CI)c | ||
| rs2732552 | 35.041168 | C/T | 0.43 | 4.00E−03 | 9.03E−08 | 0.83 (0.77–0.89) | 1.82E−09 (0.82, 0.76–0.88) | C/T | 0.27 | 4.31E−04 | 0.80 (0.71–0.90) | T/C | 0.34 | 5.36E−03 | 0.81 (0.94–0.70) |
| rs2785202 | 35.041411 | C/G | 0.42 | 8.04E−08 | 0.83 (0.77–0.89) | C/G | 0.27 | 4.09E−04 | 0.80 (0.70–0.90) | G/C | 0.36 | 8.24E−04 | 0.77 (0.66–0.90) | ||
| rs2553772 | 35.042029 | G/T | 0.43 | 7.35E−08 | 0.82 (0.77–0.88) | G/T | 0.24 | 1.05E−04 | 0.76 (0.66–0.87) | N/A | N/A | N/A | N/A | ||
| rs2732550 | 35.044894 | G/T | 0.42 | 3.21E−07 | 0.83 (0.77–0.89) | G/T | 0.24 | 1.55E−04 | 0.76 (0.66–0.88) | G/T | 0.01 | 0.060 | 0.44 (0.19–1.03) | ||
| rs2732549 | 35.044975 | A/G | 0.43 | 1.58E−07 | 0.83 (0.77–0.89) | A/G | 0.24 | 1.34E−04 | 0.76 (0.66–0.88) | N/A | N/A | N/A | N/A | ||
| rs2732547 | 35.045259 | C/T | 0.42 | 1.44E−07 | 0.83 (0.77–0.89) | C/T | 0.27 | 2.07E−04 | 0.79 (0.69–0.89) | T/C | 0.41 | 6.55E−03 | 0.78 (0.65–0.93) | ||
| rs2785201 | 35.045414 | C/G | 0.42 | 1.70E−07 | 0.83 (0.77–0.89) | C/G | 0.21 | 2.69E−03 | 0.79 (0.68–0.92) | G/C | 0.41 | 5.29E−03 | 0.77 (0.64–0.93) | ||
| rs2732546 | 35.045661 | C/T | 0.43 | 1.79E−07 | 0.83 (0.77–0.89) | C/T | 0.27 | 6.04E−04 | 0.80 (0.70–0.91) | N/A | N/A | N/A | N/A | ||
| rs1895821 | 35.046097 | C/T | 0.43 | 2.64E−07 | 0.83 (0.77–0.89) | C/T | 0.26 | 3.45E−04 | 0.79 (0.70–0.90) | N/A | N/A | N/A | N/A | ||
| rs675970 | 35.04692 | G/A | 0.42 | 1.78E−07 | 0.83 (0.77–0.89) | G/A | 0.24 | 1.44E−04 | 0.76 (0.66–0.88) | N/A | N/A | N/A | N/A | ||
| rs1834459 | 35.049346 | A/G | 0.42 | 6.13E−07 | 0.83 (0.78–0.90) | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | ||
| rs2732544 | 35.049527 | C/T | 0.43 | 3.82E−07 | 0.83 (0.77–0.89) | C/T | 0.26 | 5.98E−04 | 0.80 (0.70–0.91) | N/A | N/A | N/A | N/A | ||
| rs2785198 | 35.049605 | A/G | 0.42 | 5.90E−07 | 0.83 (0.78–0.90) | A/G | 0.26 | 6.63E−04 | 0.80 (0.70–0.91) | N/A | N/A | N/A | N/A | ||
| rs2785197 | 35.049646 | G/A | 0.43 | 3.64E−07 | 0.83 (0.77–0.89) | G/A | 0.26 | 3.95E−04 | 0.79 (0.70–0.90) | N/A | N/A | N/A | N/A | ||
| rs1116470 | 35.051067 | C/T | 0.43 | 2.86E−07 | 0.83 (0.77–0.89) | C/T | 0.25 | 1.72E−04 | 0.77 (0.67–0.88) | N/A | N/A | N/A | N/A | ||
| rs1116471 | 35.051208 | G/A | 0.43 | 3.16E−07 | 0.83 (0.77–0.89) | G/A | 0.25 | 1.96E−04 | 0.77 (0.67–0.88) | N/A | N/A | N/A | N/A | ||
| rs2785194 | 35.05183 | C/T | 0.43 | 2.75E−07 | 0.83 (0.77–0.89) | C/T | 0.25 | 1.96E−04 | 0.77 (0.67–0.88) | T/C | 0.29 | 0.015 | 0.81 (0.69–0.96) | ||
| rs2785193 | 35.052011 | C/A | 0.43 | 2.51E−07 | 0.83 (0.77–0.89) | C/A | 0.25 | 1.96E−04 | 0.77 (0.67–0.88) | N/A | N/A | N/A | N/A | ||
| rs2732540 | 35.052062 | G/A | 0.44 | 2.57E−06 | 0.84 (0.78–0.90) | G/A | 0.23 | 1.62E−04 | 0.77 (0.67–0.88) | N/A | N/A | N/A | N/A | ||
| rs1834460 | 35.052141 | C/T | 0.43 | 3.25E−07 | 0.83 (0.77–0.89) | C/T | 0.25 | 1.57E−04 | 0.77 (0.67–0.88) | T/C | 0.29 | 5.40E−03 | 0.79 (0.68–0.93) | ||
| rs2098878 | 35.052886 | G/A | 0.43 | 2.24E−07 | 0.83 (0.77–0.90) | G/A | 0.25 | 1.77E−04 | 0.77 (0.67–0.88) | A/G | 0.46 | 0.083 | 0.83 (0.67–1.02) | ||
| rs2553827 | 35.053243 | A/G | 0.43 | 4.34E−07 | 0.83 (0.78–0.89) | A/G | 0.25 | 1.52E−04 | 0.77 (0.67–0.88) | N/A | N/A | N/A | N/A | ||
| rs2553826 | 35.053334 | C/T | 0.43 | 2.49E−07 | 0.83 (0.77–0.89) | C/T | 0.25 | 1.52E−04 | 0.77 (0.67–0.88) | N/A | N/A | N/A | N/A | ||
| rs429503 | 35.053571 | T/G | 0.42 | 5.04E−07 | 0.83 (0.78–0.90) | T/G | 0.23 | 1.94E−04 | 0.77 (0.68–0.89) | N/A | N/A | N/A | N/A | ||
| rs63214363 | 35.053874 | T/A | 0.43 | 2.96E−07 | 0.83 (0.77–0.89) | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | ||
| rs387619 | 35.054769 | C/T | 0.42 | 3.00E−03 | 7.71E−07 | 0.84 (0.78–0.90) | 1.46E−08 (0.82, 0.76–0.88) | C/T | 0.23 | 1.44E−03 | 0.80 (0.70–0.92) | C/T | 0.14 | 0.485 | 0.94 (0.81–1.11) |
N/A indicates SNPs that did not meet the quality control criteria after imputation.
Lines in bold indicate observed SNPs.
Major/minor.
OR calculated according to the minor allele identified in the European data set.
Figure 2.
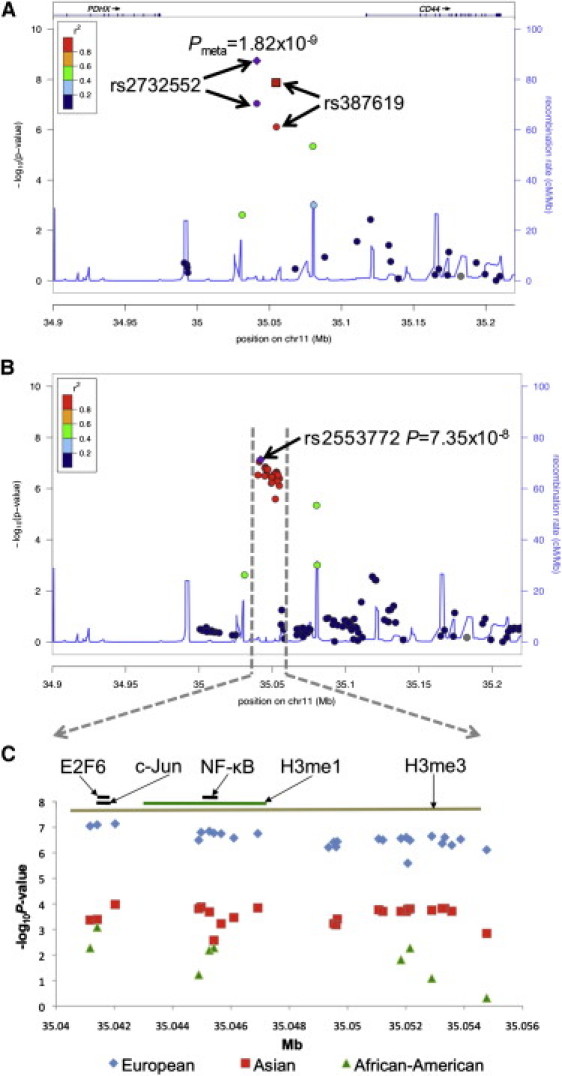
Summary of Observed and Imputed SNPs Tested at 11p13
(A and B) Regional plot of observed (A) and imputed (B) SNPs displaying –log10(p) for an ∼300 kb window at 11p13 with approximate gene locations given at top.
(A) Purple and red circles indicate results for SNPs observed in the replication study. The purple diamond and red square represent the meta-analysis results between the GWAS and the replication study. The symbol colors indicate the strength of LD with rs2732552, as given in the figure legend. Gray indicates that LD information was not available. The blue trace represents the average recombination rate for all races.
(B) The purple diamond represents the most significant SNP after imputation, rs2553772, with strength of LD to rs2553772 indicated by symbol colors as given in the legend.
(C) Expanded view of ∼14 kb haplotype associated with SLE in Europeans (blue diamonds), Asians (red squares), and African Americans (green triangles). Black lines indicate approximate location of transcription factor binding sites. Green and brown lines represent approximate location of methylated H3 histones.
The African American and Asian SLE cases also demonstrated association at rs2732552 (p = 5 × 10−3, OR = 0.81, 95% CI = 0.70–0.94 and p = 4.3 × 10−4, OR = 0.80, 95% CI = 0.70–0.91, respectively). The Asian SLE cases were associated with rs387619 (p = 0.001, OR = 0.8, 95% CI = 0.70–0.91), whereas the African American SLE cases were not, consistent with differences in the haplotype patterns between racial groups (Table 4; Figure 2A; Figure 3). Meta-analysis at rs2732552 between Europeans, Asians, African Americans, and the GWAS produced pmeta = 3.00 × 10−13. In this study, no evidence of association (p < 0.05) was observed in Hispanic, Gullah, or Amerindian subjects, possibly because of small sample sizes relative to the other races and/or ethnicities, clinical and/or genetic heterogeneity, or reduced correlation between tested markers and one or more causal variants. When evaluating all ethnic and racial groups evaluated within this study, meta-analysis yielded a pmeta = 2.36 × 10−13. Several of the subphenotypes comprising the SLE criteria, including thrombocytopenia, were also tested for association with these SNPs. No significant relationships were observed, possibly because of incomplete data.
Figure 3.
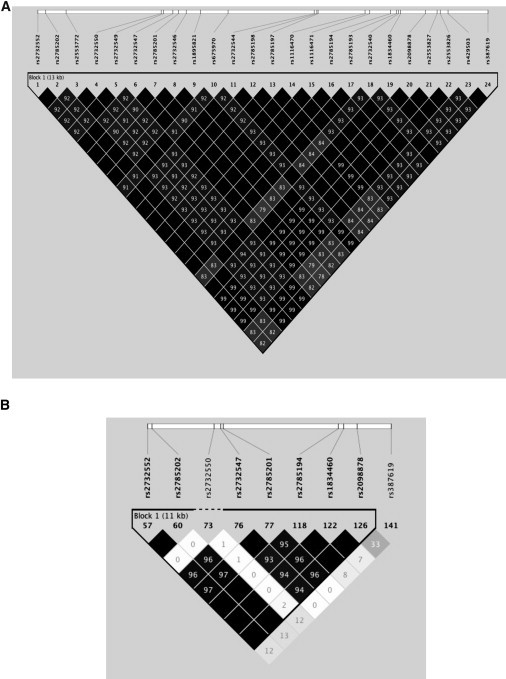
Linkage Disequilibrium Patterns for the SNPs within the Associated Region
Haplotype structure in Asians (A) and African Americans (B) from the data collected in this study. Diamonds show r2 values between markers, and those in solid black without a number are r2 = 1.0. Asian and European haplotype structure is nearly identical.
These two SNPs flank the boundaries of an ∼14 kb haplotype block observed in Europeans and Asians with r2 > 0.9, but with only r2 < 0.1 in African Americans (Figure 2A and Figure 3). To increase the informativeness of this haplotype, we conducted imputation in all three ethnic groups over a 2 Mb region. Imputation of the replication data across chromosome 11 (35–37 Mb) was performed with IMPUTE2, with the HapMap Phase III and 1000 Genomes Project as reference panels for African Americans, Asians, and Europeans (Table 5).14–16 Imputation is a method used to infer genotypes by using other correlated SNPs as proxies for those not genotyped. Subject genotypes are compared to a reference panel of all the SNPs genotyped within the study and those desired to be imputed. IMPUTE2 was selected because it can combine two reference panels in a single imputation analysis that will increase genotype-imputation accuracy. IMPUTE2 calculates posterior probabilities for the three possible genotypes (AA, AB, BB).
Table 5.
Reference Populations for Imputation
| Populations | 1000 Genomes Panel 1 | HapMap3 Panel 2 |
|---|---|---|
| African Americans | YRI | YRI + ASW + CEU + TSI |
| Asians | CHB + JPT | CHB + JPT |
| Europeans | CEU | CEU + TSI |
The following abbreviations are used: ASW, African ancestry in Southwest USA; CEU, Utah residents with Northern and Western European ancestry from the CEPH collection; CHB, Han Chinese in Beijing, China; JPT, Japanese in Tokyo, Japan; TSI, Tuscans in Italy; YRI, Yorubans in Ibadan, Nigeria.
Imputed genotypes had to meet or exceed a probability and certainty score of >0.9, and the quality control criteria described above had to be included in the analyses. However, we accepted genotype call rates for imputed SNPs at >70% in the African Americans because of the small LD blocks. After quality control, a total of 238 SNPs was imputed in the European replication cohort within a 0.5 Mb region. Because no significant associations were observed outside the 14 kb haplotype in Europeans, imputation for Asians and African American samples was performed between rs2732552 and rs387619. After quality control, imputed data were included for 24 SNPs in the Asian and 9 SNPs in the African American data sets.
Several imputed SNPs demonstrated association with disease (Table 4 and Figure 2B). The most statistically significant imputed marker among Europeans was rs2553772 (p = 7.35 × 10−8). This SNP was also significantly associated with SLE in the Asians (p = 1.05 × 10−4) but did not pass our quality control criteria in African Americans. However, a neighboring SNP, rs2785202, was found to be significant in all three populations (Table 4 and Figure 2C).
Bioinformatics database mining revealed that this region is conserved across species and has substantial regulatory potential within seven mammalian species, supported by experimental evidence using chromatin immunoprecipitation followed by sequencing (ChIP-Seq). The ENCODE project recently cataloged transcription factor binding sites and chromatin modification throughout the genome with ChIP-Seq in several different cell lines.17 The most strongly associated SNPs after imputation within our study flank two transcription factor binding sites identified in a myelogenous leukemia cell line: E2F6 (35,041,517–35,041,963 base pairs [bp]; peak binding at 35,041,776 bp) and c-Jun (35,041,681–35,042,886 bp; peak binding at 35,041,895 bp; Figure 1C).17 Another region of interest includes an NF-κB (35,044,973–35,045,701 bp) binding site identified in a lymphoblastoid cell line, with the peak binding localized at 35,045,460 bp, only 46 bases away from rs2785201 (Figure 2C). Studies of multiple cell lines have demonstrated that the regions spanning 35,043,876–35,047,875 bp (H3K4me1) and 34,998,301–35,641,975 bp (H3K27me3) are H3 histone interaction sites shown to be regulated by histone methylation, which can influence chromatin structure, resulting in transcriptional silencing or enhanced activity (Figure 2C). Further evaluation will be required to determine whether one or more of these regulatory elements contributes to SLE risk.
The genetic association with SLE established in this study lies between PDHX and CD44, both of which have been previously implicated in autoimmune or inflammatory conditions. PDHX (MIM 608769) is ∼79 kb in length with 11 exons and encodes for the E3 subunit of the pyruvate dehydrogenase (PDH) complex involved in the conversion of pyruvate to acetyl coenzyme A, which links glycolysis and the Krebs cycle.18 Interestingly, approximately 95% of primary biliary cirrhosis patients produce autoantibodies that recognize various components of the PDH complex.19
CD44 is an ∼95 kb gene with 20 exons that encodes a cell-surface glycoprotein expressed on most immunological cell types. Alternative splicing of this gene is exceptionally complex. Two constant regions of five exons flank a central variable region consisting of ten exons, potentially resulting in hundreds of protein isoforms (reviewed in 20). These alternate splice variants are differentially expressed across hematopoietic cells types, but what governs this complex process is not understood. The protein is important in lymphocyte activation, recirculation and homing, apoptosis, hematopoiesis, and tumor metastasis.20,21 CD44 can also heterodimerize with other proteins on the cell surface and bind a diverse repertoire of ligands (e.g., hyaluronic acid, osteopontin, collagens, and matrix metalloproteinases).
There are data supporting a role for CD44 in the pathogenesis of SLE and other inflammatory diseases. A genome-wide linkage scan of multiplex SLE families with thrombocytopenia has previously shown linkage (LODmax = 5.72) with the region that includes CD44 at 11p13.4 Linkage was independently confirmed, but analysis of four SNPs within CD44 showed no association, which is consistent with the negative results from SNPs within CD44 evaluated in the current study.22 Several reports have shown differential protein expression and complex alternative splicing of the CD44 mRNA to be associated with disease. Li et al. reported that T cells from SLE patients overexpressed CD44 and demonstrated increased adhesion and chemotactic migration when compared to patients with rheumatoid arthritis (RA [MIM 180300]) or healthy controls.23 These investigators also evaluated kidney biopsies from patients with lupus nephritis and with allograft kidney rejection for expression of CD44. Although both of these tissue samples had CD3+ CD44+ T cells, the SLE cases exhibited stronger staining for CD44 than the allographed tissue.23 A recent study by Crispin et al. shows that overexpression of CD44v3 and CD44v6 isoforms in T cells was observed in SLE patients and correlates with disease activity.24 These two splice variants are of particular interest because they have been found to be sufficient for fibroblast-type synoviocytes of RA patients to become invasive.25,26
CD44 is also important in the homing of T cells to the pancreas in type 1 diabetes mellitus (MIM 222100). Interestingly, nonobese diabetic mice injected with monoclonal antibodies against CD44 were resistant to diabetes, and immunohistochemical analysis of the pancreas tissue of these mice shows no active inflammation or destruction of islet cells.27 In Sjögren syndrome (MIM 270150), CD44 was found to be overexpressed in minor salivary glands of patients in a gene expression study.28 Expression of CD44 on macrophages, T cells, and endothelial cells in an ApoE−/− murine model of cardiovascular disease mediates recruitment of inflammatory cells into atherosclerotic lesions.29
We have established genetic association with SLE to a haplotype between PDHX and CD44. Imputation and transethnic mapping focus the effect on an ∼14 kb haplotype in a region of strong regulatory potential that may influence expression of the centromeric CD44. Further functional studies of this complex locus will be required to determine the precise variant or variants influencing risk and to characterize the contribution to SLE and perhaps other immunological, inflammatory, and oncologic phenotypes.
Acknowledgments
We are grateful to all the individuals with SLE and to the controls who participated in this study. We thank the following for contributing samples: Sandra D'Alfonso, Rafaella Scorza, Peter Junker, Helle Laustrup, Marc Bijl, Emoke Endreffy, Carlos Vasconcelos, Berta Martins da Silva, Ana Suarez, Carmen Gutierrez, Iñigo Rúa-Figueroa, and Cintia Garcilazo. For Asociacion Andaluza de Enfermedades Autinmunes collaboration: Norberto Ortego-Centeno, Juan Jimenez-Alonso, Enrique de Ramon, and Julio Sanchez-Roman. For the Genomica de lupues eritematosos sistemico collaboration: Mario Cardiel, Ignacio García de la Torre, Marco Maradiaga, José F. Moctezuma, Eduardo Acevedo, Cecilia Castel, Mabel Busajm, and Jorge Musuruana. We would like to thank Mary C. Comeau, Miranda C. Marion, Paula S. Ramos, Summer Frank, Stuart Glenn, and Mai Li Zhu for their assistance in genotyping, quality control analyses, and clinical data management. Grant support information is provided in the Supplemental Acknowledgments available online.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Moser K.L., Kelly J.A., Lessard C.J., Harley J.B. Recent insights into the genetic basis of systemic lupus erythematosus. Genes Immun. 2009;10:373–379. doi: 10.1038/gene.2009.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nath S.K., Han S., Kim-Howard X., Kelly J.A., Viswanathan P., Gilkeson G.S., Chen W., Zhu C., McEver R.P., Kimberly R.P. A nonsynonymous functional variant in integrin-alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat. Genet. 2008;40:152–154. doi: 10.1038/ng.71. [DOI] [PubMed] [Google Scholar]
- 3.Graham R.R., Cotsapas C., Davies L., Hackett R., Lessard C.J., Leon J.M., Burtt N.P., Guiducci C., Parkin M., Gates C. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat. Genet. 2008;40:1059–1061. doi: 10.1038/ng.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scofield R.H., Bruner G.R., Kelly J.A., Kilpatrick J., Bacino D., Nath S.K., Harley J.B. Thrombocytopenia identifies a severe familial phenotype of systemic lupus erythematosus and reveals genetic linkages at 1q22 and 11p13. Blood. 2003;101:992–997. doi: 10.1182/blood-2002-04-1003. [DOI] [PubMed] [Google Scholar]
- 5.Hochberg M.C. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 6.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 7.McKeigue P.M., Carpenter J.R., Parra E.J., Shriver M.D. Estimation of admixture and detection of linkage in admixed populations by a Bayesian approach: Application to African-American populations. Ann. Hum. Genet. 2000;64:171–186. doi: 10.1017/S0003480000008022. [DOI] [PubMed] [Google Scholar]
- 8.Hoggart C.J., Parra E.J., Shriver M.D., Bonilla C., Kittles R.A., Clayton D.G., McKeigue P.M. Control of confounding of genetic associations in stratified populations. Am. J. Hum. Genet. 2003;72:1492–1504. doi: 10.1086/375613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoggart C.J., Shriver M.D., Kittles R.A., Clayton D.G., McKeigue P.M. Design and analysis of admixture mapping studies. Am. J. Hum. Genet. 2004;74:965–978. doi: 10.1086/420855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith M.W., Patterson N., Lautenberger J.A., Truelove A.L., McDonald G.J., Waliszewska A., Kessing B.D., Malasky M.J., Scafe C., Le E. A high-density admixture map for disease gene discovery in african americans. Am. J. Hum. Genet. 2004;74:1001–1013. doi: 10.1086/420856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Halder I., Shriver M., Thomas M., Fernandez J.R., Frudakis T. A panel of ancestry informative markers for estimating individual biogeographical ancestry and admixture from four continents: Utility and applications. Hum. Mutat. 2008;29:648–658. doi: 10.1002/humu.20695. [DOI] [PubMed] [Google Scholar]
- 12.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whitlock M.C. Combining probability from independent tests: The weighted Z-method is superior to Fisher's approach. J. Evol. Biol. 2005;18:1368–1373. doi: 10.1111/j.1420-9101.2005.00917.x. [DOI] [PubMed] [Google Scholar]
- 14.Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M., International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Howie B.N., Donnelly P., Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Via M., Gignoux C., Burchard E.G. The 1000 Genomes Project: New opportunities for research and social challenges. Genome Med. 2010;2:3. doi: 10.1186/gm124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Birney E., Stamatoyannopoulos J.A., Dutta A., Guigó R., Gingeras T.R., Margulies E.H., Weng Z., Snyder M., Dermitzakis E.T., Thurman R.E., ENCODE Project Consortium. NISC Comparative Sequencing Program. Baylor College of Medicine Human Genome Sequencing Center. Washington University Genome Sequencing Center. Broad Institute. Children's Hospital Oakland Research Institute Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harris R.A., Bowker-Kinley M.M., Wu P., Jeng J., Popov K.M. Dihydrolipoamide dehydrogenase-binding protein of the human pyruvate dehydrogenase complex. DNA-derived amino acid sequence, expression, and reconstitution of the pyruvate dehydrogenase complex. J. Biol. Chem. 1997;272:19746–19751. doi: 10.1074/jbc.272.32.19746. [DOI] [PubMed] [Google Scholar]
- 19.McHugh A., Robe A.J., Palmer J.M., Jones D.E. PDC-E3BP is not a dominant T-cell autoantigen in primary biliary cirrhosis. Liver Int. 2006;26:406–413. doi: 10.1111/j.1478-3231.2006.01253.x. [DOI] [PubMed] [Google Scholar]
- 20.Ponta H., Sherman L., Herrlich P.A. CD44: From adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 2003;4:33–45. doi: 10.1038/nrm1004. [DOI] [PubMed] [Google Scholar]
- 21.Vivers S., Dransfield I., Hart S.P. Role of macrophage CD44 in the disposal of inflammatory cell corpses. Clin. Sci. 2002;103:441–449. doi: 10.1042/cs1030441. [DOI] [PubMed] [Google Scholar]
- 22.Kaufman K.M., Rankin J., Harley I.T., Kelly J.A., Harley J.B., Scofield R.H. A genetic marker within the CD44 gene confirms linkage at 11p13 in African-American families with lupus stratified by thrombocytopenia, but genetic association with CD44 is not present. Genes Immun. 2002;3(Suppl 1):S86–S88. doi: 10.1038/sj.gene.6363887. [DOI] [PubMed] [Google Scholar]
- 23.Li Y., Harada T., Juang Y.T., Kyttaris V.C., Wang Y., Zidanic M., Tung K., Tsokos G.C. Phosphorylated ERM is responsible for increased T cell polarization, adhesion, and migration in patients with systemic lupus erythematosus. J. Immunol. 2007;178:1938–1947. doi: 10.4049/jimmunol.178.3.1938. [DOI] [PubMed] [Google Scholar]
- 24.Crispín J.C., Keenan B.T., Finnell M.D., Bermas B.L., Schur P., Massarotti E., Karlson E.W., Fitzgerald L.M., Ergin S., Kyttaris V.C. Expression of CD44 variant isoforms CD44v3 and CD44v6 is increased on T cells from patients with systemic lupus erythematosus and is correlated with disease activity. Arthritis Rheum. 2010;62:1431–1437. doi: 10.1002/art.27385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wibulswas A., Croft D., Bacarese-Hamilton I., McIntyre P., Genot E., Kramer I.M. The CD44v7/8 epitope as a target to restrain proliferation of fibroblast-like synoviocytes in rheumatoid arthritis. Am. J. Pathol. 2000;157:2037–2044. doi: 10.1016/S0002-9440(10)64842-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wibulswas A., Croft D., Pitsillides A.A., Bacarese-Hamilton I., McIntyre P., Genot E., Kramer I.M. Influence of epitopes CD44v3 and CD44v6 in the invasive behavior of fibroblast-like synoviocytes derived from rheumatoid arthritic joints. Arthritis Rheum. 2002;46:2059–2064. doi: 10.1002/art.10421. [DOI] [PubMed] [Google Scholar]
- 27.Weiss L., Slavin S., Reich S., Cohen P., Shuster S., Stern R., Kaganovsky E., Okon E., Rubinstein A.M., Naor D. Induction of resistance to diabetes in non-obese diabetic mice by targeting CD44 with a specific monoclonal antibody. Proc. Natl. Acad. Sci. USA. 2000;97:285–290. doi: 10.1073/pnas.97.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pérez P., Anaya J.M., Aguilera S., Urzúa U., Munroe D., Molina C., Hermoso M.A., Cherry J.M., Alliende C., Olea N. Gene expression and chromosomal location for susceptibility to Sjögren's syndrome. J. Autoimmun. 2009;33:99–108. doi: 10.1016/j.jaut.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 29.Zhao L., Lee E., Zukas A.M., Middleton M.K., Kinder M., Acharya P.S., Hall J.A., Rader D.J., Puré E. CD44 expressed on both bone marrow-derived and non-bone marrow-derived cells promotes atherogenesis in ApoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2008;28:1283–1289. doi: 10.1161/ATVBAHA.108.165753. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.