

Abstract
Waardenburg anophthalmia syndrome, also known as microphthalmia with limb anomalies, ophthalmoacromelic syndrome, and anophthalmia-syndactyly, is a rare autosomal-recessive developmental disorder that has been mapped to 10p11.23. Here we show that this disease is heterogeneous by reporting on a consanguineous family, not linked to the 10p11.23 locus, whose two affected children have a homozygous mutation in SMOC1. Knockdown experiments of the zebrafish smoc1 revealed that smoc1 is important in eye development and that it is expressed in many organs, including brain and somites.
Main Text
Anophthalmia describes a rare developmental anomaly in which the eye is absent as the result of a deficiency in the development of the primary optic vesicles. Severe microphthalmia occurs at a prevalence of 3 in 10,000. Microphthalmia can be isolated or syndromic. Genetic studies of isolated cases have identified mutations in, among others, RAX,1 VSX22 (MIM 142993) PAX63 (MIM 607106), MITF4 (MIM 156845), MAF5 (MIM 177075), OTX26 (MIM 600037), SOX27 (MIM 184429), and SIX68 (MIM 606326). All these genes play a role in early ocular development. Mutations in members of the Wnt signaling and TGF-β1 (MIM 190180) superfamily, including members of the bone morphogenetic proteins and growth-differentiation factors, have also been implicated.9,10 For recent reviews, see 11–13. More recently, TMX3, a member of the thioredoxin family of proteins, was shown to be mutated in unilateral microphthalmia.14
At least ten forms of syndromic microphthalmia have been reported so far: MCOPS1 (MIM 309800), Lenz microphthalmia;15 MCOPS2 (MIM 300166), oculo-facio-cardio-dental syndrome;16 MCOPS3 (MIM 206900), anophthalmia-esophageal-genital syndrome;7 MCOPS4 (MIM 301590), microphthalmia-ankyloblepharon-mental retardation;17 MCOPS5 (MIM 610125), microphthalmia-optic nerve agenesis, corpus callosum agenesis, joint laxity;6 MCOPS6 (MIM 607932), microphthalmia-pituitary anomalies;18 MCOPS7 (MIM 309801), microphthalmia-dermal aplasia-sclerocornea;19 MCOPS8 (MIM 601349), microcephaly-microphthalmia-ectrodactyly of lower limbs-prognathism;20 and MCOPS9 (MIM 601186), pulmonary agenesis, microphthalmia-diaphragmatic defect.21 Several of these entities have overlapping phenotypes, and whether they all represent different syndromes remains to be shown.
Forty years ago, Waardenburg reported an autosomal-recessive anophthalmia with hand and foot malformations.22 This syndrome (Waardenburg anophthalmia syndrome, also known as microphthalmia with limb anomalies, ophthalmoacromelic syndrome, and anophthalmia-syndactyly [MIM 206920]) is characterized by unilateral or bilateral microphthalmia, clinical anophthalmia, syndactyly, polydactyly, synostosis, and/or oligodactyly. In addition, other organs may be affected (long-bone hypoplasia; renal, venous, and vertebral anomalies). Most patients are mentally retarded and are born from consanguineous parents. Since the first description by Waardenburg, more than 35 cases have been identified.22–30 Recently, Hamanoue et al. used homozygosity mapping to identify a 433 kb homozygous region on chromosome 10p11.23 between STS9 and STS12, but no mutated gene was identified.31
We report on a consanguineous family of Egyptian origin with four children, two of whom are affected with Waardenburg anophtalmia syndrome (Figure 1). The first child (V.2 in Figure 1), born to healthy parents, was a 12-yr-old girl when last seen at our clinics. At birth, her father (IV.7) was 33 yr old and her mother (IV.8) 30 yr old. The second affected child (V.4) was 8 yr old. Both were born at full term after an uneventful pregnancy and an uncomplicated vaginal delivery. Birth weights were within normal limits, but lengths are unknown. Both had global delay in developmental milestones: responsive smile was first seen at 10 and 8 mo in patients V.2 and V.4, respectively, and they walked at 3 yr. A Binet test performed on V.4 at 7 yr of age gave a score of 80, and her older sister was similarly mentally retarded. On clinical examination, both had broad lateral eyebrows, sparse eyelashes, short palpebral fissures, and bilateral anophthalmia (Figure 1A). Transpalpebral ultrasonography (12 MHz) (Figure 1B) and ultrasound biomicroscopy (50 MHz) confirmed the absence of an eye globe in both affected individuals, and no cysts were observed. Clinically observed skeletal abnormalities included malar flattening, a high palate, bilateral proximal placement of the thumb, bilateral F45 osseous syndactyly, bilateral F5 radial clinodactyly, bilateral camptodactyly of F15 (Figures 1C and 1D), an absent ray in both feet with sandal gap and pes planum (Figure 1e), and mild scoliosis in both affected children. Hand and wrist X-rays (Figure 1D) revealed fusion of the carpal bones (capitate and hamate). Elbow X-rays were normal. X-rays of the feet (Figure 1F) and legs showed bilateral partial fusion of both middle and medial cuneiform bones, an absent ray, and normal tibia and fibula. These features were common to both patients. Orbital and brain MRI scans were performed in both girls as well. No eye globe or cysts were reported on orbital MRI, but extraocular muscles normal in signal and size were present. On brain MRI, complete absence of the optic nerves, chiasma, and optic tracts was observed, and no other malformations were noted, especially of the corpus callosum and pituitary gland. The karyotype was normal (46,XX) in both patients, as was the renal ultrasound examination. The pedigree is described in Figure 2. This study was approved by the ethics committees of the Faculty of Medicine, University of Alexandria and the Faculty of Biology and Medicine, University of Lausanne.
Figure 1.
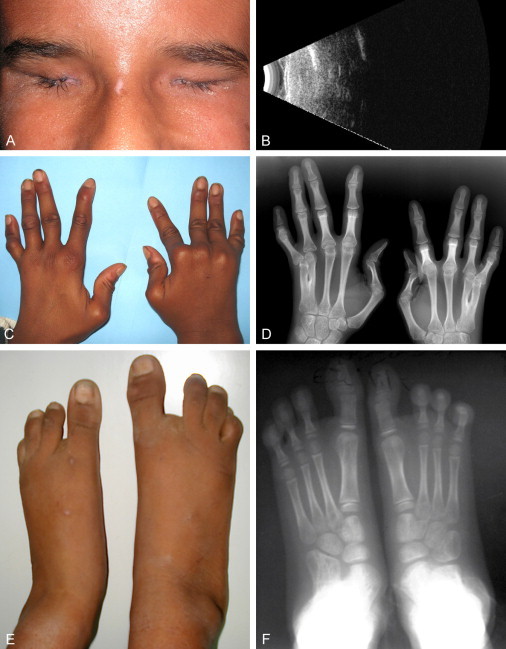
Clinical and X-Ray Evaluation of the Patients
(A) Image of the orbit of patient V.2. Note the short palpebral fissure, broad lateral eyebrows, and sparse eyelashes.
(B) Transpalpebral ultrasonography (12 MHz) in the same child, showing the absence of eye or cystic remnants.
(C) Hands of the same patient, with proximal placement of thumb, and F5 radial clinodactyly.
(D) X-ray images of the hands, highlighting proximal placement of the thumb, F45 osseous syndactyly, F5 radial clinodactyly, and fusion of the capitate and hamate carpal bones.
(E) Feet of the same child, showing an absent ray with sandal gap and pes planum.
(F) X-rays of the feet showing, in addition, bilateral partial fusion of both the middle and the medial cuneiform bones.
Figure 2.
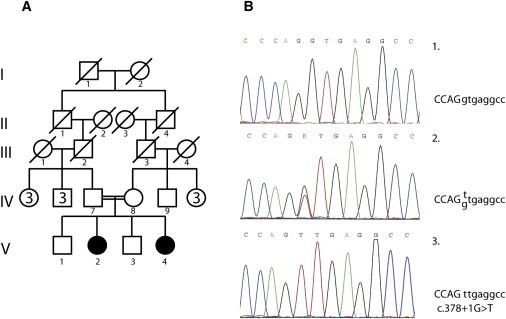
Pedigree and Mutation Analysis
(A) Pedigree showing consanguinity.
(B) Electropherogram of part of SMOC1 exon 3 showing normal (1), heterozygous (2), and homozygous (3) c.378+1G>T mutation.
Homozygous mapping via the Human660W-Quad DNA Analysis BeadChip (Illumina) was performed on individuals IV.7, IV.8, V.2, V.3, and V.4. We first concentrated on the region around the membrane protein palmitoylated 7 gene (MPP7 [MIM 610973]) that was reported to be a potential candidate on chromosome 10.31 However, the largest homozygous stretch of DNA in that region was only 60 kb. Therefore, the molecular anomaly involved in the family that we describe must be located elsewhere. Analysis of the rest of the data identified several homozygous regions in the genome of the patients, but many were also present in the nonaffected sibling and were therefore discarded. Three regions of homozygosity larger than 1 Mb and specific to the two patients were identified (Table S1, available online). The largest region was located between rs11158442 (position 62,198,792) and rs4899594 (position 76,031,821), defining a 14 Mb region on chromosome 14q23. Two genes in which mutations had already been implicated in microphthalmia and anophthalmia, BMP4 (MIM 112262) and OTX2 (MIM 600037), were located close to the homozygous region but were clearly outside of it.6,32 According to Ensembl, this interval contains more than 100 protein-coding sequences. As its name implies, the ophthalmo-acromelic syndrome shows aberration in the development of the eyes and bones. We therefore restricted the list of candidate genes to those expressed in the eyes, bones, and connective tissue. Expression of each candidate was checked in the EST profile of the UniGene database. Thirty-three genes passed this filter. Among these genes, MPP5 (MIM 606958), RDH11 (MIM 607849), ZFP36L1 (MIM 601064), SRSF5 (MIM 600914), SMOC1 (MIM 608488), and ZFYVE1 (MIM 605471) were further evaluated. MPP5 (ENSG00000072415) is a member of the peripheral membrane-associated guanylate kinase (MAGUK) family and may play a role in tight junction formation and cell-polarity establishment. Interestingly, MPP7, another member of the MAGUK family, was the only gene identified in the locus reported by Hamanoue et al.31 RDH11 (ENSG00000072042) is implicated in the reduction of all-trans-retinal, a molecule with high relevancy to the eye. The zinc finger protein 36, C3H type-like 1 gene (ZFP36L1 [ENSG00000185650]) is a member of the TIS11 family of early-response genes and is induced by, among others, the polypeptide mitogen EGF. It could thus regulate the response to growth factors. SRSF5 (ENSG00000100650) is a member of the serine- and arginine-rich family of pre-mRNA splicing factors. Mutations in splicing factors have been implicated in retinal pigmentosa. Zinc finger FYVE domain-containg protein 1 (ENSG00000165861) is a member of the FYVE domain proteins. They mediate the recruitment of proteins involved in membrane trafficking and cell signaling to phophatidylinositol 3-phosphate-containing membranes. Mutations in PIKFYVE, another member of the FYVE domain proteins, are responsible for the François-Neetens fleck corneal dystrophy (MIM 121850). Sequencing of the exons and intron-exon boundaries of these genes did not show any causative variants.
However, sequencing of SMOC1 (ENSG00000198732) revealed a c.378+1G>T (IVS3+1G>T) mutation in the canonical splice donor site of intron 3 (Figure 2). Both parents were heterozygous for this mutation, and the only available non-affected child was homozygous for the normal sequence. This mutation was looked for in 556 control chromosomes from individuals of Egyptian, North African, and European descent by denaturing high-performance liquid chromatography and was never observed. For primers used in SMOC1 sequencing, see the Supplemental Data.
SMOC1 (secreted modular calcium-binding 1) is a 48 kDa secreted modular protein containing an EF-hand calcium-binding domain and is a member of the SPARC family of proteins characterized by the presence of follistatin and EF-hand calcium-binding domains.33 Analysis of the EST profile in UniGene indicates that it is widely expressed, including in the eyes and bones. SMOC1 has 12 exons and several large introns. Intron 3 has a size of 22.2 kb, and a mutation in the canonical G nucleotide of the splice donor might either greatly perturb mRNA synthesis or activate nonsense-mediated decay. Unfortunately, we were unable to amplify SMOC1 by using RT-PCR with RNA extracted from normal blood and could not therefore investigate whether nonsense-mediated decay was happening or whether a truncated protein with an aberrant C terminus was generated.
SMOC1 is well conserved among various species, including zebrafish, in which we could identify a retired contig containing the 3′ region of a putative smoc1 (LOC795519). Protein-homology analysis between human SMOC1 and the putative zebrafish smoc1 gave an overall score of 63%, with some regions having a score over 80%. Except for the missing (or undiscovered) signal peptide, the putative smoc1 has a structure very similar to that of the human (see Figure S1). It also contains an FS domain, two TY domains, and a C-terminal SPARC calcium-binding domain containing two EF hands. Compared to SMOC1, smoc1 contains an additional region coding for a 27-amino-acid-long stretch between the FS domain and the first TY domains, as well as a second, 17-amino acid-long region situated just after the first TY domain and before the first SMOC1-specific domain (Figure S1). Interestingly, the SMOC1-specific domains are also conserved in zebrafish. Database searching and PCR amplifications using various primers located in the coding regions of the gene allowed us to identify only one copy of smoc1 in the D. rerio genome.
In order to evaluate the role of smoc1 in the development of the eye, we first evaluated its expression in zebrafish by in situ hybridization and then observed the effect of a transient knockdown of its translation by using a morpholino targeting the donor splice site of intron 3, the location that is mutated in the patients.
On the basis of the sequence of LOC795519, we cloned, after RT-PCR of zebrafish embryo mRNA using primers F (5′-ATGCC CTCAT CGCTT CAT-3) and R (5′-CTAAC CCTCC TTATT GACTCC-3′), a 1359 bp cDNA coding for 453 amino acids. From this clone, we generated two dioxygenin-labeled probes for in situ hybridization that covered the 5′ and 3′ regions of smoc1 with primers F100 (5′-GGTTC AGACG GACGC AGTTATG-3′) and R628 (5′-TCTGC CTCTC CTGGT CACAA-3′ and F716 (5′-GCCAC CAATC TACAG GTTAC-3′) and R1063 (5′-CGTGA CTGCC GTTAT TATCC-3′), respectively. and examined smoc1 expression in zebrafish embryos as described previously.34 Expression of smoc1 by whole-mount in situ hybridization was first evident at the 10-somite stage (10 ss). In the optic vesicles, transcripts were first detected in the ventrolateral diencephalon (Figures 3A and 3B), then in the most anterior part of the retina at 18 ss (Figures 3C and 3D). At 24 and 48 hr postfertilization (hpf), smoc1 was expressed in the ventral retina, on both sides of the choroid fissure (Figure 3E and 3F). Signals were seen in the brain at 10 ss and 18 ss as well as in pharyngeal arches at 48 hpf (Figure 3F). Functional analysis of smoc1 was assessed with the use of morpholino injections. Microinjections were performed with a Femtojet (Eppendorf). The smoc1 morpholino covering the exon 3-intron 3 splice site (5′-CCGGA ACTCT GACAG ACCTG AGCAA-3′) was synthesized by Gene Tools. For control injections, the standard control morpholino provided by Gene Tools was used. The stock solutions were diluted to 750 μM in H2O with 0.1% phenol red (Acros Organics, Brunschwig AG). Two nanoliters were injected in the yolk at 1- to 2-cell stage. First, efficiency of the morpholino targeting the exon 3-intron 3 splice site of smoc1 was controlled by RT-PCR with the use of primers F100 and R628 (Figure 4B). Additional amplicons larger and smaller than the normal 660 bp product were observed. They may correspond to fragments containing part of intron 3 or abnormal splice variants. Knockdown of smoc1 revealed microphthalmia associated with defects, especially in the ventral retina at 2 days postfertilization (dpf): the choroid fissure remained widely open compared to the untreated or control morpholino-injected larvae (Figures 4A and 4B). The coloboma was no longer detectable at 5 dpf, but microphthalmia was still severe, and the retina was especially underdeveloped at the ventral and nasal parts of the eye (Figure 4C). Smoc1 may also be important for brain development, because defects in the forebrain were observed at 5 dpf (Figure 4C). Pharyngeal arches seemed to be underdeveloped as well, but additional studies will be needed to confirm this.
Figure 3.
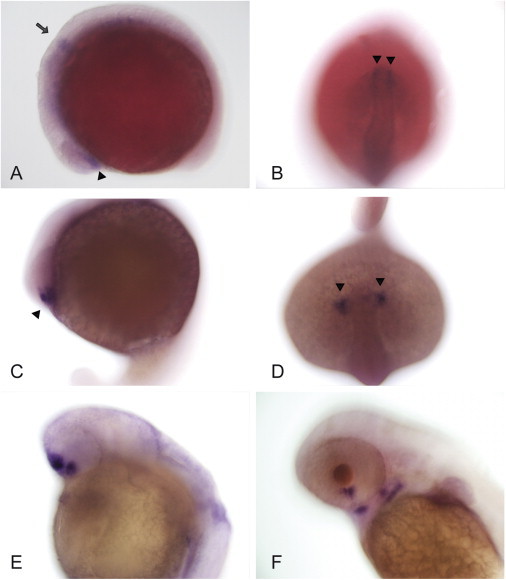
Expression of smoc1 in Zebrafish
(A–D) Whole-mount in situ hybridization experiments showing early expression of smoc1 in the brain (arrow) and in the anterior retina (arrowhead) at 10 ss and 18 ss, respectively.
(E and F) smoc1 is localized in the ventral retina, on both sides of the choroid fissure at 24 hr (E), and in the pharyngeal arches at 48 hpf (F).
(A, C, E, and F) Top and left represent dorsal and frontal parts of the animal. (B) and (D) are top views. Magnification: 200×.
Figure 4.
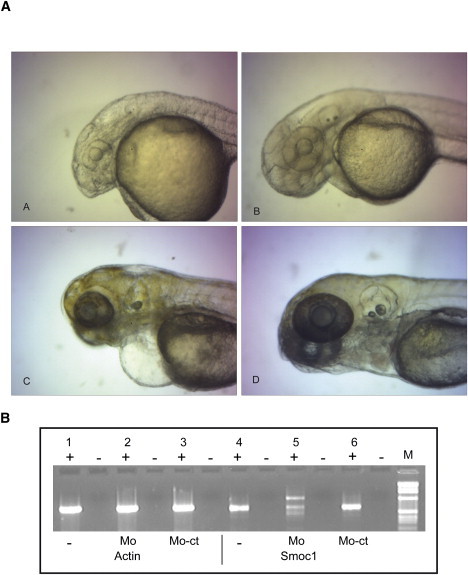
smoc1 Morpholino-Treated Embryos
(A and C) smoc1 morpholino-injected embryos at 2 and 5 dpf, respectively.
(B and D) Control morpholino-injected embryo at 2 dpf and wild-type embryo at 5 dpf. (B) RT-PCR (+ indicates with reverse transcriptase; - indicates without reverse transcriptase) on mRNA of noninjected embryos (-), embryos injected with smoc1 morpholino (MO), or those injected with control morpholino (MO-Ct) at 1 dpf. smoc1 knockdown revealed microphthalmia with defects in the ventral retina associated with abnormalities in brain development. Control for RNA extraction and amplification was performed with actin (PCR primers: 5′-GGGAG TGATG GTTGG CATGG-3′ and 5′-AGGAA GGAAG GCTGG AAGAG-3′).
The exact role of SMOC1 is still poorly understood. It was identified through its homology with secreted protein acidic and rich in cysteine (SPARC [MIM 182120]), also known as osteonectin, a protein important for bone calcification.35 Sparc-deficient mice show cataract and rupture of the lens capsule around the age of 6 mo.36 SMOC1 is localized in many tissues, where it associates with basement membranes, and interaction between the FS domain and the EC domain influences calcium binding.37 Calcium is critical for many developmental programs and is required for continuous function of the visual cycle, in which, among other roles, it regulates the guanylyl cyclase activating protein (GUCA1A [MIM 600364]), a calcium-binding protein with four EF-hand domains. Interestingly, Jalili syndrome (MIM 217080), another malformation syndrome affecting ocular development and the formation of the tooth, a tissue closely related to bone, is due to mutations in CNNM4, a gene implicated in Ca2+ control through Mg2+ exchange at GUCA1A.34 Novinec et al. also showed that SMOC1 binds many proteins, including C-reactive protein (CRP [MIM 123260]), fibulin-1 (MIM 135820), and vitronectin (MIM 193190).37 Fibulins are proteins involved in elastic-fiber assembly, cell proliferation, migration, adhesion, and angiogenesis, in which fibulin-1 stabilizes newly formed blood vessels.38,39 SMOC1 has also been involved in osteoblast differentiation and SMOC1 knockdown by shRNA-inhibited mineralization and expression of osteoblast-differentiation markers in human bone-marrow-derived mesenchymal stem cells.40 Interestingly, the same authors also identified EFEMP1 (MIM 601548), a member of the fibulin family, as implicated in osteoblast differentiation. Mutation in EFEMP1 causes malattia leventinese or Doyne honeycomb retinal dystrophy (MIM 126600), a disease characterized by the early development of drusen, similar to age-related macular degeneration.41 It is not known how SMOC1 participates in the development of bones, but on the basis of the phenotype associated with Waardenburg anophthalmia, it may be required for proper individualization of hand and foot rays.
In the zebrafish, smoc1 is highly expressed in the brain, choroidal fissure, pharyngeal arches, and somites. Knockdown experiments showed a coloboma at 2 dpf. At 5 dpf, the eye was microphthalmic and the brain showed gross malformations. Additional studies need to be done to see whether it is also implicated in the development of fin rays. Our analysis showed that Waardenburg anophthalmia syndrome is genetically heterogeneous. In addition to the previously described locus on chromosome 10, we identified a second locus on chromosome 14 and showed that mutations in SMOC1 also cause this syndrome.
Acknowledgments
The authors would like to thank the patients and their families. We also thank the staff of IRO and Isabelle Durussel for technical assistance.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl, http://www.ensembl.org
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/omim
UniGene, http://www.ncbi.nlm.nih.gov/unigene
Accession Numbers
LOC795519 information was obtained from NCBI. Zebrafish smoc1 accession number: HQ665031.
References
- 1.Voronina V.A., Kozhemyakina E.A., O'Kernick C.M., Kahn N.D., Wenger S.L., Linberg J.V., Schneider A.S., Mathers P.H. Mutations in the human RAX homeobox gene in a patient with anophthalmia and sclerocornea. Hum. Mol. Genet. 2004;13:315–322. doi: 10.1093/hmg/ddh025. [DOI] [PubMed] [Google Scholar]
- 2.Ferda P.E., Ploder L.A., Yu J.J., Arici K., Horsford D.J., Rutherford A., Bapat B., Cox D.W., Duncan A.M., Kalnins V.I. Human microphthalmia associated with mutations in the retinal homeobox gene CHX10. Nat. Genet. 2000;25:397–401. doi: 10.1038/78071. [DOI] [PubMed] [Google Scholar]
- 3.Hever A.M., Williamson K.A., van Heyningen V. Developmental malformations of the eye: the role of PAX6, SOX2 and OTX2. Clin. Genet. 2006;69:459–470. doi: 10.1111/j.1399-0004.2006.00619.x. [DOI] [PubMed] [Google Scholar]
- 4.Tachibana M., Perez-Jurado L.A., Nakayama A., Hodgkinson C.A., Li X., Schneider M., Miki T., Fex J., Francke U., Arnheiter H. Cloning of MITF, the human homolog of the mouse microphthalmia gene and assignment to chromosome 3p14.1-p12.3. Hum. Mol. Genet. 1994;3:553–557. doi: 10.1093/hmg/3.4.553. [DOI] [PubMed] [Google Scholar]
- 5.Kim J.I., Li T., Ho I.C., Grusby M.J., Glimcher L.H. Requirement for the c-Maf transcription factor in crystallin gene regulation and lens development. Proc. Natl. Acad. Sci. USA. 1999;96:3781–3785. doi: 10.1073/pnas.96.7.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ragge N.K., Brown A.G., Poloschek C.M., Lorenz B., Henderson R.A., Clarke M.P., Russell-Eggitt I., Fielder A., Gerrelli D., Martinez-Barbera J.P. Heterozygous mutations of OTX2 cause severe ocular malformations. Am. J. Hum. Genet. 2005;76:1008–1022. doi: 10.1086/430721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fantes J., Ragge N.K., Lynch S.A., McGill N.I., Collin J.R., Howard-Peebles P.N., Hayward C., Vivian A.J., Williamson K., van Heyningen V. Mutations in SOX2 cause anophthalmia. Nat. Genet. 2003;33:461–463. doi: 10.1038/ng1120. [DOI] [PubMed] [Google Scholar]
- 8.Gallardo M.E., Rodriguez De C.S., Schneider A.S., Dwyer M.A., Ayuso C., Bovolenta P. Analysis of the developmental SIX6 homeobox gene in patients with anophthalmia/microphthalmia. Am. J. Med. Genet. A. 2004;129A:92–94. doi: 10.1002/ajmg.a.30126. [DOI] [PubMed] [Google Scholar]
- 9.Asai-Coakwell M., French C.R., Ye M., Garcha K., Bigot K., Perera A.G., Staehling-Hampton K., Mema S.C., Chanda B., Mushegian A. Incomplete penetrance and phenotypic variability characterize Gdf6-attributable oculo-skeletal phenotypes. Hum. Mol. Genet. 2009;18:1110–1121. doi: 10.1093/hmg/ddp008. [DOI] [PubMed] [Google Scholar]
- 10.Ye M., Berry-Wynne K.M., Asai-Coakwell M., Sundaresan P., Footz T., French C.R., Abitbol, M, Fleisch V.C., Corbett N., Allison W.T. Mutation of the bone morphogenetic protein GDF3 causes ocular and skeletal anomalies. Hum. Mol. Genet. 2010;19:287–298. doi: 10.1093/hmg/ddp496. [DOI] [PubMed] [Google Scholar]
- 11.Fuhrmann S. Eye morphogenesis and patterning of the optic vesicle. Curr. Top. Dev. Biol. 2010;93:61–84. doi: 10.1016/B978-0-12-385044-7.00003-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fuhrmann S. Wnt Signaling in Eye Organogenesis. Organogenesis. 2008;4:60–67. doi: 10.4161/org.4.2.5850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Graw J. The genetic and molecular basis of congenital eye defects. Nat. Rev. Genet. 2003;4:876–888. doi: 10.1038/nrg1202. [DOI] [PubMed] [Google Scholar]
- 14.Chao R., Nevin L., Agarwal P., Riemer J., Bai X., Delaney A., Akana M., Jimenez Lopez N., Bardakjian T., Schneider A. A male with unilateral microphthalmia reveals a role for TMX3 in eye development. PLoS ONE. 2010;5:e10565. doi: 10.1371/journal.pone.0010565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lenz W. Recessive, sex-limited microphthalmia with multiple abnormalities. Z. Kinderheilkd. 1955;77:384–390. [PubMed] [Google Scholar]
- 16.Ng D., Thakker N., Corcoran C.M., Donnai D., Perveen R., Schneider A., Hadley D.W., Tifft C., Zhang L., Wilkie A.O. Oculofaciocardiodental and Lenz microphthalmia syndromes result from distinct classes of mutations in BCOR. Nat. Genet. 2004;36:411–416. doi: 10.1038/ng1321. [DOI] [PubMed] [Google Scholar]
- 17.Graham C.A., Redmond R.M., Nevin N.C. X-linked clinical anophthalmos. Localization of the gene to Xq27-Xq28. Ophthalmic Paediatr. Genet. 1991;12:43–48. doi: 10.3109/13816819109023084. [DOI] [PubMed] [Google Scholar]
- 18.Bennett C.P., Betts D.R., Seller M.J. Deletion 14q (q22q23) associated with anophthalmia, absent pituitary, and other abnormalities. J. Med. Genet. 1991;28:280–281. doi: 10.1136/jmg.28.4.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.al Gazali L.I., Sabarinathan D.K., Khidir A. Microphthalmia and distal limb abnormalities in a child of consanguineous parents. Clin. Dysmorphol. 1994;3:258–262. [PubMed] [Google Scholar]
- 20.Viljoen D.L., Smart R. Split-foot anomaly, microphthalmia, cleft-lip and cleft-palate, and mental retardation associated with a chromosome 6;13 translocation. Clin. Dysmorphol. 1993;2:274–277. [PubMed] [Google Scholar]
- 21.Ostor A.G., Stillwell R., Fortune D.W. Bilateral pulmonary agenesis. Pathology. 1978;10:243–248. doi: 10.3109/00313027809063507. [DOI] [PubMed] [Google Scholar]
- 22.Waardenburg P.J. Genetics and Ophthalmology. Royal Van Gorcum; Assen, The Netherlands: 1961. Autosomally-recessive anophthalmia with malformations of the hands and feet; p. 773. [Google Scholar]
- 23.Richieri-Costa A., Gollop T.R., Otto P.G. Brief clinical report: autosomal recessive anophthalmia with multiple congenital abnormalities-type Waardenburg. Am. J. Med. Genet. 1983;14:607–615. doi: 10.1002/ajmg.1320140403. [DOI] [PubMed] [Google Scholar]
- 24.Traboulsi E.I., Nasr A.M., Fahd S.D., Jabbour N.M., Der K.V. Waardenburg's recessive anophthalmia syndrome. Ophthalmic Paediatr. Genet. 1984;4:13–18. doi: 10.3109/13816818409009888. [DOI] [PubMed] [Google Scholar]
- 25.Le Merrer M., Nessmann C., Briard M.L., Maroteaux P. Ophthalmo-acromelic syndrome. Ann. Genet. 1988;31:226–229. [PubMed] [Google Scholar]
- 26.Sayli B.S., Akarsu A.N., Altan S. Anophthalmos-syndactyly (Waardenburg) syndrome without oligodactyly of toes. Am. J. Med. Genet. 1995;58:18–20. doi: 10.1002/ajmg.1320580105. [DOI] [PubMed] [Google Scholar]
- 27.Suyugul Z., Seven M., Hacihanefioglu S., Kartal A., Suyugul N., Cenani A. Anophthalmia-Waardenburg syndrome: a report of three cases. Am. J. Med. Genet. 1996;62:391–397. doi: 10.1002/(SICI)1096-8628(19960424)62:4<391::AID-AJMG12>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 28.Megarbane A., Souraty N., Tamraz J. Ophthalmo-acromelic syndrome (Waardenburg) with split hand and polydactyly. Genet. Couns. 1998;9:195–199. [PubMed] [Google Scholar]
- 29.Tekin M., Tutar E., Arsan S., Atay G., Bodurtha J. Ophthalmo-acromelic syndrome: report and review. Am. J. Med. Genet. 2000;90:150–154. doi: 10.1002/(sici)1096-8628(20000117)90:2<150::aid-ajmg12>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 30.Garavelli L., Pedori S., Dal Z.R., Franchi F., Marinelli M., Croci G.F., Bellato S., Ammenti A., Virdis R., Banchini G. Anophthalmos with limb anomalies (Waardenburg opththalmo-acromelic syndrome): report of a new Italian case with renal anomaly and review. Genet. Couns. 2006;17:449–455. [PubMed] [Google Scholar]
- 31.Hamanoue H., Megarbane A., Tohma T., Nishimura A., Mizuguchi T., Saitsu H., Sakai H., Miura S., Toda T., Miyake N. A locus for ophthalmo-acromelic syndrome mapped to 10p11.23. Am. J. Med. Genet. A. 2009;149A:336–342. doi: 10.1002/ajmg.a.32656. [DOI] [PubMed] [Google Scholar]
- 32.Bakrania P., Efthymiou M., Klein J.C., Salt A., Bunyan D.J., Wyatt A., Ponting C.P., Martin A., Williams S., Lindley V. Mutations in BMP4 cause eye, brain, and digit developmental anomalies: overlap between the BMP4 and hedgehog signaling pathways. Am. J. Hum. Genet. 2008;82:304–319. doi: 10.1016/j.ajhg.2007.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vannahme C., Smyth N., Miosge N., Gosling S., Frie C., Paulsson M., Maurer P., Hartmann U. Characterization of SMOC-1, a novel modular calcium-binding protein in basement membranes. J. Biol. Chem. 2002;277:37977–37986. doi: 10.1074/jbc.M203830200. [DOI] [PubMed] [Google Scholar]
- 34.Polok B., Escher P., Ambresin A., Chouery E., Bolay S., Meunier I., Nan F., Hamel C., Munier F.L., Thilo B. Mutations in CNNM4 cause recessive cone-rod dystrophy with amelogenesis imperfecta. Am. J. Hum. Genet. 2009;84:259–265. doi: 10.1016/j.ajhg.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Termine J.D., Kleinman H.K., Whitson S.W., Conn K.M., McGarvey M.L., Martin G.R. Osteonectin, a bone-specific protein linking mineral to collagen. Cell. 1981;26:99–105. doi: 10.1016/0092-8674(81)90037-4. [DOI] [PubMed] [Google Scholar]
- 36.Gilmour D.T., Lyon G.J., Carlton M.B., Sanes J.R., Cunningham J.M., Anderson J.R., Hogan B.L., Evans M.J., Colledge W.H. Mice deficient for the secreted glycoprotein SPARC/osteonectin/BM40 develop normally but show severe age-onset cataract formation and disruption of the lens. EMBO J. 1998;17:1860–1870. doi: 10.1093/emboj/17.7.1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Novinec M., Kovacic L., Skrlj N., Turk V., Lenarcic B. Recombinant human SMOCs produced by in vitro refolding: calcium-binding properties and interactions with serum proteins. Protein Expr. Purif. 2008;62:75–82. doi: 10.1016/j.pep.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 38.Zhang H., Gao X., Weng C., Xu Z. Interaction between angiogenin and fibulin 1: evidence and implication. Acta Biochim. Biophys. Sin. (Shanghai) 2008;40:375–380. doi: 10.1111/j.1745-7270.2008.00420.x. [DOI] [PubMed] [Google Scholar]
- 39.Zhang H.Y., Timpl R., Sasaki T., Chu M.L., Ekblom P. Fibulin-1 and fibulin-2 expression during organogenesis in the developing mouse embryo. Dev. Dyn. 1996;205:348–364. doi: 10.1002/(SICI)1097-0177(199603)205:3<348::AID-AJA13>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 40.Choi Y.A., Lim J., Kim K.M., Acharya B., Cho J.Y., Bae Y.C., Shin H.I., Kim S.Y., Park E.K. Secretome analysis of human BMSCs and identification of SMOC1 as an important ECM protein in osteoblast differentiation. J. Proteome Res. 2010;9:2946–2956. doi: 10.1021/pr901110q. [DOI] [PubMed] [Google Scholar]
- 41.Stone E.M., Lotery A.J., Munier F.L., Heon E., Piguet B., Guymer R.H., Vandenburgh K., Cousin P., Nishimura D., Swiderski R.E. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat. Genet. 1999;22:199–202. doi: 10.1038/9722. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.