

Abstract
Indian hedgehog (IHH) is a secreted signaling molecule of the hedgehog family known to play important roles in the regulation of chondrocyte differentiation, cortical bone formation, and the development of joints. Here, we describe that copy-number variations of the IHH locus involving conserved noncoding elements (CNEs) are associated with syndactyly and craniosynostosis. These CNEs are able to drive reporter gene expression in a pattern highly similar to wild-type Ihh expression. We postulate that the observed duplications lead to a misexpression and/or overexpression of IHH and by this affect the complex regulatory signaling network during digit and skull development.
Main Text
Submicroscopic copy-number variations (CNVs) are structural variations of the human genome, i.e., deletions, duplications, or insertions, which account for genetic diversity between individuals as well as conditions referred to as genomic disorders.1 In the past years studies have sought to determine the frequency and location of CNVs in the genome and to assess how CNVs relate to variation in phenotypes.2–4 Deletions and/or duplications that affect entire genes can be expected to modify the dosage of gene expression. However, CNVs may influence expression levels of genes not only within but also far away from their boundaries, thereby altering tissue transcriptomes.5 Such long-range regulatory effects appear to be of particular importance for developmental genes, probably because of their often complex spatial and temporal expression patterns. Genome-wide screening methods such as array CGH are a suitable tool for identifying aberrations in such noncoding regulatory regions.6–8 Here, we show that microduplications at the IHH (Indian hedgehog [MIM 600726]) locus containing highly conserved noncoding sequence elements are associated with craniosynostosis and syndactyly.
We investigated families with syndactyly type 1 (SD1 [MIM 185900]) and craniosynostosis Philadelphia type (MIM 601222), a rare form of premature fusion of cranial sutures in combination with syndactyly. Both conditions were mapped to a locus at 2q35, indicating that they may share a common etiology.9,10 Blood sampling and extraction of DNA was performed by standard methods. All participants gave their consent for molecular testing. The study was approved by the Charité Universitätsmedizin Berlin ethics committee. Family 1 consisted of 77 affected members in eight generations featuring cutaneous syndactyly with variable expressivity.9 Family 2 showed variable degrees of cutaneous and distal osseous syndactyly together with craniosynostosis, mainly affecting the sagittal suture. This condition has previously been described as craniosynostosis Philadelphia type.11 Family 3 presented with variable degrees of cutaneous and distal osseous syndactyly, mainly affecting the feet (preaxial polydactyly of the feet in one affected individual [IV-1]), and variable degrees of craniosynostosis, affecting the sagittal suture in one affected individual (IV-1) and resulting in a cloverleaf skull in another (III-5) (Figures 1B–1E; for pedigree, see Figure S1 available online).
Figure 1.
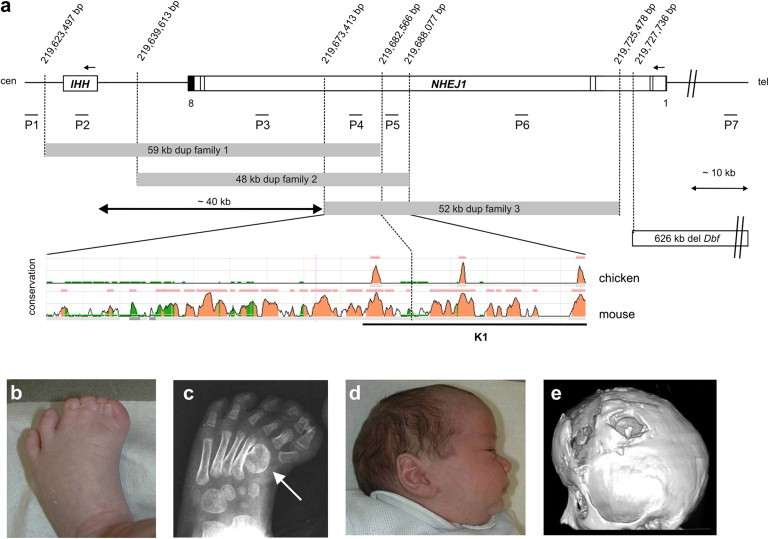
CNVs at the IHH Locus on 2q35 and the Associated Clinical Phenotype
(A) The duplicated sequences observed in the families with syndactyly and craniosynostosis are shown as gray boxes. The localization of the Dbf deletion converted from the mm8 assembly is indicated as an open box. The conservation of the overlapping duplicated sequence is depicted in the ECR browser plot. Within this sequence are three regions highly conserved between human and chicken (colored peaks). The underlined orthologous mouse sequence was cloned for in vivo experiments (K1). The approximate positions of qPCR amplicons used are indicated (P1–P7). Black bars represent the NHEJ1 exon structure. Nucleotide numbering refers to the Human Genome version hg18.
(B–D) Clinical phenotype of family 3 (pedigree information see Figure S1). (B) Preaxial synpolydactyly of feet. (C) X-rays showing preaxial polydactyly and abnormal shape of first metatarsal (arrow). (D) Dolichocephaly due to premature fusion of sagittal suture (individual IV:1).
(E) Three-dimensional CT scan of individual III:5 after surgical intervention. Note synostosis of all sutures.
Array comparative genomic hybridization (array CGH) was carried out with the use of a whole-genome 244K Oligonucleotide Array (Agilent) and a custom-designed array (NimbleGen/Roche) that covers the critical region from 219.5–222.2 Mb on human chromosome 2 at a high density (average probe spacing: 8 bp). Analysis was performed with Feature Extraction and CGH Analytics software (Agilent), with the following settings used: Aberration Algorithm: ADM-2; Threshold: 6.0; Window Size: 0.2 Mb; Filter: 5probes, log2ratio = 0.29. Custom arrays were analyzed with the NimbleScan software and the circular binary segmentation algorithm for aberration detection. The genomic profile was visualized with SignalMap software (SignalMap v1.9.0.03, NimbleGen). Using this protocol, we detected duplications of varying sizes on chromosome 2q35 involving the IHH locus (Figure 1A). In family 1, we identified a 59 kb duplication involving the entire IHH coding sequence plus 49 kb upstream of the IHH start site (DECIPHER BER254250). Family 2 (DECIPHER BER254251) and family 3 (DECIPHER BER254252) showed duplications of 48 kb and 52 kb, respectively, upstream of IHH (Figure 1A). After the array CGH analyses, we designed PCR primers for breakpoint analysis (primer sequences are available on request) and sequenced the amplified junction fragments in both directions. All duplications were arranged in direct tandem orientation, and the distal breakpoints were localized within introns of the neighboring NHEJ1 (MIM 611290) (Figure 1A). Mutations in NHEJ1, the gene disrupted by the duplications, cause severe combined immunodeficiency with microcephaly, growth retardation, and sensitivity to ionizing radiation (MIM 611291).12 Because this condition is recessive and no expression could be identified in the developing limb (data not shown), we conclude that NHEJ1 is unlikely to be involved in the condition described here. The three duplications overlap in a ∼9.1 kb region 40 kb 5′ of IHH that contains one highly conserved noncoding element (CNE). The smallest region of overlap between duplications in families 2 and 3 is ∼14.6 kb and encompasses three CNEs. Cosegregation of the genomic duplications with the phenotype was studied and confirmed in all three families by quantitative real-time PCR (Figure S2; primer sequences and positions given in Table S1 and Figure 1A). Affected individuals in all three families carried microduplications, whereas the unaffected siblings showed normal copy number. Similar duplications were not observed in 685 control samples analyzed via array CGH.
The duplication of IHH plus most of the gene's regulatory region (family 1) is likely to result in an increase in IHH expression. Given the very similar limb phenotypes in the three families, we conclude that the duplications that do not involve the IHH coding region have a similar effect, i.e., increased IHH expression. To further evaluate the functional relevance of the duplicated CNEs, we cloned the 6.2 kb orthologous highly conserved mouse sequence (K1, black bar, Figure 1A) into a LacZ reporter gene vector as described previously.8 The CNE containing the orthologous mouse sequence (mm8, chr1: 74,940,512–74,946,807) was amplified by conventional PCR from mouse DNA (BLI6J) with the following primer combination including NotI restriction sites: 5′-gtatGCGGCCGCtctccaccctctgtgctctt-3′ (forward) and 5′-gtatGCGGCCGCaagttgaggtttggggttttt-3′ (reverse) (construct K1, Figure 1A). After pronuclear injection and subsequent oviduct transfer, transgenic embryos were harvested at different stages of development (embryonic day 13.5 [E13.5], E15.5, and E17.5) and analyzed for LacZ expression as described previously8 (Figure 2, Figure 3). At developmental stage E13.5, we detected LacZ positive cells in the mesenchymal condensations of the humerus and femur, as well as in the digit tips (Figure 2). At later time points, we observed X-gal staining in the cartilage growth plates. For the visualization of staining on a cellular level, stained limbs were embedded in glycol methacrylat at room temperature (Technovit 7100, Heraeus-Kulzer) and sectioned in longitudinal orientation with a hard-tissue microtome (RM 2255, Leica). LacZ and Ihh stainings of 5 μm serial sections were analyzed by light microscopy after counterstaining with nuclear fast red. As shown in Figure 2 (bottom right) LacZ-positive cells were present only in prehypertrophic chondrocytes (PreHC) and hypertrophic chondrocytes (HC). Staining in hypertrophic chondrocytes is due to persistence of LacZ in these cells and does not necessarily reflect promotor activity. Thus, the observed LacZ pattern recapitulated the known endogenous Ihh expression in the developing skeleton.13 On the basis of these results, it is conceivable that the critical duplicated region serves as a long-range enhancer of Ihh specifically regulating Ihh expression during endochondral bone formation.
Figure 2.

Expression Profiles of LacZ in Transgenic Mice Compared to WT Ihh Expression
LacZ expression of transgenic mice carrying the orthologous mouse sequence to the duplicated region (K1). At stages E13.5 and E15.5, X-Gal staining is present in the condensations of long bones and at the finger tips (arrow). The E13.5 WT embryo shows additional Ihh staining in the condensations of the phalanges (arrow head). At E17.5, strong staining is detectable in the growth plates. Note specific staining in prehypertrophic chondrocytes (PreHC) and hypertrophic chondrocytes (HC) in growth plate sections with X-Gal (upper right) compared to WT Ihh (lower right).
Figure 3.

Expression Profiles of LacZ in the Skull of Transgenic Mice Compared to WT Ihh and Ptch1 Expression
(A) LacZ expression in transgenic mice (left) compared to Ihh (middle) and Ptch1 (right) expression patterns in the skull of E17.5 WT embryos. Expression of lacZ is observed at the sutures (arrowhead). Strong expression of Ptch1 is detected along the developing cranial sutures (arrow), which overlaps with Ihh expression in WT embryos.
(B) Skull preparations of WT and Ihh−/− mice at E18.5. Ihh−/− embryos have a smaller skull. Staining with Alizarin red reveals significantly less bone in the skull of Ihh−/− than in WT mice. In contrast to the WT embryos, Ptch1 expression is undetectable along the sutures of Ihh−/− embryos at stage E17.5.
X-gal staining was also observed in the skull, first in the suture area of the nasal and the metopic bones, and later also in and around other sutures (Figure 3). So far, Ihh has not been recognized as a major factor in the development of the cranial vault, a region where bone is directly formed from mesenchymal progenitor cells without an intermediate cartilaginous template (desmal bone formation). To study the role of Ihh in skull growth in more detail, we analyzed Ihh expression and expression of its downstream target, the hedgehog receptor patched1 (ptch1 [MIM 601309]) in calvarial bone. In situ hybridization (ISH) for Ihh and Ptch1 was carried out on wild-type (WT) embryos (C57/Bl6J) at embryonic stages E15.5 and E17.5. In addition, Ptch1 expression was investigated in Ihh−/− skulls (Figure 3). Calvaria were excised from the head together with the underlying brain, and, after removal of the skin, treated for 5 min with proteinase K (20 μg/ml) prior to ISH. Skeletal preparations and alizarin red staining of E18.5 WT and Ihh knockout embryos were performed as previously described.14
We identified strong expression of Ptch1 along the developing cranial sutures, indicating that hedgehog signal is present in these cells (Figure 3). Ihh expression was detected in the same area, but the intensity of expression was much weaker than in the growth plate cartilage. Ihh−/− skulls did not show any Ptch1 expression (data not shown). To investigate the significance of hedgehog signaling in the growth of calvarial bones, we compared the skulls of Ihh−/− and WT mice at E18.5. Ihh−/− mice had a much smaller skull compared to WT mice. They showed significantly less calvarial ossification than WT mice at this stage, indicating a growth-promoting effect of Ihh on calvarial bone (Figure 3B).
IHH is a signaling molecule expressed predominantly in prehypertrophic chondrocytes. It plays a major role in endochondral bone formation by regulating the proliferation and differentiation of chondrocytes via a parathyroid hormone-related peptide (PTHrP [MIM 168470])-controlled feedback loop.13 At the same time, it signals via its receptor patched 1 (Ptch1) to osteoblasts of the bone shaft, thereby promoting cortical bone formation. Accordingly, Ihh−/− mice show markedly reduced chondrocyte proliferation, highly abnormal growth plates, and a failure of osteoblast development in endochondral bones.15 In humans, mutations in IHH cause brachydactyly type A1 (MIM 112500), a dominantly inherited condition with short or missing middle phalanges, and acrocapitofemoral dysplasia (MIM 607778), an autosomal-recessive skeletal dysplasia characterized by cone-shaped epiphyses, short stature, and brachydactyly.
The patients described here do not show any overlap with the known IHH-associated phenotypes. In particular the skull deformity, due to premature fusion of cranial sutures, has so far not been reported in association with IHH. Typically, craniosynostosis syndromes have been associated with activating mutations in the FGF pathway, such as in Apert (MIM 101200), Saethre-Chotzen (MIM 101400), or Muenke (MIM 602849) syndrome, indicating that the regulation of FGF activity is crucial for cranial suture development.16 Likewise, activating mutations of MSX2 (MIM 123101), a transcription factor involved in osteoblast differentiation, result in craniosynostosis.17 Interestingly, Msx2 is implicated in endochondral as well as membranous ossification and was shown to induce Ihh expression in mouse primary chondrocytes.18
The involvement of hedgehog (Hh) signaling in suture development is also indicated by mutations in RAB23 (Ras associated protein RAB23 [MIM 606144]), which are associated with Carpenter syndrome (acrocephalopolysyndactyly type II [MIM 201000]).19 RAB23 negatively regulates the Hh pathway by promoting the production of Gli3 repressor.20 In line with this hypothesis are recent findings showing that mutations in Gli3 (MIM 165240), an important mediator of Hh signaling, are associated with craniosynostosis in the extra-toes (Xt) mouse mutant21 and in patients with metopic craniosynostosis.22 In the presence of Hh signal, Gli3 is maintained in its activated form, Gli3A, and positively regulates Hh downstream target genes. The authors postulate an increased proliferation and enhanced osteoblastic differentiation due to ectopic Ptch1 expression, loss of Twist1 (MIM 601622), and elevated Runx2 (MIM 600211) expression in the sutural mesenchyme as the underlying cause for the craniosynostosis.21 Twist1 negatively regulates osteogenesis to maintain suture patency, and loss-of-function mutations in TWIST1 are associated with Saethre-Chotzen syndrome, whereas Runx2 functions as a positive regulator of osteoblast differentiation and mutations in RUNX2 result in delayed closure of sutures.23 These observations link aberrant Hh signaling with suture development and craniosynostosis. The observed delay in calvarial bone formation in Ihh−/− mice further supports a role of Ihh in the regulation of osteoblast proliferation and suture formation. It is a likely scenario that the duplications reported here cause a moderate increase in IHH expression, similar to the situation proposed for regulatory duplications of BMP2 (MIM 112261).8 Such an increase in Hh signal is likely to induce osteoblast proliferation and consecutive calvarial overgrowth that in turn results in premature closure of sutures.
Hh signaling has been shown to play essential roles in the determination of digit number and identity. This, however, has been solely attributed to Shh (MIM 600725), a signaling molecule expressed in the zone of polarizing activity (ZPA) at the posterior margin of the limb bud, and the interaction between Shh and Gli3.24 Mutations in the SHH regulatory region ZRS (ZPA regulatory sequence) located within an intron of the neighboring gene LMBR1 (MIM 605522) and duplications encompassing the ZRS are known to cause syndactyly type IV (MIM 186200) and polydactyly (MIM 174500) due to ectopic misexpression of Shh at the anterior margin of the limb bud.6,25,26 Polydactyly as observed in SHH regulatory mutations is also a feature of the mouse mutant Doublefoot (Dbf).27 In this case, however, Ihh and not Shh is ectopically expressed in the anterior limb bud mesenchyme.28,29 This ectopic expression leads to an anterior expansion of positive Hh regulators such as Ptch1 and Gli1 (MIM 165220) and at the same time to a reduction of Gli3 expression.27 The Dbf mutation was identified as a large deletion located 5′ of Ihh and encompassing ∼600 kb of sequence, including parts of the Ihh flanking gene Nhej1 and 22 other genes.27 The Dbf deletion is thus telomeric to the duplications described here. It has been hypothesized that the deletion removes a long-range regulatory element, most likely a repressor, and by this causes ectopic Ihh expression. Syndactyly of the fingers and toes was also observed in a case with a balanced translocation (2;7)(q26;p22) disrupting NHEJ1 in intron 5.30 The translocation separates any regulatory sequences located distal to the breakpoint, including the hypothetical repressor, and may thus have a pathogenetic effect similar to that of the deletion. In the Hoxd cluster it was shown that the distance of repressor and/or enhancer elements to their target gene is important for precise gene regulation.31 Tandem duplications result in the expansion of distance between regulator and target and may thus result in imprecision or even inactivation of the regulator.32 The duplications presented here may have a similar effect and by this cause misexpression of IHH as described in the Dbf mice. The presence of bilateral preaxial polydactyly, as observed in individual IV-1 from family 3, is a typical abnormality in patients with mutations in the ZRS leading to SHH misexpression, thus supporting this hypothesis.
Our results identify a putative distant regulator of IHH that is located within a large intron of the neighboring gene NHEJ1. A very similar situation exists for SHH, in which the limb control region ZRS is also located in an intron of a flanking gene (LMBR1). Such similarities may reflect the common evolutionary past of these Hh genes und underscore the importance of long-range regulation in development. Our findings show that developmental defects can be caused by CNVs in noncoding regions. The resulting phenotype may be completely different from those caused by mutations within the coding sequence of the corresponding gene.
Acknowledgments
This work was supported by a grant from the Deutsche Forschungsgemeinschaft to E.K., K.D., and S.M. (KL 2158/2-1) and by funding from the University Grants Council (AoE 04/04) and the General Research Grant (HKU760608M) of Hong Kong. We thank Douglas P. Mortlock for generously providing us with the vector pSfi-Hsp68lacZ. We acknowledge Fabienne Trotier for excellent technical assistance.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Decipher, https://decipher.sanger.ac.uk/
Ensembl Genome Browser, http://www.ensembl.org/index.html
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Lupski J.R. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/s0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- 2.Redon R., Ishikawa S., Fitch K.R., Feuk L., Perry G.H., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sebat J., Lakshmi B., Troge J., Alexander J., Young J., Lundin P., Månér S., Massa H., Walker M., Chi M. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- 4.Iafrate A.J., Feuk L., Rivera M.N., Listewnik M.L., Donahoe P.K., Qi Y., Scherer S.W., Lee C. Detection of large-scale variation in the human genome. Nat. Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- 5.Henrichsen C.N., Vinckenbosch N., Zöllner S., Chaignat E., Pradervand S., Schütz F., Ruedi M., Kaessmann H., Reymond A. Segmental copy number variation shapes tissue transcriptomes. Nat. Genet. 2009;41:424–429. doi: 10.1038/ng.345. [DOI] [PubMed] [Google Scholar]
- 6.Klopocki E., Ott C.E., Benatar N., Ullmann R., Mundlos S., Lehmann K. A microduplication of the long range SHH limb regulator (ZRS) is associated with triphalangeal thumb-polysyndactyly syndrome. J. Med. Genet. 2008;45:370–375. doi: 10.1136/jmg.2007.055699. [DOI] [PubMed] [Google Scholar]
- 7.Kurth I., Klopocki E., Stricker S., van Oosterwijk J., Vanek S., Altmann J., Santos H.G., van Harssel J.J., de Ravel T., Wilkie A.O. Duplications of noncoding elements 5′ of SOX9 are associated with brachydactyly-anonychia. Nat. Genet. 2009;41:862–863. doi: 10.1038/ng0809-862. [DOI] [PubMed] [Google Scholar]
- 8.Dathe K., Kjaer K.W., Brehm A., Meinecke P., Nürnberg P., Neto J.C., Brunoni D., Tommerup N., Ott C.E., Klopocki E. Duplications involving a conserved regulatory element downstream of BMP2 are associated with brachydactyly type A2. Am. J. Hum. Genet. 2009;84:483–492. doi: 10.1016/j.ajhg.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bosse K., Betz R.C., Lee Y.A., Wienker T.F., Reis A., Kleen H., Propping P., Cichon S., Nöthen M.M. Localization of a gene for syndactyly type 1 to chromosome 2q34-q36. Am. J. Hum. Genet. 2000;67:492–497. doi: 10.1086/303028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jain M., Wallis D., Robin N.H., De Vrieze F.W., Hardy J.A., Ghadami M., Bosse K., Betz R.C., Nöthen M.M., Arcos-Burgos M., Muenke M. Locus homogeneity between syndactyly type 1A and craniosynostosis Philadelphia type? Am. J. Med. Genet. A. 2008;146A:2308–2311. doi: 10.1002/ajmg.a.32445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robin N.H., Segel B., Carpenter G., Muenke M. Craniosynostosis, Philadelphia type: a new autosomal dominant syndrome with sagittal craniosynostosis and syndactyly of the fingers and toes. Am. J. Med. Genet. 1996;62:184–191. doi: 10.1002/(SICI)1096-8628(19960315)62:2<184::AID-AJMG13>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 12.Buck D., Malivert L., de Chasseval R., Barraud A., Fondanèche M.C., Sanal O., Plebani A., Stéphan J.L., Hufnagel M., le Deist F. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell. 2006;124:287–299. doi: 10.1016/j.cell.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 13.Vortkamp A., Lee K., Lanske B., Segre G.V., Kronenberg H.M., Tabin C.J. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996;273:613–622. doi: 10.1126/science.273.5275.613. [DOI] [PubMed] [Google Scholar]
- 14.Albrecht A.N., Schwabe G.C., Stricker S., Böddrich A., Wanker E.E., Mundlos S. The synpolydactyly homolog (spdh) mutation in the mouse — a defect in patterning and growth of limb cartilage elements. Mech. Dev. 2002;112:53–67. doi: 10.1016/s0925-4773(01)00639-6. [DOI] [PubMed] [Google Scholar]
- 15.St-Jacques B., Hammerschmidt M., McMahon A.P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999;13:2072–2086. doi: 10.1101/gad.13.16.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morriss-Kay G.M., Wilkie A.O. Growth of the normal skull vault and its alteration in craniosynostosis: insights from human genetics and experimental studies. J. Anat. 2005;207:637–653. doi: 10.1111/j.1469-7580.2005.00475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jabs E.W., Müller U., Li X., Ma L., Luo W., Haworth I.S., Klisak I., Sparkes R., Warman M.L., Mulliken J.B. A mutation in the homeodomain of the human MSX2 gene in a family affected with autosomal dominant craniosynostosis. Cell. 1993;75:443–450. doi: 10.1016/0092-8674(93)90379-5. [DOI] [PubMed] [Google Scholar]
- 18.Amano K., Ichida F., Sugita A., Hata K., Wada M., Takigawa Y., Nakanishi M., Kogo M., Nishimura R., Yoneda T. MSX2 stimulates chondrocyte maturation by controlling Ihh expression. J. Biol. Chem. 2008;283:29513–29521. doi: 10.1074/jbc.M803681200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jenkins D., Seelow D., Jehee F.S., Perlyn C.A., Alonso L.G., Bueno D.F., Donnai D., Josifova D., Josifiova D., Mathijssen I.M. RAB23 mutations in Carpenter syndrome imply an unexpected role for hedgehog signaling in cranial-suture development and obesity. Am. J. Hum. Genet. 2007;80:1162–1170. doi: 10.1086/518047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eggenschwiler J.T., Bulgakov O.V., Qin J., Li T., Anderson K.V. Mouse Rab23 regulates hedgehog signaling from smoothened to Gli proteins. Dev. Biol. 2006;290:1–12. doi: 10.1016/j.ydbio.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 21.Rice D.P., Connor E.C., Veltmaat J.M., Lana-Elola E., Veistinen L., Tanimoto Y., Bellusci S., Rice R. Gli3Xt-J/Xt-J mice exhibit lambdoid suture craniosynostosis which results from altered osteoprogenitor proliferation and differentiation. Hum. Mol. Genet. 2010;19:3457–3467. doi: 10.1093/hmg/ddq258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McDonald-McGinn D.M., Feret H., Nah H.D., Bartlett S.P., Whitaker L.A., Zackai E.H. Metopic craniosynostosis due to mutations in GLI3: A novel association. Am. J. Med. Genet. A. 2010;152A:1654–1660. doi: 10.1002/ajmg.a.33495. [DOI] [PubMed] [Google Scholar]
- 23.Mundlos S., Otto F., Mundlos C., Mulliken J.B., Aylsworth A.S., Albright S., Lindhout D., Cole W.G., Henn W., Knoll J.H. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell. 1997;89:773–779. doi: 10.1016/s0092-8674(00)80260-3. [DOI] [PubMed] [Google Scholar]
- 24.Litingtung Y., Dahn R.D., Li Y., Fallon J.F., Chiang C. Shh and Gli3 are dispensable for limb skeleton formation but regulate digit number and identity. Nature. 2002;418:979–983. doi: 10.1038/nature01033. [DOI] [PubMed] [Google Scholar]
- 25.Furniss D., Lettice L.A., Taylor I.B., Critchley P.S., Giele H., Hill R.E., Wilkie A.O. A variant in the sonic hedgehog regulatory sequence (ZRS) is associated with triphalangeal thumb and deregulates expression in the developing limb. Hum. Mol. Genet. 2008;17:2417–2423. doi: 10.1093/hmg/ddn141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lettice L.A., Hill A.E., Devenney P.S., Hill R.E. Point mutations in a distant sonic hedgehog cis-regulator generate a variable regulatory output responsible for preaxial polydactyly. Hum. Mol. Genet. 2008;17:978–985. doi: 10.1093/hmg/ddm370. [DOI] [PubMed] [Google Scholar]
- 27.Babbs C., Furniss D., Morriss-Kay G.M., Wilkie A.O. Polydactyly in the mouse mutant Doublefoot involves altered Gli3 processing and is caused by a large deletion in cis to Indian hedgehog. Mech. Dev. 2008;125:517–526. doi: 10.1016/j.mod.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang Y., Guillot P., Boyd Y., Lyon M.F., McMahon A.P. Evidence that preaxial polydactyly in the Doublefoot mutant is due to ectopic Indian Hedgehog signaling. Development. 1998;125:3123–3132. doi: 10.1242/dev.125.16.3123. [DOI] [PubMed] [Google Scholar]
- 29.Crick A.P., Babbs C., Brown J.M., Morriss-Kay G.M. Developmental mechanisms underlying polydactyly in the mouse mutant Doublefoot. J. Anat. 2003;202:21–26. doi: 10.1046/j.1469-7580.2003.00132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cantagrel V., Lossi A.M., Lisgo S., Missirian C., Borges A., Philip N., Fernandez C., Cardoso C., Figarella-Branger D., Moncla A. Truncation of NHEJ1 in a patient with polymicrogyria. Hum. Mutat. 2007;28:356–364. doi: 10.1002/humu.20450. [DOI] [PubMed] [Google Scholar]
- 31.Tarchini B., Duboule D. Control of Hoxd genes' collinearity during early limb development. Dev. Cell. 2006;10:93–103. doi: 10.1016/j.devcel.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 32.Kantaputra P.N., Klopocki E., Hennig B.P., Praphanphoj V., Le Caignec C., Isidor B., Kwee M.L., Shears D.J., Mundlos S. Mesomelic dysplasia Kantaputra type is associated with duplications of the HOXD locus on chromosome 2q. Eur. J. Hum. Genet. 2010 doi: 10.1038/ejhg.2010.116. [epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.