

Abstract
Discovering and following up on genetic associations with complex phenotypes require large patient cohorts. This is particularly true for patient cohorts of diverse ancestry and clinically relevant subsets of disease. The ability to mine the electronic health records (EHRs) of patients followed as part of routine clinical care provides a potential opportunity to efficiently identify affected cases and unaffected controls for appropriate-sized genetic studies. Here, we demonstrate proof-of-concept that it is possible to use EHR data linked with biospecimens to establish a multi-ethnic case-control cohort for genetic research of a complex disease, rheumatoid arthritis (RA). In 1,515 EHR-derived RA cases and 1,480 controls matched for both genetic ancestry and disease-specific autoantibodies (anti-citrullinated protein antibodies [ACPA]), we demonstrate that the odds ratios and aggregate genetic risk score (GRS) of known RA risk alleles measured in individuals of European ancestry within our EHR cohort are nearly identical to those derived from a genome-wide association study (GWAS) of 5,539 autoantibody-positive RA cases and 20,169 controls. We extend this approach to other ethnic groups and identify a large overlap in the GRS among individuals of European, African, East Asian, and Hispanic ancestry. We also demonstrate that the distribution of a GRS based on 28 non-HLA risk alleles in ACPA+ cases partially overlaps with ACPA- subgroup of RA cases. Our study demonstrates that the genetic basis of rheumatoid arthritis risk is similar among cases of diverse ancestry divided into subsets based on ACPA status and emphasizes the utility of linking EHR clinical data with biospecimens for genetic studies.
Introduction
Genome-wide association studies (GWASs) have successfully identified hundreds of genetic risk factors predisposing individuals to many complex diseases.1,2 Most common DNA variants by themselves, however, confer relatively small increments of risk. This poses a challenge for genetic studies of individual risk alleles because achieving sufficient statistical power in a genetic association study requires thousands of case-control samples. The problem is amplified in patients of diverse ancestry and for clinically relevant phenotypes within a given disease because creating subsets of patients further reduces sample size.
Electronic health records (EHRs) represent a rich source of clinical data and might make it possible to efficiently identify large and diverse patient cohorts for translational genetic research.3 Because EHR data have been collected as part of routine clinical care over many years, EHRs could make it possible to rapidly procure patient data across a broad range of clinical phenotypes. Recent reports indicate that in 2010 approximately 20% of physicians in the US use a basic EHR system4 (see Web Resources). EHR adoption rates are expected to grow because the US government has called for every American to have an EHR by 2014, making this a growing opportunity for genetics research.5
Few studies have demonstrated that EHR clinical data linked with biospecimens are suitable for genetic research. Two genetic studies have used EHR data to conduct case-control association studies,6,7 but they did not specifically explore genetic associations of disease across different ethnic groups or within clinically relevant subsets of cases. Our group8 and others3 have defined clinical phenotypes on the basis of EHR data but have not conducted genetic research with EHR clinical data.
Rheumatoid arthritis (RA [MIM 180300]) is a complex disease that provides an appropriate test case for the utility of genetic studies using EHR clinical data. It is a relatively rare disease, occurring in approximately 0.5% of the adult population,9 making it difficult to collect large, multi-ethnic patient cohorts. There is a clear genetic basis to RA: approximately 60% of the disease variability is inherited.10,11 To date, more than 30 loci, which explain approximately 20% of variance in disease risk, have been identified.12–23 The vast majority of RA risk alleles have been identified and validated in patients who are of European ancestry and are seropositive for disease-specific autoantibodies (either anti-citrullinated protein antibodies [ACPAs] or rheumatoid factor [RF]). Accordingly, it is largely unknown whether these alleles contribute to risk in other ethnic groups or in seronegative disease (and ACPA− disease in particular).
The purpose of our study was to investigate the relevance of known RA risk alleles in a multi-ethnic case-control cohort that leverages clinical data from the EHR and biospecimens collected as part of routine clinical care. We used RA cases identified from EHR data to obtain a large cohort suitable for genetic studies of RA risk8 and created subsets of patients on the basis of ACPA status and ethnic group. In comparing the effect sizes of individual risk alleles and aggregate genetic risk scores (GRS) among these patient subgroups, we provide a deeper understanding of the genetic basis of ACPA+ and ACPA− RA risk in a multi-ethnic cohort.
Subjects and Methods
EHR Case-Control Cohort
We have previously described an algorithm that uses codified EHR data and narrative EHR data to define RA cases with high accuracy.8 This approach allows the user to select sensitivity and specificity thresholds to maximize power and minimize misclassification bias depending upon the research question. For our genetic study, we performed simulations that varied by sample size, rates of case-control status, misclassification, and statistical power to select a specificity threshold of 95% (Figure S1). At this threshold, we defined a cohort of 4,575 potential RA cases who had received medical care within our healthcare system. For each case, we identified three potential controls (n = 13,725) matched for age, gender, self-reported ethnicity, and number of observations of codified data entries in the EHR. Matching by the number of observations (facts) gives a rough approximation to hospital activity. To assess how well cases and controls were matched, we compared ranking of concepts in cases and controls. The top ten diagnoses (not related to original selection) consistently give Spearman rank-order correlations of the two sets >0.9. We excluded controls with any diagnostic code of the following autoimmune diseases: 714.x RA and other inflammatory polyarthropathies, 710.x diffuse diseases of connective tissue, 720.x ankylosing spondylitis and other inflammatory spondyloarthropathies, 711.2x arthropathy in Behcet syndrome, 135 sarcoidosis, 425.8 dilated cardiomyopathy 2/2 dermatomyositis, scleroderma, vasculitis, 446 polyarteritis nodosa, 447.6 arteritis unspecified, 725 polymyalgia rheumatica, 136.1 Behcet syndrome, 286.5 antiphospholipid antibody syndrome, 446.21 Goodpasture syndrome, 446.4 Wegener granulomatosis, 446.5 giant cell aortitis/temporal arteritis, 446.7 Takayasu arteritis, and 696.0 psoriatic arthropathy.
Biospecimen Collection
To collect biospecimens on cases and controls, we submitted cohorts of unique medical record numbers linked to the project-specific subject ID to the Brigham and Women's Hospital (BWH) Specimen Bank, which handles prospective collection of discarded samples across five clinical laboratories within Partners Healthcare in the Boston Metropolitan area (USA). The BWH Specimen Bank operates under an approved Institutional Review Board (IRB) protocol. It acts as an “Honest Broker” for the collection and release of anonymous and de-identified samples to investigators with IRB-approved protocol for the use of retrospective and/or prospectively collected materials.
After receipt of the cohorts, Specimen Bank staff verified and loaded the cohorts into the Crimson LIMS, which identified clinical samples at their point of discard after completion of all clinical diagnostic testing. Additional filters were added to the queries so that discarded EDTA-anticoagulated whole blood from patients who had not received blood or platelet transfusions in the past 5 days could be found. Over the course of approximately 1 year, discarded samples from a total of 1552 RA cases and 1504 matched controls were collected in this manner (Table 1). Discarded blood remained at room temperature for up to 12 hr until clinical laboratory testing was complete, and it was then stored at 4°C until the point of discard, which varied at supplying labs from 24–72 hr after its initial collection. Prior work conducted on discarded samples showed no effects on the quality or amount of genomic DNA obtained from 1– 5 days after collection (L. Bry, personal communication).
Table 1.
Characteristics of Cases and Controls Included in the EHR Study
| Characteristic | Cases (n = 1552) | Controls (n = 1504) |
|---|---|---|
| Age, mean (SD) | 60.1 (13.8) | 63.5 (13.8) |
| Female, n (%) | 1258 (81.1) | 1207 (80.3) |
| ACPA+, n (%) | 1051 (67.7) | NA |
| Methotrexate, n (%) | 978 (63.0) | 18 (1.20) |
| Anti-TNF, n (%) | 589 (37.9) | 10 (0.66) |
| Reported EHR Ethnicity | ||
| European (%) | 1118 (72.0) | 1139 (75.7) |
| African (%) | 127 (8.2) | 135 (9.0) |
| Asian (%) | 30 (1.9) | 25 (1.7) |
| Hispanic (%) | 98 (6.3) | 108 (7.2) |
| Other (%) | 14 (0.9) | 5 (0.3) |
| Unreported (%) | 165 (11) | 92 (6) |
“NA” indicates that ACPA (Anti-citrullinated protein antibodies) were not measured in controls. Reported EHR Race: individuals who could not be classified under the four broad ethnic groups of European, African, East Asian, or Hispanic ancestries were classified as “other.” Individuals for whom we had no EHR-reported ethnicities were classified as “unreported.” Age and gender were derived from the codified EHR; ACPA status was derived by direct measurement; medications were obtained from the codified EHR data (when the medication was prescribed by a treating physician).
Samples were centrifuged so that Buffy coat cells would be separated from plasma. Aliquots consisting of 1 ml Buffy coat and 2 aliquots of up to 1 ml of plasma were created and stored at −80°C. DNA was extracted from frozen blood with the Gentra Puregene DNA extraction kit from QIAGEN. DNA concentration was determined with the Quant-IT Picogreen dsDNA reagent kit from Invitrogen. Of the 3090 blood samples processed, we were able to retrieve genomic DNA at >50 ng/μl from 2626 samples. Samples with <50 ng/μl of DNA (n = 464) were whole-genome amplified with the REPLI-g kit from QIAGEN. In total, 98.9% of the 3090 samples were of sufficient final concentration (≥50 ng/μl) for our genetic studies. Once the biospecimen was selected for research purposes, all personal health information was removed so that patient confidentiality was maintained. The Institutional Review Board of Partners HealthCare approved our protocol.
Genotyping
One hundred and ninety-two ancestry-informative markers (AIMs)24,25 and 29 SNPs from 27 RA risk loci were genotyped at the Broad Institute according to the BeadExpress manufacturer's protocol (www.illumina.com). Ninety-six-well plates were prepared with DNA at a uniform concentration of 50 ng/μl. BeadExpress raw data were processed with Illumina's BeadStudio software suite (genotyping module 3.3.7), producing report files containing normalized intensity data and SNP genotypes. All SNP genotypes were inferred via a genotyping cluster file automatically generated by BeadStudio. This file normalizes the intensities and identifies clusters. After genotyping, a manual review of clusters ensured high-quality data. Quality-control filters for SNPs included a missing-genotype rate of <10% and a minor-allele frequency of >1%. At this stage in our analysis, we did not exclude SNPs on the basis of deviation from Hardy-Weinberg equilibrium because of the multi-ethnic component of the study. We have, however, applied this quality control at a later time point after assigning our ethnicities on the basis of genetic markers (see population structure assessment). Out of the 221 SNPs genotyped, seven SNPs had >10% missing genotypes. We excluded individuals (n = 61) who were missing >10% of SNPs passing quality control.
ACPA Measurement
We used the plasma collected to measure RA disease-specific autoantibodies against ACPAs by using the second-generation kit from Inova Diagnostic. Positivity was defined according to the manufacturer's protocol. The kappa statistic for subjects with ACPA checked directly in our lab and ACPA checked in the hospital was 0.76 (where kappa > 0.75 indicates excellent reproducibility).
Assessment of Population Structure
Of the 192 AIMs, 185 passed our quality-control filters (see above); one was in high LD with another SNP (r2 ≥ 0.80) and was excluded from the analysis. Using these 184 SNPs, we calculated principal components (PCs) by using EIGENSTRAT without outlier removal.26 Of the 184 AIMs, 144 AIMs overlapped with SNPs genotyped in 11 Phase 3 HapMap populations. We used two methods to correct for population stratification in our multi-ethnic cohort. First, we developed a naive Bayes classifier to assign genetic ancestries. Second, we used PCs to correct for residual stratification within each broad category of ancestry. For our naive Bayes approach, we performed the following analysis. We grouped 11 HapMap populations consisting of CEU (Utah residents with ancestry from northern and western Europe), TSI (Tuscans in Italy), YRI (Yorubans in Ibadan, Nigeria), CHB (Han Chinese in Beijing, China), JPT (Japanese in Tokyo, Japan), CHD (Chinese in Denver, CO, USA), MKK (Maasai in Kinyawa, Kenya), LWK (Luhya in Webuye, Kenya), ASW (African Americans from Southwestern USA), GIH (Gujarati Indians in Houston, TX, USA), and MEX (Mexicans in Los Angeles, CA, USA) into four broad categories of African, East Asian, European, and other ancestries. We estimated the allele frequency of the 144 AIMs in our four aggregated populations. For each individual, we computed the probability of generating those genotypes across 144 SNPs on the condition of their having been sampled from each of our populations. Normalizing these values yielded the probability that those genotypes were drawn from a particular population. For each individual, we report the classification corresponding to the most likely population of origin (Figure 1). Individuals clustering with CEU/TSI populations along the top two principal components (orange filled circles) were classified as being of European origin on the basis of AIMs; individuals clustering with CHB/JPT/CHD were classified as having East Asian ancestry (purple filled circles); and individuals clustering with YRI/ASW/MKK/LWK were classified as having African ancestry (green filled circles). The remaining individuals were classified as Hispanics (gray filled circles) and correlated predominantly with EHR-reported Hispanic ethnicity (Table S1A). We also analyzed the correlation between self-reported ancestry and ancestries we classified with high confidence (probability of assigned ancestry > 0.9999) (Table S1B). We performed structured analyses within each broad category of classified ancestries separately.
Figure 1.
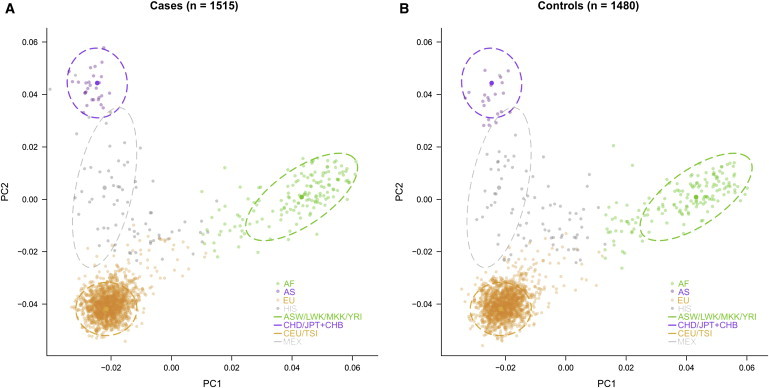
Assessment of Population Structure and Assignment of EHR Subjects into Four Ancestry Groups of European, African, East Asian, and Hispanic Descent
Population structure of the EHR (A) cases and (B) controls are plotted on PCs 1 and 2 (filled circles). These subjects were projected onto reference populations from Phase 3 of the HapMap Project via 144 ancestry-informative markers (dotted lines). The centroids indicate the outer bounds of the three major HapMap continental populations, and the darker filled circles indicate the center of each centroid. Individuals who were not assigned to these major continental populations were classified as “other” and were predominantly of EHR-reported Hispanic origin (gray circles).
Single-SNP Analysis
We performed single SNP analysis for 29 SNPs that have previously been reported to exceed genome-wide significance (p < 5 × 10−8) in at least one GWAS or in a recent meta-analysis of GWA studies.22 Some SNPs in our study were proxies of previously reported associations. SNP rs6679677 at the PTPN22 (MIM 600716) locus has an r2 of 1 with rs2476601;22 rs1160542 at the AFF3 (MIM 601464) locus has an r2 of 0.97 with rs10865035;22 rs13277113 at the BLK (MIM 191305) locus has an r2 of 0.88 with rs2736340;22 rs10118357 at the TRAF1-C5 (TRAF1 [MIM 601711], C5 [MIM 120900]) locus has an r2 of 0.97 with rs376184716 and an r2 of 1.0 with rs10818488;17 and rs10040327 at ANKRD55 (MIM not available) locus has an r2 of 0.33 with rs6850219.22
We performed single-SNP analysis on 1515 RA cases and 1480 controls that passed quality-control genotyping filters. We performed SNP associations with RA risk with a logistic regression model as implemented in the PLINK v. 1.06 software package.27 Each model consisted of one SNP as an independent variable and included the top five PCs as covariates. All reported p values are two tailed.
We compared the odds ratios from our EHR cohort to odds ratios derived from a recent meta-analysis of 5539 autoantibody-positive RA cases and 20,169 controls, all of European descent.22 To formally test this comparison, we performed a heterogeneity test across the 29 SNPs. For each SNP, we calculated the difference between the natural log of the ORs (βdiff), the SEdiff (square root of the sum of the two variances), and a Z score (βdiff/SEdiff). We then summed the squared Z scores across the 29 SNPs and determined an overall p value from a chi-square distribution with 29 degrees of freedom.
For the individuals of non-European ancestry, we observed differences in population allele frequency in healthy controls (Table S2). The statistical power to detect a significant association of SNPs with RA was calculated on the basis of the following: the ORs for association with RA in the European population; the sample size of the current population under study; and the allele frequencies in the population (Table S2).
Aggregate Genetic-Risk Score
We calculated a cumulative aggregate genetic-risk score,21 which is the sum of the weighted risk-allele counts for (i) all previously known RA risk alleles, including one HLA and 28 non-HLA SNPs and (ii) 28 non-HLA risk alleles in all datasets, including the EHR cohorts stratified by classified ancestries (see assessment of population structure) and the GWAS-meta-analysis dataset. As a result of a different analytical procedure (i.e., PCA correction and no case-control matching), the GWAS dataset in the current study consisted of slightly different sample numbers than our published GWAS.22 The weights for each SNP were derived from the recently published GWAS meta-analysis22 and were calculated as the natural log of the odds ratio for each allele (Table 2). These same weights were used in our comparison of GRS in the GWAS meta-analysis and ACPA+ versus ACPA− disease. Any individual with missing genotypes for a particular SNP was assigned the expected value of twice the risk-allele frequency for that SNP (missing genotypes were inferred for cases and controls separately). We calculated GRS across n SNPs according to the following formula:
where n is the number of SNPs, i is the SNP, wi is the weight for SNP i, and Xi is the number of risk alleles (0, 1, or 2).
Table 2.
Association at Known RA Loci in Four Major Continental Populations of European, African, East Asian, and Hispanic Descent in ACPA+ Cases versus Controls
|
Previous (European Ancestry) |
EHR EU (871 Cases, 1212 Controls) |
EHR AF (100 Cases, 150 Controls) |
EHR AS (23 Cases, 21 Controls) |
EHR HIS (57 Cases, 74 Controls) |
|||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
SNP |
Alleles |
Allele Frequency |
Allele Frequency |
Allele Frequency |
Allele Frequency |
||||||||||||||||
| SNP ID | Locus | Gene (s) | A1/A2 | Risk | OR (95% CI) | Cases | Controls | OR (95% CI) | P | Cases | Controls | OR (95% CI) | P | Cases | Controls | OR (95% CI) | P | Cases | Controls | OR (95% CI) | P |
| rs6679677 | 1p13 | PTPN22 | C/A | A | 1.94 (1.81,2.08) | 0.15 | 0.07 | 2.06 (1.68,2.53) | 7.19 × 10−12 | 0.01 | 0.01 | 1.03 (0.22, 4.77) | 0.97 | 0.00 | 0.00 | NA | NA | 0.03 | 0.04 | 0.54 (0.11, 2.74) | 0.46 |
| rs11586238 | 1p13 | CD2, CD58 | C/G | G | 1.13 (1.07,1.19) | 0.24 | 0.20 | 1.20 (1.04,1.40) | 0.02 | 0.10 | 0.12 | 0.89 (0.50,1.58) | 0.69 | 0.04 | 0.05 | 0.16 (0.01,3.17) | 0.23 | 0.21 | 0.21 | 1.12 (0.62, 2.02) | 0.71 |
| rs13031237 | 2p16 | REL | G/T | T | 1.13 (1.07,1.18) | 0.37 | 0.35 | 1.10 (0.97,1.25) | 0.15 | 0.13 | 0.09 | 1.61 (0.88,2.96) | 0.12 | 0.04 | 0.10 | 0.13 (0.01,1.51) | 0.10 | 0.18 | 0.23 | 0.62 (0.33, 1.17) | 0.14 |
| rs934734 | 2p14 | SPRED2 | A/G | G | 1.13 (1.08,1.18) | 0.53 | 0.52 | 1.05 (0.93,1.19) | 0.43 | 0.51 | 0.47 | 1.09 (0.77,1.52) | 0.64 | 0.25 | 0.24 | 1.71 (0.39,7.47) | 0.48 | 0.44 | 0.40 | 1.25 (0.76,2.07) | 0.38 |
| rs1160542 | 2q11 | AFF3 | A/G | G | 1.12 (1.07,1.17) | 0.49 | 0.47 | 1.11 (0.98,1.26) | 0.10 | 0.79 | 0.79 | 0.89 (0.57,1.40) | 0.62 | 0.43 | 0.33 | 3.25 (0.77,13.7) | 0.11 | 0.60 | 0.61 | 1.05 (0.59,1.86)∗ | 0.88 |
| rs7574865 | 2q32 | STAT4 | G/T | T | 1.16 (1.10,1.23) | 0.23 | 0.25 | 0.93 (0.80,1.08) | 0.33 | 0.14 | 0.12 | 1.24 (0.71,2.15) | 0.45 | 0.35 | 0.50 | 0.62 (0.19,2.02) | 0.43 | 0.48 | 0.22 | 2.81 (1.53,5.18) | 1.00E-03 |
| rs1980422 | 2q33 | CD28 | T/C | C | 1.12 (1.06,1.18) | 0.27 | 0.24 | 1.18 (1.02,1.36) | 0.02 | 0.25 | 0.25 | 1.00 (0.67,1.50) | 0.99 | 0.15 | 0.10 | 1.45 (0.26,8.08) | 0.67 | 0.27 | 0.20 | 1.68 (0.86,3.28) | 0.13 |
| rs3087243 | 2q33 | CTLA4 | G/A | G | 1.15 (1.10,1.20) | 0.60 | 0.53 | 1.28 (1.13,1.45) | 1.19E-04 | 0.80 | 0.76 | 1.19 (0.76,1.85) | 0.46 | 0.78 | 0.69 | 1.01 (0.28,3.62) | 0.98 | 0.60 | 0.64 | 0.83 (0.48,1.45) | 0.52 |
| rs13315591 | 3p14 | PXK | T/C | C | 1.29 (1.17,1.43) | 0.09 | 0.07 | 1.39 (1.11,1.75) | 4.79E-03 | 0.31 | 0.30 | 1.08 (0.73,1.60) | 0.71 | 0.00 | 0.00 | NA | NA | 0.09 | 0.10 | 1.18 (0.46,3.02)∗ | 0.73 |
| rs874040 | 4p15 | RBPJ | G/C | C | 1.14 (1.09,1.20) | 0.33 | 0.29 | 1.21 (1.06,1.38) | 0.01 | 0.32 | 0.37 | 0.73 (0.49,1.09) | 0.13 | 0.00 | 0.00 | NA | NA | 0.27 | 0.22 | 1.80 (0.92,3.51) | 0.09 |
| rs6822844 | 4q27 | IL2,IL21 | G/T | G | 1.11 (1.05,1.19) | 0.85 | 0.86 | 0.97 (0.81,1.15) | 0.70 | 0.96 | 0.99 | 0.29 (0.08,1.03) | 0.06 | 1.00 | 1.00 | NA | NA | 0.92 | 0.95 | 0.67 (0.24,1.88) | 0.44 |
| rs10040327 | 5q11 | ANKRD55, IL6ST | C/A | C | 1.33 (1.23,1.47) | 0.90 | 0.88 | 1.28 (1.05,1.57) | 0.02 | 0.90 | 0.89 | 1.07 (0.59,1.93) | 0.82 | 1.00 | 1.00 | NA | NA | 0.92 | 0.92 | 1.16 (0.42,3.18) | 0.78 |
| rs26232 | 5q21 | C5orf13 | C/T | C | 1.14 (1.09,1.19) | 0.69 | 0.69 | 1.04 (0.90,1.19) | 0.62 | 0.72 | 0.72 | 0.98 (0.66,1.45) | 0.92 | 0.74 | 0.88 | 0.50 (0.10,2.45) | 0.39 | 0.81 | 0.76 | 1.38 (0.69,2.74) | 0.36 |
| rs6457620 | 6p21 | HLA∗04 tag | G/C | C | 2.35 (2.25,2.46) | 0.68 | 0.52 | 2.03 (1.77,2.32) | 4.44E-25 | 0.63 | 0.49 | 1.89 (1.29,2.76) | 1.09E-03 | 0.72 | 0.52 | 1.98 (0.65,6.07) | 0.23 | 0.75 | 0.61 | 1.92 (1.03,3.57) | 0.04 |
| rs548234 | 6q21 | PRDM1 | T/C | C | 1.10 (1.05,1.16) | 0.33 | 0.30 | 1.08 (0.94,1.24) | 0.27 | 0.12 | 0.11 | 1.23 (0.69,2.20) | 0.48 | 0.39 | 0.29 | 2.08 (0.61,7.07) | 0.24 | 0.19 | 0.22 | 0.80 (0.42,1.52) | 0.49 |
| rs10499194 | 6q23 | TNFAIP3 | C/T | C | 1.10 (1.04,1.15) | 0.74 | 0.68 | 1.33 (1.16,1.54) | 5.88E-05 | 0.85 | 0.79 | 1.38 (0.86,2.22) | 0.18 | 0.96 | 0.98 | 0.37 (0.02,8.11) | 0.53 | 0.71 | 0.72 | 1.02 (0.56,1.88)∗ | 0.95 |
| rs6920220 | 6q23 | TNFAIP3 | G/A | A | 1.22 (1.16,1.29) | 0.23 | 0.19 | 1.25 (1.08,1.46) | 3.55E-03 | 0.11 | 0.12 | 0.92 (0.50,1.68) | 0.79 | 0.00 | 0.00 | NA | NA | 0.11 | 0.12 | 1.00 (0.42,2.38)∗ | 1.00 |
| rs394581 | 6q25 | TAGAP | T/C | T | 1.10 (1.04,1.15) | 0.71 | 0.67 | 1.16 (1.01,1.33) | 0.04 | 0.49 | 0.57 | 0.74 (0.51,1.08) | 0.12 | 0.95 | 0.88 | 7.24 (0.43,123) | 0.17 | 0.78 | 0.80 | 0.66 (0.34,1.30) | 0.23 |
| rs3093023 | 6q27 | CCR6 | G/A | A | 1.13 (1.08,1.19) | 0.46 | 0.42 | 1.16 (1.02,1.31) | 0.02 | 0.22 | 0.16 | 1.47 (0.93,2.33) | 0.10 | 0.54 | 0.40 | 2.12 (0.66,6.77) | 0.21 | 0.38 | 0.30 | 1.74 (0.97,3.11) | 0.06 |
| rs10488631 | 7q32 | IRF5 | T/C | C | 1.19 (1.11,1.28) | 0.13 | 0.11 | 1.28 (1.06,1.55) | 0.01 | 0.05 | 0.03 | 1.76 (0.70,4.44) | 0.23 | 0.00 | 0.00 | NA | NA | 0.15 | 0.16 | 0.91 (0.44,1.87) | 0.79 |
| rs13277113 | 8p23 | BLK | G/A | A | 1.12 (1.07,1.18) | 0.26 | 0.23 | 1.15 (1.00,1.33) | 0.05 | 0.14 | 0.12 | 1.21 (0.71,2.08) | 0.49 | 0.76 | 0.69 | 1.08 (0.36,3.28) | 0.89 | 0.60 | 0.43 | 1.78 (1.01,3.14) | 0.05 |
| rs2812378 | 9p13 | CCL21 | A/G | G | 1.10 (1.05,1.16) | 0.35 | 0.33 | 1.08 (0.95,1.23) | 0.24 | 0.38 | 0.39 | 0.99 (0.68,1.45) | 0.95 | 0.11 | 0.07 | 10.5 (0.57,192) | 0.11 | 0.39 | 0.32 | 1.57 (0.89,2.76) | 0.12 |
| rs951005 | 9p13 | CCL21 | A/G | A | 1.19 (1.11,1.27) | 0.83 | 0.80 | 1.18 (1.00,1.38) | 0.05 | 0.68 | 0.69 | 0.92 (0.62,1.36) | 0.66 | 0.96 | 0.90 | 2.47 (0.22,27.2) | 0.46 | 0.81 | 0.83 | 0.82 (0.42,1.60) | 0.56 |
| rs10118357 | 9q33 | TRAF1, C5 | A/G | G | 1.13 (1.08,1.18) | 0.44 | 0.39 | 1.19 (1.05,1.34) | 0.01 | 0.85 | 0.87 | 0.75 (0.45,1.24) | 0.26 | 0.57 | 0.60 | 0.85 (0.29,2.53) | 0.77 | 0.29 | 0.47 | 0.41 (0.23,0.76) | 4.00E-03 |
| rs706778 | 10p15 | IL2RA | C/T | T | 1.14 (1.08,1.19) | 0.43 | 0.40 | 1.10 (0.97,1.25) | 0.14 | 0.53 | 0.47 | 1.26 (0.87,1.82) | 0.22 | 0.57 | 0.62 | 0.77 (0.26,2.30) | 0.64 | 0.52 | 0.51 | 0.78 (0.45,1.37)∗ | 0.39 |
| rs4750316 | 10p15 | PRKCQ | G/C | G | 1.15 (1.09,1.22) | 0.82 | 0.81 | 1.08 (0.92,1.27) | 0.32 | 0.66 | 0.60 | 1.34 (0.92,1.96) | 0.13 | 0.87 | 0.86 | 0.91 (0.16,5.18)∗ | 0.92 | 0.89 | 0.82 | 1.55 (0.72,3.33) | 0.26 |
| rs1678542 | 12q13 | KIF5A, PIP4K2C | C/G | C | 1.10 (1.04,1.15) | 0.63 | 0.63 | 1.05 (0.92,1.19) | 0.46 | 0.52 | 0.58 | 0.77 (0.53,1.12) | 0.17 | 0.30 | 0.19 | 2.96 (0.73,12.0) | 0.13 | 0.56 | 0.57 | 1.09 (0.64,1.85)∗ | 0.75 |
| rs4810485 | 20q13 | CD40 | G/T | G | 1.18 (1.11,1.25) | 0.76 | 0.72 | 1.23 (1.06,1.43) | 0.01 | 0.95 | 0.94 | 1.13 (0.48,2.61) | 0.78 | 0.72 | 0.55 | 3.99 (0.92,17.4) | 0.07 | 0.82 | 0.81 | 0.97 (0.51,1.87)∗ | 0.94 |
| rs3218253 | 22q12 | IL2RB | G/A | A | 1.09 (1.03,1.15) | 0.26 | 0.27 | 0.92 (0.80,1.06) | 0.26 | 0.14 | 0.14 | 1.00 (0.58,1.74) | 1.00 | 0.09 | 0.10 | 1.80 (0.26,12.6)∗ | 0.56 | 0.15 | 0.22 | 0.77 (0.41,1.44) | 0.41 |
Previously known SNPs associated with rheumatoid arthritis risk among European populations are shown above. Listed are SNP ID, chromosome, position, and candidate gene(s) in the region. A1 refers to the major allele, and A2 refers to the minor allele based on the frequency in the controls in the GWAS meta-analysis. The risk allele refers to the allele that has previously been associated with risk of RA.22 Abbreviations are as follows: EU, Individuals of European ancestry; AF, individuals of African ancestry (including admixed African Americans); AS, individuals of East Asian ancestry; and HIS, individuals of Hispanic origin. An asterisk indicates the SNPs for which an inverse direction of association was obtained after PC correction (none of those SNPs were significant prior to or after PC correction).
We plotted the distribution of the GRS separately for cases with RA and controls. Using a regression model, we determined the relationship between case-control status and GRS and adjusted for the top five PCs. Using PC correction in the current analysis of the GWAS meta-analysis sample set as compared to outlier removal and PC case-control matching in the previously published study resulted in a modest difference in sample numbers (Table 3). Using the adjusted predicted values from the regression model, we calculated the area under the receiver operating characteristic (ROC) curves by plotting the sensitivity of the GRS against 1 specificity by using the library (pROC) in the statistical package R 2.10.28
Table 3.
Aggregate Genetic-Risk Scores, Effect Sizes, and AUC for ACPA+ Cases in the EHR Cohort and GWAS Meta-analysis Datasets
|
Sample Set |
Aggregate Genetic-Risk Score Derived from 29 RA Risk Alleles |
||||||
|---|---|---|---|---|---|---|---|
|
Controls |
ACPA+ Cases |
Effect Size and AUC in Cases versus Controlsa |
|||||
| Mean | SD | Mean | SD | OR (95% CI) | p | AUC (95% CI) | |
| EU (871 cases, 1229 controls) | 4.37 | 0.81 | 4.93 | 0.77 | 2.43 (2.29,2.59) | 5.55 × 10−46 | 0.71 (0.68–0.73) |
| AF (100 cases, 153 controls) | 4.35 | 0.72 | 4.62 | 0.73 | 1.74 (1.44,1.85) | 0.003 | 0.63 (0.56–0.70) |
| AS (23 cases, 21 controls) | 4.22 | 0.72 | 4.68 | 0.58 | 3.12 (1.65,3.31) | 0.075 | 0.74 (0.59–0.89) |
| HIS (57 cases, 77 controls) | 4.62 | 0.75 | 4.96 | 0.65 | 1.91 (1.43,2.03) | 0.026 | 0.66 (0.56–0.76) |
| GWAS (5500 cases, 22,619 controls) | 4.42 | 0.79 | 5.07 | 0.76 | 1.87 (1.85,1.99) | <10−300 | 0.73 (0.72–0.73) |
We calculated a GRS score derived from 29 known RA risk alleles for each individual in the EHR cohort and the GWAS meta-analysis dataset. Shown are the unadjusted mean and standard deviation (SD) of the GRS scores in controls and ACPA+ cases across all ethnic groups. Abbreviations are as follows: EU, individuals of European ancestry; AF, individuals of African ancestry (including admixed African Americans); AS, individuals of East Asian ancestry; HIS, individuals of Hispanic origin; GWAS, GWAS meta-analysis study.
A logistic-regression model adjusting for the top five PCs was used for calculating odds ratios (ORs) for each unit increase in GRS and corresponding p values. AUC (95% CI) represents the area under the receiver operating curve with a 95% CI interval.
Results
Multi-ethnic Case-Control Cohort
The clinical characteristics of the case-control samples used in our genetic study are shown in Table 1. The characteristics of our RA cases were similar to those observed in traditional patient registries.8 Here, however, cases and matched controls were of diverse ancestries, reflecting the demographics of the patient population served by hospitals in the Boston metropolitan area. Approximately 75% of the cases and controls were of self-described European ancestry, and the remainder were of African, East Asian, Hispanic, or other ancestry. Approximately 10% of cases did not have ancestry information reported in the EHR.
Population stratification due to genetic ancestry is known to bias genetic association studies.29 Because self-reported ancestry can differ from genetic ancestry, and because ancestry was not reported in 11.3% of cases and 6.4% of controls in our EHR cohort, we determined genetic ancestry in our EHR cohort by using a panel of 192 AIMs, which we selected to differentiate continental ancestry and European ancestry.24,25 Using a naive Bayes classifier, we assigned genetic ancestry to each case-control subject from our EHR cohort (see Subjects and Methods). As shown in Figure 1, the ancestries assigned by this approach were consistent with genetic ancestry observed via a principal-components method26 among four broad HapMap groups and were as follows: European ancestry, African ancestry (including admixed African Americans), East Asian ancestry, and Hispanic ancestry (individuals who were not classified according to the three groups mentioned above but who clustered with Hispanic individuals from HapMap).
We compared our genetically assigned ancestries to the EHR-reported ancestries (Table S1A). We observed 98% and 94% concordance between genetic and EHR-reported ancestry for individuals of European and African ancestry; concordance was 78% and 52% for East Asian and Hispanic ancestry, respectively. In individuals with highly confident predictions of genetic ancestry, concordance rates were close to 100% in individuals of European, East Asian, and African ancestry (Table S1B). On the basis of these data, we used AIMs to assign genetic ancestries to all individuals, rather than relying solely on ancestries reported in the EHR. In our subsequent analyses, we conducted structured statistical tests of association with risk of RA within these genetically defined ancestry subgroups, and we also used principal components to correct for residual population stratification within each subgroup by using 184 AIMs.
Single-SNP Analysis in ACPA+ Cases of Diverse Ancestries
Even though the clinical characteristics of the RA cases in our EHR cohort are similar to those of traditional RA cohorts,8 we sought to empirically validate previous associations to demonstrate the feasibility of human genetic studies in our newly collected case-control cohort. We reasoned that if our case-control definitions are precise, then the odds ratios of RA risk alleles measured in our cohort should be similar to those derived from other cohorts. To test this specifically, we conducted a case-control association study in a subset of cases that were ACPA+ and of European genetic ancestry. We chose this subgroup in order to compare odds ratios to a recent GWAS meta-analysis of 5539 autoantibody-positive RA cases and 20,169 controls, all of European ancestry.22
Of the 29 SNPs tested in 871 ACPA+ RA cases and 1212 controls of European ancestry, 16 achieved p < 0.05 in our EHR cohort, and the most significant SNPs demonstrated p = 4.4 × 10−25 (HLA-DRB1∗04 [MIM 142857] tag SNP, rs6457620, OR = 2.03) and p = 7.19 × 10−12 (PTPN22, rs6679677, OR = 2.06) (Table 2). As shown in Figure 2, the direction and magnitude of point estimates of the odds ratios for 26 of 29 SNPs were consistent between our EHR cohort and the GWAS meta-analysis; the remaining three SNPs (STAT4 [MIM 600558], rs7574865; IL2/21 (IL2 [MIM 147680] and IL21 [MIM 605384]), rs6822844; and IL2RB [MIM 146710], rs3218253) have point estimates close to 1. Considering the odds-ratio distribution of all 29 SNPs, there was no statistical difference between those observed in our EHR cohort and those observed in the GWAS meta-analysis (overall heterogeneity p = 0.18).
Figure 2.
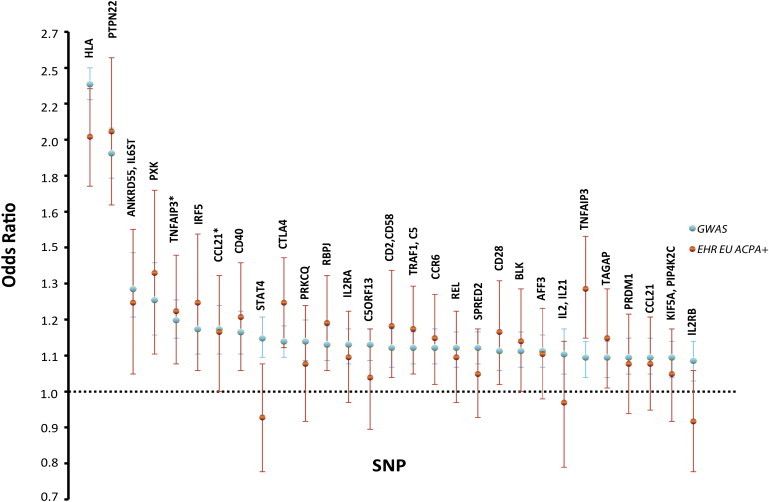
Overlap of Odds Ratio and 95% Confidence Intervals between Previous GWAS Meta-Analysis Dataset and ACPA+ European Subset from EHR Cohort
Asterisks indicate TNFAIP3 SNP rs6920220 and CCL21 SNP rs951005. EU indicates individuals of European descent from our EHR cohort. GWAS represents samples from the previously published GWAS meta-analysis.22
As a first step to determine whether these 29 SNPs also contribute to risk of ACPA+ RA in cases of non-European ancestry, we analyzed single SNPs for association within case-control samples of African, East Asian, and “other” (predominantly Hispanic) genetic ancestries (Table 2). Our multi-ethnic subgroups consisted of 100 ACPA+ cases and 150 controls of African ancestry, 23 ACPA+ cases and 21 controls of East Asian ancestry, and 57 ACPA+ cases and 74 controls of predominantly Hispanic ancestry. Despite the small sample size, we observed significant association at the HLA-DRB1∗04 tag SNP (rs6679677, OR 1.89, 95% confidence interval [CI] 1.29–2.76, p = 0.0011) in the individuals of African ancestry. Individuals of East Asian and Hispanic ancestry had a similar trend of association at this HLA-DRB1∗04 SNP (OR 1.98 and p = 0.23; and OR 1.92 and p = 0.04, respectively).
Aggregate Genetic-Risk Score in ACPA+ Cases across Diverse Ancestries
Although single-SNP analyses are required for confirmation of the contribution of individual risk alleles, aggregate SNP analyses provide a useful summary of risk across all alleles. This approach has the added benefit of a single statistical test (which reduces the multiple-hypothesis testing burden such that a p < 0.05 can be considered significant) that is useful for comparing genetic-risk profiles across multi-ethnic groups of small sample sizes.
To compare the RA genetic-risk profiles of our EHR cases to those of controls, we used a weighted genetic-risk score (GRS), which considers each individual's aggregate number of risk alleles weighted by the effect size of the allele21 (see Subjects and Methods). The larger the GRS number, the greater the number of risk alleles. As shown in Figure 3A, the distribution of the GRS in European ACPA+ cases significantly differs from that in controls (pEU = 5.6 × 10−46). As expected from our single-SNP analysis, the GRS in our EHR cohort was nearly identical to the GRS derived from the same 29 SNPs in a recent GWAS meta-analysis. We observed a mean (±SD) GRS of 5.1 (±0.8) in the GWAS cases (n = 5500) and 4.9 (±0.8) in the EHR cases of European ancestry (n = 871). In controls, the mean GRS was 4.4 (±0.8) in the GWAS dataset (n = 22,619) and 4.4 (±0.8) in the EHR dataset (n = 1229).
Figure 3.

Distribution of the Aggregate Genetic-Risk Score from 29 RA Risk Alleles in ACPA+ Cases versus Controls
Samples represented in the respective panels are (A) controls and ACPA+ cases in our EHR study and controls (orange lines) and seropositive (ACPA+ and or RF+) and healthy individuals from the GWAS meta-analysis, all of European descent (EU) (blue lines); (B) controls and ACPA+ cases of African descent (AF) (green lines), (C) controls and ACPA+ cases of East Asian descent (AS) (purple lines), and (D) controls and ACPA+ cases of Hispanic descent (HIS) (gray lines).
Despite small sample sizes, the distribution of an aggregate GRS for individuals of African (100 cases, 150 controls) and Hispanic (57 cases, 74 controls) descent was also significantly different between ACPA+ cases and controls in our EHR cohort (pAF = 0.003, pHIS = 0.026; Table 3 and Figure 3). We observed a similar nonsignificant trend in individuals of East Asian ancestry (23 cases, 21 controls; pAS = 0.075). In the admixed African American group, similar results were obtained when our analysis was limited to those with the highest probability of being of African origin (individuals who clustered most strongly with YRI samples from HapMap; data not shown), indicating that the result in African Americans is not driven by European admixture.
As a complementary method to compare the GRS across all ancestry groups, we calculated the ORs for each unit increase in GRS and AUC. Although the latter is most often used to discriminate cases from controls, we used it as a measure of the contribution of the GRS in each of the ethnic subgroups. All genetic ancestry subgroups displayed similar effect sizes (Table 3). We note that the OR and AUC in cases of African ancestry are less than in cases of non-African ancestry.
Taken together, these data indicate that, despite some individual locus heterogeneity, common SNPs derived from association testing in ACPA+ Europeans also contribute to risk in ACPA+ cases of non-European ancestry.
Aggregate Risk Score in ACPA− Cases of European Ancestry
It is unknown whether the majority of risk alleles discovered in ACPA+ disease also contribute to risk in ACPA− disease. One exception is the HLA region, in which distinct alleles are associated with risk in ACPA+ but not ACPA− disease.30,31 We evaluated the genetic basis of ACPA+ versus ACPA− disease among patients of European ancestry in two ways: by using weights derived from a previous meta-analysis in autoantibody positive RA to compare the GRS derived from 28 non-HLA risk alleles in ACPA− individuals to that from controls; and by comparing the GRS derived from 28 non-HLA risk alleles in ACPA− individuals to that in ACPA+ individuals in our EHR cohort. (There were too few ACPA− cases in the non-European individuals to permit a meaningful comparison; an exploratory single-SNP analysis can be found in Table S3).
Among ACPA− cases of European ancestry (n = 378), we found a significant difference in the distribution of the GRS derived from 28 non-HLA risk alleles in ACPA− RA cases compared to controls (p = 8.42 × 10−4, mean GRS = 3.59, AUC = 0.55). The effect was driven by SNPs in aggregate rather than individual SNPs; only 1 out of 29 SNPs had a p < 0.05, but 20 out of 29 had an OR in the same direction as that for the previous GWAS meta-analysis of autoantibody-positive disease (Table S3, Figure 4). Because the weights for the GRS were derived from studies investigating autoantibody-positive RA, these results demonstrate that there is some overlap between the genetic basis of ACPA+ and ACPA− disease.
Figure 4.
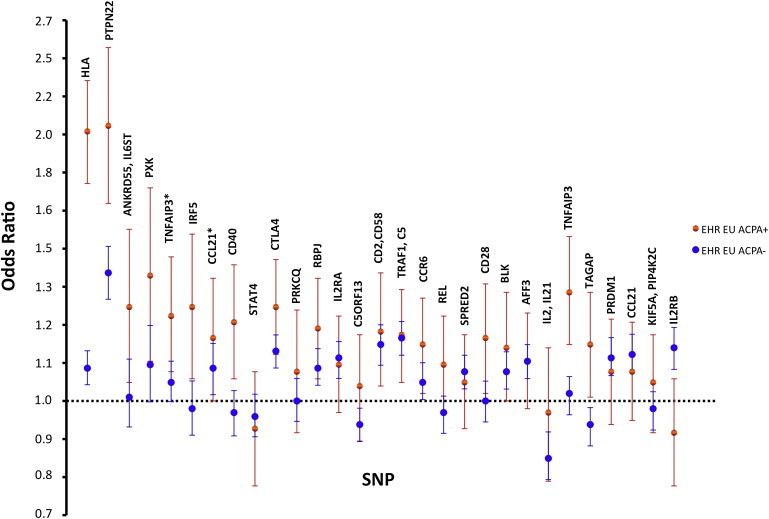
Overlap of Odds Ratio and 95% Confidence Intervals between European ACPA+ and ACPA− Subsets from the EHR Cohort
Asterisks indicate TNFAIP3 SNP rs6920220 and CCL21 SNP rs951005. EU indicates individuals of European descent from our EHR cohort.
To investigate risk alleles in ACPA+ and ACPA− cases more directly, we compared the GRS derived from 28 non-HLA risk alleles of the two subsets in cases of European ancestry from our EHR cohort. The effect sizes of these individual risk alleles and the distribution of the GRS were lower ACPA− disease than in ACPA+ disease (p = 7.29 × 10−7, Table S4, Figure 5). This result is consistent with the lower AUC in ACPA− versus ACPA+ individuals when compared to controls (AUC = 0.55 versus 0.66, respectively).
Figure 5.
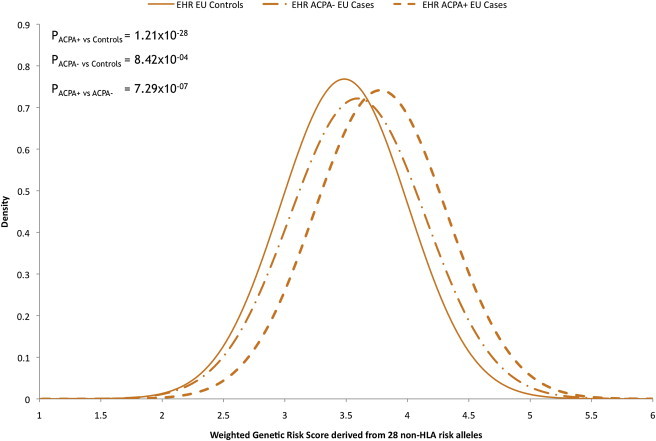
Distribution of the Aggregate Genetic-Risk Score from 28 non-HLA RA Risk Alleles in Controls, ACPA+ Cases, and ACPA− Cases in Individuals of European Ancestry
On the basis of these results, we conclude that although there is overlap between the genetic basis of ACPA+ and ACPA− disease, the overlap is only partial.
Discussion
Our study utilizes a valuable resource by linking EHR data with biospecimens to conduct discovery genetic research. For a complex autoimmune disease, RA, we have demonstrated empirically that the effect sizes of individual RA risk alleles and aggregate genetic-risk scores are similar in our EHR cohort and traditional cohorts. What is notable about our study, however, is that we used EHR-derived data and a genetic-risk score to demonstrate that (a) risk alleles derived from cases of European ancestry also contribute to risk among cases of East Asian, African, and Hispanic ancestries and (b) ACPA+ risk alleles also confer risk in ACPA− disease (although the genetic overlap is partial and incomplete).
To demonstrate that our case-control collection is valid for genetic discovery research, we compared the effect sizes of 29 validated RA risk alleles in our EHR case-control cohort to those of a recently published GWAS meta-analysis involving 5,539 autoantibody-positive RA cases and 20,169 controls and using subjects enrolled in traditional cohorts.22 If there were substantial misclassification of case-control status in our EHR cohort, then the OR in our EHR study would be consistently less than the OR from traditional registries. In contrast, we found that the effect sizes were quite similar. This was true not just at the single-SNP level, but also for analysis of all SNPs in aggregate (weighted GRS). Our study demonstrates a direct comparison of genetic findings from an EHR-derived cohort, as opposed to traditional collections.
With few exceptions,12–14,16,23 most non-HLA validated RA risk alleles have emerged from GWAS of seropostive RA cases of European ancestry. This raises the important issue of whether these alleles contribute to risk in cases of non-European ancestry. Although our sample size in non-Europeans was small, we show that an aggregate GRS significantly predicted risk in ACPA+ cases of African and Hispanic ancestry (pAF = 0.003, pHIS = 0.026), and there was a similar trend in those with East Asian ancestry (pAS = 0.075; Table 3 and Figure 3). We note that the mean GRS and AUC values were lower among individuals of non-European ancestry, especially those of African ancestry, than among those of European ancestry. Although it is possible that the significant GRSs were driven solely by the presence of European admixture, this seems unlikely because the mean GRS among the case-control samples that clustered most strongly with the YRI samples from HapMap was higher in cases than in controls. The lower GRS and AUCs could instead reflect differences in patterns of linkage disequilibrium at these loci between the common tag SNP and the underlying causal allele (which is unknown) across the different ethnic populations. The lower GRS and AUCs might also indicate that some of the underlying causal alleles at these loci are absent among cases of non-European ancestry, as is the case for the PTPN22 risk allele among cases of African and East Asian ancestry.32
An important consequence of shared genetic risk across diverse ancestries is that it supports the hypothesis that the underlying causal alleles are common, rather than rare and specific to only one ethnic group. If the underlying causal alleles were rare, we would not expect tag SNPs in Europeans to predict risk in other genetic ancestries. Our results support the utility of fine-mapping of RA risk loci across diverse ancestries to localize the causal allele.
Outside of the HLA region,30,31 it is largely unknown whether the genetic basis of ACPA− RA is distinct from or overlaps with the genetic basis of ACPA+ RA. A major reason for this uncertainty is that no study has yet systematically investigated the contribution of all known RA risk alleles in a collection of both ACPA− and ACPA+ cases. In general, ACPA+ cases have more severe disease than ACPA− cases; ACPA− cases are thought to represent a more heterogeneous group of cases.33,34 Despite this clinical heterogeneity, a small study of twins indicated that the heritability in ACPA+ disease was similar to that of ACPA− disease.11 Our EHR-based study, which ascertained 378 ACPA− cases of European ancestry, provides evidence that there is overlap between the two subsets, as a distribution of the GRS derived from SNPs associated with autoantibody-positive disease is significantly higher in ACPA− cases than in controls (Figure 3). However, our study also demonstrates that the overall GRS distribution for these susceptibility SNPs is lower in ACPA− disease than in ACPA+ disease (Figure 5). Whether this difference is due to the effect size of individual risk alleles, the subset of alleles that contribute to risk in both diseases, or both needs to be explored further.
In our EHR study, we used discarded blood to target specific patient populations (RA cases and matched controls) as one approach to acquiring large sample sizes at an affordable cost. Using discarded blood samples meant that no direct patient consent was obtained. We did, however, undergo a thorough IRB-review of our research protocol, and we ensured that all clinical data linked to a discarded biospecimen was completely anonymous to protect patient confidentiality (see Subjects and Methods). In approximately one year, we were able to acquire DNA and plasma for over 3,000 case-control samples. There are also other options for linking EHR data to biospecimens,35 including large biobanks at academic health centers36 and in the United Kingdom.37 Regardless of the method of procuring biospecimens, the approach outlined here demonstrates that EHRs are a reliable resource for discovery research. Such an approach should be particularly attractive within large health care centers such as the Veteran's Administration (VA) hospital system or within multiple health care systems that can integrate data derived from EHR for a common research question.
Currently, approximately 20% of physicians in the United States use EHRs.38 It is inevitable that this number will increase substantially in the very near future. As the informatics tools to mine EHRs improve, as biobanks grow, and as genomic information is systematically gathered, secondary use of EHR data will serve as a mainstay for genomic research. Our study demonstrates that EHR clinical data linked with biospecimens represent a valuable resource for genetics research in rheumatoid arthritis.
Acknowledgments
The project was supported by award number U54-LM008748 from the National Library of Medicine. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Library of Medicine or the National Institutes of Health. R.M.P. was supported by grants from the National Institutes of Health (R01-AR057108, R01-AR056768, and U01-GM092691) and holds a Career Award for Medical Scientists from the Burroughs Wellcome Fund.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
NCHS Health E-Stat, http://www.cdc.gov/nchs/data/hestat/emr_ehr/emr_ehr.htm
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
R project, http://www.r-project.org/
References
- 1.Hindorff L.A., Sethupathy P., Junkins H.A., Ramos E.M., Mehta J.P., Collins F.S., Manolio T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kruglyak L. The road to genome-wide association studies. Nat. Rev. Genet. 2008;9:314–318. doi: 10.1038/nrg2316. [DOI] [PubMed] [Google Scholar]
- 3.Murphy S., Churchill S., Bry L., Chueh H., Weiss S., Lazarus R., Zeng Q., Dubey A., Gainer V., Mendis M. Instrumenting the health care enterprise for discovery research in the genomic era. Genome Res. 2009;19:1675–1681. doi: 10.1101/gr.094615.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DesRoches C.M., Campbell E.G., Vogeli C., Zheng J., Rao S.R., Shields A.E., Donelan K., Rosenbaum S., Bristol S.J., Jha A.K. Electronic health records' limited successes suggest more targeted uses. Health Aff. (Millwood) 2010;29:639–646. doi: 10.1377/hlthaff.2009.1086. [DOI] [PubMed] [Google Scholar]
- 5.Blumenthal D., Tavenner M. The “meaningful use” regulation for electronic health records. N. Engl. J. Med. 2010;363:501–504. doi: 10.1056/NEJMp1006114. [DOI] [PubMed] [Google Scholar]
- 6.Denny J.C., Ritchie M.D., Basford M.A., Pulley J.M., Bastarache L., Brown-Gentry K., Wang D., Masys D.R., Roden D.M., Crawford D.C. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics. 2010;26:1205–1210. doi: 10.1093/bioinformatics/btq126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ritchie M.D., Denny J.C., Crawford D.C., Ramirez A.H., Weiner J.B., Pulley J.M., Basford M.A., Brown-Gentry K., Balser J.R., Masys D.R. Robust replication of genotype-phenotype associations across multiple diseases in an electronic medical record. Am. J. Hum. Genet. 2010;86:560–572. doi: 10.1016/j.ajhg.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liao K.P., Cai T., Gainer V., Goryachev S., Zeng-Treitler Q., Raychaudhuri S., Szolovits P., Churchill S., Murphy S., Kohane I. Arthritis Care Res; Hoboken: 2010. Electronic medical records for discovery research in rheumatoid arthritis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silman A.J., Pearson J.E. Epidemiology and genetics of rheumatoid arthritis. Arthritis Res. 2002;4(Suppl 3):S265–S272. doi: 10.1186/ar578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.MacGregor A.J., Snieder H., Rigby A.S., Koskenvuo M., Kaprio J., Aho K., Silman A.J. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. 2000;43:30–37. doi: 10.1002/1529-0131(200001)43:1<30::AID-ANR5>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 11.van der Woude D., Houwing-Duistermaat J.J., Toes R.E., Huizinga T.W., Thomson W., Worthington J., van der Helm-van Mil A.H., de Vries R.R. Quantitative heritability of anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis. Arthritis Rheum. 2009;60:916–923. doi: 10.1002/art.24385. [DOI] [PubMed] [Google Scholar]
- 12.Begovich A.B., Carlton V.E., Honigberg L.A., Schrodi S.J., Chokkalingam A.P., Alexander H.C., Ardlie K.G., Huang Q., Smith A.M., Spoerke J.M. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am. J. Hum. Genet. 2004;75:330–337. doi: 10.1086/422827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suzuki A., Yamada R., Chang X., Tokuhiro S., Sawada T., Suzuki M., Nagasaki M., Nakayama-Hamada M., Kawaida R., Ono M. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat. Genet. 2003;34:395–402. doi: 10.1038/ng1206. [DOI] [PubMed] [Google Scholar]
- 14.Plenge R.M., Padyukov L., Remmers E.F., Purcell S., Lee A.T., Karlson E.W., Wolfe F., Kastner D.L., Alfredsson L., Altshuler D. Replication of putative candidate-gene associations with rheumatoid arthritis in >4,000 samples from North America and Sweden: Association of susceptibility with PTPN22, CTLA4, and PADI4. Am. J. Hum. Genet. 2005;77:1044–1060. doi: 10.1086/498651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomson W., Barton A., Ke X., Eyre S., Hinks A., Bowes J., Donn R., Symmons D., Hider S., Bruce I.N. Rheumatoid arthritis association at 6q23. Nat. Genet. 2007;39:1431–1433. doi: 10.1038/ng.2007.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plenge R.M., Seielstad M., Padyukov L., Lee A.T., Remmers E.F., Ding B., Liew A., Khalili H., Chandrasekaran A., Davies L.R. TRAF1-C5 as a risk locus for rheumatoid arthritis—A genomewide study. N. Engl. J. Med. 2007;357:1199–1209. doi: 10.1056/NEJMoa073491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurreeman F.A., Padyukov L., Marques R.B., Schrodi S.J., Seddighzadeh M., Stoeken-Rijsbergen G., van der Helm-van Mil A.H., Allaart C.F., Verduyn W., Houwing-Duistermaat J. A candidate gene approach identifies the TRAF1/C5 region as a risk factor for rheumatoid arthritis. PLoS Med. 2007;4:e278. doi: 10.1371/journal.pmed.0040278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhernakova A., Alizadeh B.Z., Bevova M., van Leeuwen M.A., Coenen M.J., Franke B., Franke L., Posthumus M.D., van Heel D.A., van der Steege G. Novel association in chromosome 4q27 region with rheumatoid arthritis and confirmation of type 1 diabetes point to a general risk locus for autoimmune diseases. Am. J. Hum. Genet. 2007;81:1284–1288. doi: 10.1086/522037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gregersen P.K., Amos C.I., Lee A.T., Lu Y., Remmers E.F., Kastner D.L., Seldin M.F., Criswell L.A., Plenge R.M., Holers V.M. REL, encoding a member of the NF-kappaB family of transcription factors, is a newly defined risk locus for rheumatoid arthritis. Nat. Genet. 2009;41:820–823. doi: 10.1038/ng.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raychaudhuri S., Remmers E.F., Lee A.T., Hackett R., Guiducci C., Burtt N.P., Gianniny L., Korman B.D., Padyukov L., Kurreeman F.A. Common variants at CD40 and other loci confer risk of rheumatoid arthritis. Nat. Genet. 2008;40:1216–1223. doi: 10.1038/ng.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karlson E.W., Chibnik L.B., Kraft P., Cui J., Keenan B.T., Ding B., Raychaudhuri S., Klareskog L., Alfredsson L., Plenge R.M. Cumulative association of 22 genetic variants with seropositive rheumatoid arthritis risk. Ann. Rheum. Dis. 2010;69:1077–1085. doi: 10.1136/ard.2009.120170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stahl E.A., Raychaudhuri S., Remmers E.F., Xie G., Eyre S., Thomson B.P., Li Y., Kurreeman F.A., Zhernakova A., Hinks A. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet. 2010;42:508–514. doi: 10.1038/ng.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kochi Y., Okada Y., Suzuki A., Ikari K., Terao C., Takahashi A., Yamazaki K., Hosono N., Myouzen K., Tsunoda T. A regulatory variant in CCR6 is associated with rheumatoid arthritis susceptibility. Nat. Genet. 2010;42:515–519. doi: 10.1038/ng.583. [DOI] [PubMed] [Google Scholar]
- 24.Kosoy R., Nassir R., Tian C., White P.A., Butler L.M., Silva G., Kittles R., Alarcon-Riquelme M.E., Gregersen P.K., Belmont J.W. Ancestry informative marker sets for determining continental origin and admixture proportions in common populations in America. Hum. Mutat. 2009;30:69–78. doi: 10.1002/humu.20822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Price A.L., Butler J., Patterson N., Capelli C., Pascali V.L., Scarnicci F., Ruiz-Linares A., Groop L., Saetta A.A., Korkolopoulou P. Discerning the ancestry of European Americans in genetic association studies. PLoS Genet. 2008;4:e236. doi: 10.1371/journal.pgen.0030236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 27.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.R Development Core Team . R Foundation for Statistical Computing; Vienna, Austria: 2008. R: A Language and Environment for Statistical Computing. [Google Scholar]
- 29.Freedman M.L., Reich D., Penney K.L., McDonald G.J., Mignault A.A., Patterson N., Gabriel S.B., Topol E.J., Smoller J.W., Pato C.N. Assessing the impact of population stratification on genetic association studies. Nat. Genet. 2004;36:388–393. doi: 10.1038/ng1333. [DOI] [PubMed] [Google Scholar]
- 30.Irigoyen P., Lee A.T., Wener M.H., Li W., Kern M., Batliwalla F., Lum R.F., Massarotti E., Weisman M., Bombardier C. Regulation of anti-cyclic citrullinated peptide antibodies in rheumatoid arthritis: Contrasting effects of HLA-DR3 and the shared epitope alleles. Arthritis Rheum. 2005;52:3813–3818. doi: 10.1002/art.21419. [DOI] [PubMed] [Google Scholar]
- 31.Huizinga T.W., Amos C.I., van der Helm-van Mil A.H., Chen W., van Gaalen F.A., Jawaheer D., Schreuder G.M., Wener M., Breedveld F.C., Ahmad N. Refining the complex rheumatoid arthritis phenotype based on specificity of the HLA-DRB1 shared epitope for antibodies to citrullinated proteins. Arthritis Rheum. 2005;52:3433–3438. doi: 10.1002/art.21385. [DOI] [PubMed] [Google Scholar]
- 32.Mori M., Yamada R., Kobayashi K., Kawaida R., Yamamoto K. Ethnic differences in allele frequency of autoimmune-disease-associated SNPs. J. Hum. Genet. 2005;50:264–266. doi: 10.1007/s10038-005-0246-8. [DOI] [PubMed] [Google Scholar]
- 33.De Rycke L., Peene I., Hoffman I.E., Kruithof E., Union A., Meheus L., Lebeer K., Wyns B., Vincent C., Mielants H. Rheumatoid factor and anticitrullinated protein antibodies in rheumatoid arthritis: diagnostic value, associations with radiological progression rate, and extra-articular manifestations. Ann. Rheum. Dis. 2004;63:1587–1593. doi: 10.1136/ard.2003.017574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van der Helm-van Mil A.H., Verpoort K.N., Breedveld F.C., Toes R.E., Huizinga T.W. Antibodies to citrullinated proteins and differences in clinical progression of rheumatoid arthritis. Arthritis Res. Ther. 2005;7:R949–R958. doi: 10.1186/ar1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ginsburg G.S., Burke T.W., Febbo P. Centralized biorepositories for genetic and genomic research. JAMA. 2008;299:1359–1361. doi: 10.1001/jama.299.11.1359. [DOI] [PubMed] [Google Scholar]
- 36.Roden D.M., Pulley J.M., Basford M.A., Bernard G.R., Clayton E.W., Balser J.R., Masys D.R. Development of a large-scale de-identified DNA biobank to enable personalized medicine. Clin. Pharmacol. Ther. 2008;84:362–369. doi: 10.1038/clpt.2008.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Palmer L.J. UK Biobank: Bank on it. Lancet. 2007;369:1980–1982. doi: 10.1016/S0140-6736(07)60924-6. [DOI] [PubMed] [Google Scholar]
- 38.Jha A.K., DesRoches C.M., Kravolec P.D., Joshi M.S. A progress report on electronic health records in U.S. hospitals. Health Affairs. 2010;29:1951–1957. doi: 10.1377/hlthaff.2010.0502. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.