

Abstract
Defects of ciliogenesis have been implicated in a wide range of human phenotypes and play a crucial role in signal transduction and cell-cycle coordination. We used homozygosity mapping in two families with autosomal-recessive short-rib polydactyly syndrome Majewski type to identify mutations in NEK1 as an underlying cause of this lethal osteochondrodysplasia. NEK1 encodes a serine/threonine kinase with proposed function in DNA double-strand repair, neuronal development, and coordination of cell-cycle-associated ciliogenesis. We found that absence of functional full-length NEK1 severely reduces cilia number and alters ciliar morphology in vivo. We further substantiate a proposed digenic diallelic inheritance of ciliopathies by the identification of heterozygous mutations in NEK1 and DYNC2H1 in an additional family. Notably, these findings not only increase the broad spectrum of ciliar disorders, but suggest a correlation between the degree of defective microtubule or centriole elongation and organization and the severity of the resulting phenotype.
Main Text
Many key developmental processes are maintained by regulation of cell-signaling pathways. A variety of the involved regulatory proteins localize to the cilium or the basal body of an individual cell and are important for correct body tissue patterning.1 In principle, cilia are highly conserved, dynamic organelles initiated by the location of a basal body, composed of a pair of centrioles embedded in the pericentriolar material (PCM) and the axoneme to the cytoplasmatic side of the cell membrane.2 During ciliogenesis, new axonemal subunits elongate the cilia to the distal tip.3 The assembly of the axonemal units divides the cilia into two main classes: motile cilia, with two central microtubules (9+2), and nonmotile or primary cilia, without central microtubules (9+0).3 The formation and maintenance of the cilium is basically mediated by the dynamic, bidirectional process of intraflagellular transport (IFT).4 Anterograde movement is driven by the heterotrimeric kinesin 2 motor, whereas retrograde transport is mediated by cytoplasmatic dynein 2.5,6 Although most vertebrate cells form at least one single, nonmotile primary cilium, only recently have the functions of cilia become widely recognized. Whereas motile cilia are involved in fluid flow, especially in epithelial and ependymal tissues,7 nonmotile, primary cilia play an important role in signal transduction of the Hedgehog, Wnt planar cell polarity, and the platelet-derived growth factor (PDGF) pathways.1–3,8,9 Additionally, as the primary cilium is reabsorbed before the cell is entering mitosis and the basal body breaks down into the centrioles, absence of ciliary signaling is observed in uncontrolled cell division and cancer.10
Several disorders have been associated with defects of a variety of proteins involved in cilia formation, maintenance, and function.2,11 These ciliopathies affect the IFT, components of the cilia, the basal body, or the centrosome, and many of the associated phenotypes include brain malformations, polydactyly, kidney cysts, retinal degeneration, and skeletal abnormalities (Table S1 available online).12,13 In particular, this phenotypic spectrum is present among the patients with short-rib polydactyly syndromes (SRPS, Tables S2A and S2B).14 This group constitutes the most frequent lethal autosomal-recessive osteochrondodysplasia and was formerly classified into four distinct types (type I, Saldino-Noonan [MIM 263530]; type II, Majewski [MIM 263520]; type III, Verma-Neumhoff [MIM 263510]; type IV, Beemer [MIM 269860]).15 Types I and III have been recently grouped together. In addition, asphyxiating thoracic dystrophy (ATD [MIM 208500]) and Ellis-van Crefeld syndrome (EVC [MIM 225500]) are phenotypically related to the group of SRPS. From this group of disorders, the underlying cause has been identified for EVC (mutations in EVC1 or EVC2 in two-thirds of patients), and only recently have mutations in IFT80 (MIM 611177) and DYNC2H1 (MIM 603297) been identified in a small subset of patients with ATD and SRPS type III.16–18 Both proteins are involved in anterograde and retrograde IFT.
We investigated probands (IV-4 in Figure 1O, IV-10 in Figure 1P, and III-2 in Figure 1Q) of three independent families: two consanguineous families (families 1 and 2) of Turkish and Bedouin (Arab Moslem nomadic group) origin and one nonconsanguineous family (family 3) of German origin, with one affected child each (Figure 1). This study was approved by the ethical review board of the Friedrich-Alexander University Erlangen-Nuremberg, and clinical classification was performed according to Majewski et al.19 and a personal communication with Frank Majewski.
Figure 1.
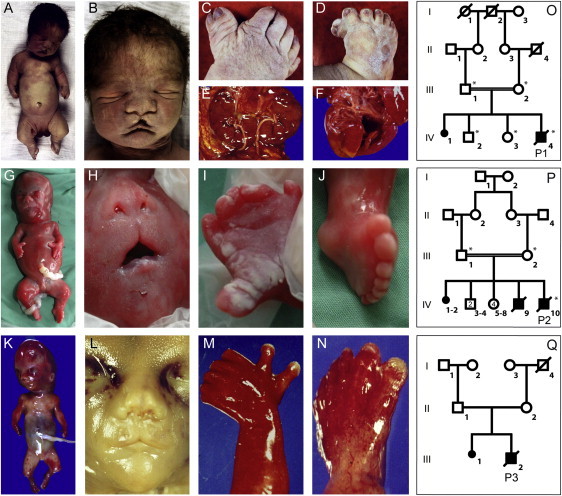
Morphological Features of the Investigated Patients with SRPS Type Majewski
(A–F) The affected individual of the consanguineous family 1 was born at 36 wk of gestation and deceased 1 hr postpartum. Clinical evaluation showed severe extreme shortening of the ribs with a narrow thorax, a bulging forehead, depressed nasal bridge, and median cleft (B); extreme pre- and postaxial polysyndactyly with seven fingers and toes (C, D); horseshoe configuration with fused lower parts of both kidneys (E); and a small ventricular septal defect and open foramen ovale of the heart (F).
(G–N) Similar clinical features were observed in the affected individual of family 2 (G–J) at 21 wk of gestation and the affected individual of family 3 (K–N) at 19 wk of gestation, with hydropic fetus, flat nose and median cleft, high and broad forehead, and mild microretrognathia, as well as pre- and postaxial polysyndactyly in both hands and feet.
(O–Q) Pedigrees of the investigated family 1 (O), family 2 (P), and family 3 (Q). Individuals marked with asterisks were included in the linkage analysis.
All affected individuals had a narrow thorax with hypoplastic lungs, extreme polysyndactyly, dysproportionate dwarfism, and median cleft lip and palate (Figure 1). In addition, the affected individual of family 1 (IV-4 in Figure 1O) presented with a ventriculoseptal defect and cystic kidneys. In accordance with the classification of SRPS type II (Majewski), radiographic hallmarks of all probands included shortened and horizontal ribs, squared scapulae and elevated clavicles with lateral kinking, normal spine and pelvis configuration, and shortening of the bones of all four extremities, with extreme reduction of tibial bone length (Figure 2, Table S2B).
Figure 2.
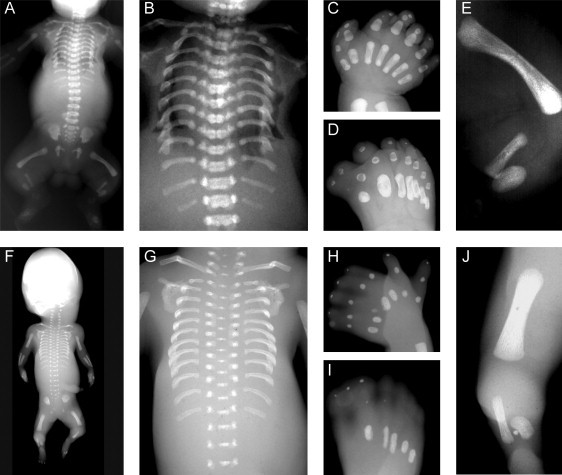
Radiographic Features of the Investigated Patients with SRPS Type Majewski
Both affected individuals, from family 1 (A–E) and family 3 (F–J), presented with severely shortened and horizontal ribs (A, B, F, G), squared scapulae and elevated clavicles with lateral kinking (A, F), normal vertebral column and pelvis and regular metaphyseal margins of the tubular bones, incompletely calcified and irregularly shaped metacarpal and metatarsal bones (C, D, H, I), shortened humerus and femurs, and extreme shortening of the tibial bones.
By histological examination of probands 1 (IV-4 in Figure 1O) and 3 (III-2 in Figure 1Q) we found retardation in endochondral ossification with persistence of hypertrophic chondrocytes within the trabecular bone and almost complete lack of column formation of the epiphyseal zone, as well as heterotopic endochondral ossification in the tibial bone in probands 1 and 3 (Figure 3). In addition, rib bones of the affected individual of family 1 showed a thin and highly disorganized growth zone arranged in a semicircle.
Figure 3.
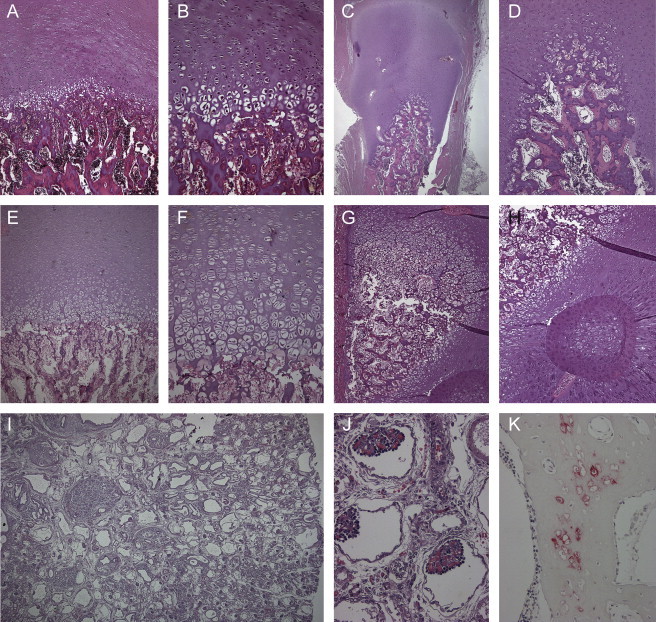
Histological and Immunohistochemical Analysis
(A, B, E, F) Normal resting cartilage and growth plates with disorganization of endochondral ossification in the affected individuals P1 (A, B) and P3 (E, F). Note the reduced number of chondrocytes in the epiphyseal zone of proliferation and the diminished cartilage-column formation of the growth zone.
(C, D) Additional lack of columnization of the enchondral ossification of the epiphyses of the ribs. Here, the growth zone was lined in a semicircle around the osseous center of the rib bone.
(G, H) The growing cartilage of the tibial bone formed a semicircle around the metadiaphyseal zone, with heterotopic ossification.
(I, J) Polycystic kidney disease was found in affected individual P1 with dilatated collecting tubules, glomerules of differing size, and well developed glomerules with dense convolutes of capillary vessels. (K) Immunohistochemical analysis showed small islands of persistent hypertrophic chondrocytes with expression of collagen X within osseous trabecules.
After excluding mutations in IFT80 (NM_001190241.1, NM_020800.2) and DYNC2H1 (NM_001080463.1) in families 1 and 2 by Sanger sequencing, we aimed at identifying the underlying molecular basis of SRPS type II through genome-wide linkage analysis and homozygosity mapping by using Affymetrix 6.0 SNP arrays in these two consanguineous families (Figure 4A). For parametric multipoint linkage analysis and haplotype-structure analysis, we used the programs Allegro,20,21 Merlin,22 and easyLinkage-Plus23 under the assumption of an autosomal-recessive mode of inheritance with 100% penetrance, disease-allele frequency of 0.01%, and at least 0.01 cM intermarker distance. Haplotypes were visualized with the HaploPainter program.24 We identified one linked interval with a maximum LOD score (HLOD) of 2.95 representing 17.36 Mb/18.65 cM on chromosome 4, encompassing 38 genes (Figure 4A; see also Figure S1). To prioritize genes under the hypothesis of a defect in cilia function, we used known genes of the cilia proteome database25 and compared them with the genes in our candidate interval using the ENDEAVOUR software26,27 (Table S3). Additional phenotype information about orthologous genes highlighted the NIMA-related kinase 1 gene (NEK1) as a highly interesting candidate, because mutant mice (kat/kat-2J) homozygous for the orthologuous gene Nek1 show polycystic kidney disease, craniofacial anomalies, and growth reduction.28–30 NEK1 is composed of 34 exons encoding a 1258-amino-acid-long protein localized to the cilium basal body and is suggested to be involved in the coordination between the cilium and the cell cycle (Figures 4B and 4C).29 Through Sanger sequencing analysis of NEK1 (NM_012224.1) we identified the homozygous nonsense mutation c.379C>T (p.Arg127X) resulting in a premature stop codon in the affected individual of family 1 (IV-4 in Figure 1O) and the homozygous splice-site mutation c.869-2A>G in the affected individual of the second consanguineous family (IV-10 in Figure 1P) (Table 1, Figure S2B). In agreement with autosomal-recessive segregation, parents and unaffected siblings were heterozygous for the identified mutations. Both mutations were excluded in 378 and 370 control chromosomes of the corresponding ethnic origin. In addition to the splice-site mutation c.869-2A>G, we identified in the affected individual of family 2 (IV-10 in Figure 1P) a second homozygous variant, c.2680+5G>A, in which splice-site prediction was not conclusive, suggesting that c.869-2A>G is the main underlying cause of SRPS in this family. Because of the deleterious phenotype, RNA was available only for the affected individual of family 1. Quantitative real-time PCR analysis of NEK1 (Taqman probe Hs01591486_m1, Applied Biosystems) demonstrated a 64% reduction of cDNA levels in lymphoblastoid cell lines in the affected individual and 30%–40% reduction in the heterozygous parents, indicating only a partial nonsense-mediated mRNA decay of the c.379C>T nonsense mutant allele (Figure S3).
Figure 4.
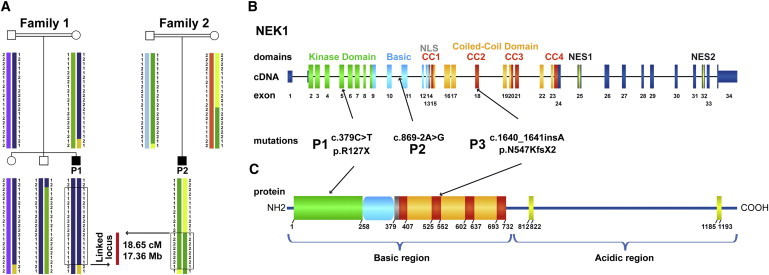
Identification of NEK1 Mutations as Underlying Cause of SRPS Majewski Type by Positional Cloning
(A) Haplotype structure analysis in the two consanguineous families 1 and 2 refined a locus to 18.65 cM / 17.36 Mb by homozygosity mapping including 54 RefSeq and hypothetical genes on chromosome 4q32.1-q34.3.
(B) Sanger sequencing of the prioritized functional candidate gene NEK1 revealed a homozygous nonsense mutation in P1 (p.R127X), a homozygous splice site mutation in P2 (c.869-2A>G), and a heterozygous insertion in P3 (c.1640_1641_insA).
(B, C) Schematic drawing of domain-coding exons according to the prediction of the domain structure in mice and protein mice-human homology. Abbreviations are as follows: kinase domain, N-terminal kinase domain (green); basic, basic domain (light blue); NLS, nuclear localization signal (gray); CC1-4, coiled-coil domains (red and orange); NES1-2, nuclear export sequence (yellow).
Table 1.
Mutations Identified in Families with SRPS Type II
| Family | Gene | Nucleotide Change | Status | Protein | Location | Mutation Type | Domain |
|---|---|---|---|---|---|---|---|
| Family 1 | NEK1 | c.379C>T | homozygous | p.Arg127X | exon 5 | nonsense | kinase |
| Family 2 | NEK1 | c.869-2A>G | homozygous | intron 10 | splice site | basic | |
| Family 3 | NEK1 | c.1640 dup | heterozygous | p.Asn547LysfsX2 | exon18 | insertion, frameshift | coiled-coil 2 |
| DYNC2H1 | c.11747G>A | heterozygous | p.Gly3916Asp | exon 82 | missense, splice site | C domain |
We were not able to prove the effect of the c.869-2A>G splice-site mutation in family 2, but several splice-site-modeling programs predict aberrant splicing of this mutant allele (Table S4). The position is highly conserved throughout different species and is part of the 3′ splice-site motif. In addition, there is no evidence for alternative splicing of exon 11, and skipping of this exon by the mutant allele is predicted to cause a premature stop codon.
Of note, the affected individual of the nonconsanguineous family 3 (III-2 in Figure 1Q) was heterozygous only for an insertion of one nucleotide: c.1640dup (p.Asn547LysfsX2), resulting in a premature stop codon. No second mutation within the genomic and the promoter region of NEK1 could be identified. Copy-number variation of the genomic NEK1 region was excluded by molecular karyotyping with the use of the Affymetrix Cytogenetics Whole-Genome 2.7M Array and the Affymetrix Chromosome Analysis Suite software. Even though a second, undetected mutation or smaller deletion of NEK1 could not ultimately be excluded, sequencing of genes known to be mutated in SRPS disclosed the additional heterozygous missense variation of DYNC2H1: c.11747G>A (p.Gly3916Asp), affecting the exon 82 splice-acceptor site (Table 1, Figure S2B). Each parent was a carrier of one of these mutations, which were absent in 382 population-matched control chromosomes. Again, no second mutation or deletion was identified in DYNC2H1. Given that digenic diallelic inheritance has already been proposed for retinitis pigmentosa and Bardet-Biedl syndrome and that digenic triallelic inheritance has been observed in other ciliopathies, these two aberrations might be disease causing in this affected individual through combined haploinsufficiency of cilia formation and IFT.31–33 Apart from the lack of morphological cardiac and kidney abnormalities, this affected individual was otherwise clinically, histologically, and radiographically indistinguishable from the other two individuals with homozygous NEK1 mutations. Homozygous Dync2h1 genetrap or ENU-induced alleles in mice appeared to be null alleles leading to an abnormal brain morphology and frequent heart-looping reversal and are not viable.34,35 Given that heterozygous Dync2h1 and Nek1 mice each seem not to show a specific phenotype, a knockout intercross mouse model in further studies might lead to a recognizable phenotype and therefore confirm digenic diallelic inheritance.
NEKs (NIMA-related kinases) represent an ancient family of serine/threonine kinases with proposed function in cell-cycle control and cilia regulation.36 NEK1 in particular has been proposed to be involved in DNA double-strand repair, neuronal development, and coordination of cell-cycle-associated ciliogenesis in vitro.29,37,38 The development of kidney cysts in the Nek1 knockout mice suggested aberrant primary cilia. Furthermore, Nek1 is associated with the centrosomes and translocates from the nucleus during mitosis to the basal body, initiating cilia formation. In the nucleus, Nek1 may additionally influence nuclear gene expression.39
In order to analyze protein levels and location, we evaluated three different commercially available NEK1 antibodies (sc-7437, Santa Cruz; AP15020PU-N, Acris; ab98237, abcam) for immunofluorescence and immunoblotting. Immunoblot analysis failed to detect any full-length NEK1 in control cell lysates, which has been reported before in human NEK1 and might be explained by rapid proteolysis of this large and unstable protein in cell lysates.40 The AP15020PU-N antibody, which targets amino acids 1173–1202 located downstream of the predicted mutated stop codon, detected a shorter protein (approximately 60 kDa; wild-type protein would be 142 kDa) in control fibroblasts. Genescan gene prediction identified an open reading frame that might lead to a 571-amino-acid-long protein (63 kDa) that harbors the antibody binding site. The AP15020PU-N antibody detected the same shorter protein in the patient cells. None of the antibodies were able to detect either the wild-type or the predicted truncated NEK1 of an expected size of only 14.9 kDa, which lacks most of the functional domains in the patient (Figures 4B and 4C).
On the basis of the suggested NEK1 localization to the basal body and the phenotype of the mouse mutants,28–30 we expected loss of functional full-length NEK1 to lead to morphological abnormal or missing cilia in humans. The AP15020PU-N antibody was used for immunofluorescence in cultured fibroblasts of patient P1, and we indeed demonstrated not only a significant decrease to 27% ciliated cells but, more importantly, structural abnormal remaining cilia with severely reduced length, a broad base, and a thin apex in the three-dimensional reconstruction (Figures 5A, 5B, 5F, 5H, and 5I). This is in marked contrast to DYNC2H1-deficient fibroblasts, in which formation of the primary cilium through elongation and maintenance of the axoneme by IFT is altered.29 Cells were analyzed with the use of a Zeiss Axio Imager Z1 with an ApoTome, an Axiocam MRm camera, and the Axiovision 4.6.3 program (Zeiss). Further three-dimensional reconstruction of cilia was performed with the Imaris software (Bitplane). Detection of a signal in fibroblasts of the control and patient P1 by the antibody raised against the C-terminal end of NEK1 (amino acids 1173–1202) suggests the presence of a shorter isoform lacking the N-terminal region of NEK1. This hypothesis is supported by the observation that the N-terminal kinase domain (amino acids 1–258) is necessary for cilia formation, whereas the cilium localization signal is represented by the coiled-coil domain (amino acids 379–732).29,37 The affected cells of patient P1 do not show regular-shaped cilia of any kind but a considerable increase in acetylated α-tubulin, confirming a central defect of cilia formation and cytoskeleton microtubule architecture (Figure 5). Furthermore, Aspergillus nidulans lacking the functional NEK1 ortholog nimA forms bipolar spindles that fail to separate nucleoplasm. Analysis of the cell cycle of fibroblasts of our affected individual P1 did not reveal any impairment in the mitotic spindle formation (Figure S4).
Figure 5.
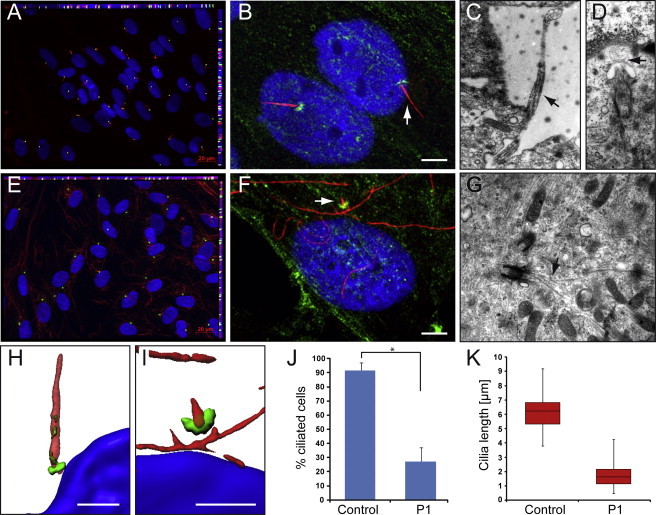
SRPS Majewski Type Fibroblasts Have a Severely Reduced Number and Structurally Abnormal Cilia
The cells were grown on coverslips or on cell-culture dishes with gas-permeable base to confluence and then serum starved for 5 days, fixed, and treated for immunofluorescence analyses or TEM.
(A, E) Immunofluorescence for acetylated α-tubulin (red), pericentrin (green), and DAPI (blue) shows normal presentation of cilia in control fibroblast cells (A), but missing or severely shortened cilia with normal appearance of the basal body and increased cytoplasmatic acetylated α-tubulin in P1 fibroblast cells (E).
(J) Only 27% of P1 cells have verifiable cilia, in contrast to 92% of control cells (t test, p value 4 × 10−10).
(B, F) Immunofluorescence for α-tubulin (red), NEK1 (green), and DAPI (blue) suggests the presence of the 63 kDa isoform NEK1 seen by immunoblotting in the PCM in control and P1 cells (white bar represents 5μm). The latter indicates presence of a physiological NEK1 isoform not affected by the nonsense mutation. Such an isoform lacking the N-terminal region would not be able to maintain cilial formation because of lack of the kinase domain.
(K) Cilia length was measured at 6.2 ± 1.2 μm on average (median 6.2 μm, 25th centile 5.3 μm, 75th centile 6.8 μm) in control cells but only 1.7 ± 0.7 μm (median 1.6 μm, 25th centile 1.2 μm, 75th centile 2.2 μm) in P1 fibroblast cells (t test, p value 1.7 × 10−69).
(C, D, G) Electron micrographs illustrating representative images from normal control fibroblast cells (C, D) and P1 fibroblast cells (F). (C) shows a primary cilia in full length, whereas in (D) the TEM image illustrates a primary cilia at the beginning of ciliogenesis (stage 2).41 In contrast, in P1 fibroblast cells (G), we found only centrioles at stage 1 of ciliogenesis with an abnormal microtubule growth (arrow) but no enveloping ciliary pocket around.
(H) Imaris three-dimensional construction of normal cilia reveals the location of NEK1 at the basal body and within the cilium, suggesting participation in the intraflagellular transport.
(I) In contrast, P1 cell cilia are shortened, with a broad base and a thin tip.
Primary cilia display distinct stages of ciliogenesis and can be divided into five stages in electron micrographs.41 For transmission electron microscopy (TEM), we grew cells on cell-culture dishes and prefixed them for 20 min with 2.5% glutaraldehyde followed by an incubation with 2.5% glutaraldehyde. Cells were washed and postfixed with 1% osmium tetroxide for 1 hr at 4°C and with 1% uranyl acetate for 1 hr at 4°C. We then dehydrated the cells in ethanol, followed by flat embedding and cutting with an ultramicrotome (Reichert Ultracut). Sections were examined and photographed with a Zeiss EM10 electron microscope (Zeiss) and a Gatan SC1000 OriusTM CCD camera in combination with the Digital Micrograph TM software (Gatan). TEM of 5-day-starved growing cells of P1 revealed abnormally cilia-like structures (Figure 5G), which was never observed in wild-type cells (Figures 5C and 5D). The NEK1 mutant centrioles had the microtubule composition and morphology of normal-size centrioles at stage 1 of ciliogenesis. Stage 1 was characterized by the presence of two vesicles at the distal end of the mother centriole (basal body). The P1 fibroblast cells never appear to initiate ciliogenesis beyond stage 1, whereas the wild-type fibroblast cell lines progress in ciliogenesis to varying stages (Figures 5C and 5D). The electron micrographs of P1 fibroblasts are examples of misshaped cilia. We illustrated a cell arrest at stage 1 and an abnormal microtubule growth, confirming a defect in centriole elongation or organization in NEK1 mutated cells. Together, these data indicate that the main defect in ciliogenesis of NEK1 mutated cells might be located in the progression from stage 1 after vesicular accumulation to stage 2 and subsequent axoneme growth.
Because primary cilia act as chemosensors for different pathways, among other functions, loss of NEK1 and therefore disturbed cilia formation can negatively affect signal transduction in the various pathways, leading to the pleiotropic clinical defects as observed in SRPSs.2 The PDGF pathway is associated with the primary cilium in fibroblasts and is involved in proliferation, survival, and migration. The disruption of these diverse developmental processes goes in line with a median cleft palate, cardiac and kidney abnormalities, polydactyly, and the other observed limb malformations in SRPS type II. Altered sonic hedgehog signaling may also explain neuronal-tube and limb-development defects. Furthermore, the basal body plays a role in the planar cell polarity (PCP) pathway by Wnt/β-catenin signal interpretation. The prenatal phenotype in NEK1-deficient humans and mice suggested a specific expression pattern. Quantitative real-time PCR analysis of different wild-type fetal and adult tissues (“Human Fetal MTCTM Panel,” “Human MTCTM Panel I,” Clonetech) confirmed a higher fetal expression in the brain and kidney, supporting the role of cilia in embryonic formation of these tissues, as becomes evident in the histological analysis in our patients and the homozygous mutant mice (kat/kat-2J)28 (Figure S3B). Low expression levels in wild-type lung tissue suggest that the observed hypoplastic lungs in SRPS type II are a secondary effect of the narrow thorax. All genes known to be mutated in SRPSs to date involve the cilia cycle. The clinical distinction in different but phenotypically overlapping SRPSs suggests different degrees of cilia disruption during embryogenesis resulting in different impairment of the involved signal-transduction pathways.
We report of a protein associated with the PCM leading to an SRPS phenotype. Further colocalization of NEK1 within the primary cilium supports an additional direct interaction with proteins of the IFT. Here, a combined effect of NEK1 and DYNC2H1 might support a diallelic digenic inheritance, as presented in one affected proband. Given that the known defects in IFT80 and DYNC2H1, which are associated with other forms of SRPS phenotypes, are involved in intraflagellar transport, our data suggest that the fundamental disruption of cilia is a premise of the pleiotropic phenotypes of all SRPSs. The phenotypic variability of the different SRPSs might be characterized by the extent of the impairment of cilia formation, elongation, and maintenance and, finally, the combined influence on different proteins of the ciliome network.
Acknowledgments
We thank Frank Majewski for helpful discussion of the phenotype, the families for giving their consent for this study, and J. Adam for technical assistance. This work was supported by the Bundesministerium für Bildung und Forschung (BMBF) network grant SKELNET GFGM01141901 to A. Rauch and A. Reis and the DFG grant TH896/4-1 to C.T.T. The study was approved by the ethical review board of the Medical Faculty of the Friedrich-Alexander University Erlangen-Nuremberg.
Supplemental Data
Web Resources
The URLs for the data presented herein are as follows:
Allegro, http://www.decode.com/software/
Berkeley Drosophila Genome Project (BDGP), http://www.fruitfly.org/seq_tools/splice.html
Ciliome database, http://www.ciliome.com
easyLinkage-Plus http://compbio.charite.de/genetik/hoffmann/easyLINKAGE/
Endeavour software, http://homes.esat.kuleuven.be/∼bioiuser/endeavour/
HaploPainter, http://haplopainter.sourceforge.net/
HSF V2.4, http://www.umd.be/HSF/
Merlin, http://www.sph.umich.edu/csg/abecasis/merlin/index.html
NetGene2, http://www.cbs.dtu.dk/services/NetGene2/
PANTHER 7.0 Beta http://www.pantherdb.org/tools/csnpScoreForm.jsp
PolyPhen, http://genetics.bwh.harvard.edu/pph/
SpliceView, http://zeus2.itb.cnr.it/∼webgene/wwwspliceview_ex.html
SIFT, http://sift.jcvi.org/
References
- 1.Rohatgi R., Milenkovic L., Scott M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317:372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- 2.D'Angelo A., Franco B. The dynamic cilium in human diseases. Pathogenetics. 2009;2:3. doi: 10.1186/1755-8417-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Satir P., Christensen S.T. Overview of structure and function of mammalian cilia. Annu. Rev. Physiol. 2007;69:377–400. doi: 10.1146/annurev.physiol.69.040705.141236. [DOI] [PubMed] [Google Scholar]
- 4.Rosenbaum J.L., Witman G.B. Intraflagellar transport. Nat. Rev. Mol. Cell Biol. 2002;3:813–825. doi: 10.1038/nrm952. [DOI] [PubMed] [Google Scholar]
- 5.Cole D.G., Snell W.J. SnapShot: Intraflagellar transport. Cell. 2009;137:784. doi: 10.1016/j.cell.2009.04.053. [DOI] [PubMed] [Google Scholar]
- 6.Pedersen L.B., Rosenbaum J.L. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr. Top. Dev. Biol. 2008;85:23–61. doi: 10.1016/S0070-2153(08)00802-8. [DOI] [PubMed] [Google Scholar]
- 7.Eley L., Yates L.M., Goodship J.A. Cilia and disease. Curr. Opin. Genet. Dev. 2005;15:308–314. doi: 10.1016/j.gde.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 8.Simons M., Gloy J., Ganner A., Bullerkotte A., Bashkurov M., Kronig C., Schermer B., Benzing T., Cabello O.A., Jenny A. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat. Genet. 2005;37:537–543. doi: 10.1038/ng1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schneider L., Clement C.A., Teilmann S.C., Pazour G.J., Hoffmann E.K., Satir P., Christensen S.T. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr. Biol. 2005;15:1861–1866. doi: 10.1016/j.cub.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 10.Christensen S.T., Pedersen S.F., Satir P., Veland I.R., Schneider L. The primary cilium coordinates signaling pathways in cell cycle control and migration during development and tissue repair. Curr. Top. Dev. Biol. 2008;85:261–301. doi: 10.1016/S0070-2153(08)00810-7. [DOI] [PubMed] [Google Scholar]
- 11.Eggenschwiler J.T., Anderson K.V. Cilia and developmental signaling. Annu. Rev. Cell Dev. Biol. 2007;23:345–373. doi: 10.1146/annurev.cellbio.23.090506.123249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bisgrove B.W., Yost H.J. The roles of cilia in developmental disorders and disease. Development. 2006;133:4131–4143. doi: 10.1242/dev.02595. [DOI] [PubMed] [Google Scholar]
- 13.Fliegauf M., Benzing T., Omran H. When cilia go bad: cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 2007;8:880–893. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- 14.Elcioglu N.H., Hall C.M. Diagnostic dilemmas in the short rib-polydactyly syndrome group. Am. J. Med. Genet. 2002;111:392–400. doi: 10.1002/ajmg.10562. [DOI] [PubMed] [Google Scholar]
- 15.Superti-Furga A., Unger S. Nosology and classification of genetic skeletal disorders: 2006 revision. Am. J. Med. Genet. A. 2007;143:1–18. doi: 10.1002/ajmg.a.31483. [DOI] [PubMed] [Google Scholar]
- 16.Dagoneau N., Goulet M., Genevieve D., Sznajer Y., Martinovic J., Smithson S., Huber C., Baujat G., Flori E., Tecco L. DYNC2H1 mutations cause asphyxiating thoracic dystrophy and short rib-polydactyly syndrome, type III. Am. J. Hum. Genet. 2009;84:706–711. doi: 10.1016/j.ajhg.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tompson S.W., Ruiz-Perez V.L., Blair H.J., Barton S., Navarro V., Robson J.L., Wright M.J., Goodship J.A. Sequencing EVC and EVC2 identifies mutations in two-thirds of Ellis-van Creveld syndrome patients. Hum. Genet. 2007;120:663–670. doi: 10.1007/s00439-006-0237-7. [DOI] [PubMed] [Google Scholar]
- 18.Merrill A.E., Merriman B., Farrington-Rock C., Camacho N., Sebald E.T., Funari V.A., Schibler M.J., Firestein M.H., Cohn Z.A., Priore M.A. Ciliary abnormalities due to defects in the retrograde transport protein DYNC2H1 in short-rib polydactyly syndrome. Am. J. Hum. Genet. 2009;84:542–549. doi: 10.1016/j.ajhg.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Majewski F., Pfeiffer R.A., Lenz W., Muller R., Feil G., Seiler R. Z. Kinderheilkd. 1971;111:118–138. [PubMed] [Google Scholar]
- 20.Gudbjartsson D.F., Jonasson K., Frigge M.L., Kong A. Allegro, a new computer program for multipoint linkage analysis. Nat. Genet. 2000;25:12–13. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- 21.Gudbjartsson D.F., Thorvaldsson T., Kong A., Gunnarsson G., Ingolfsdottir A. Allegro version 2. Nat. Genet. 2005;37:1015–1016. doi: 10.1038/ng1005-1015. [DOI] [PubMed] [Google Scholar]
- 22.Rampersaud E., Scott W.K., Hauser E.R., Speer M.C. Potential for expanded power in linkage studies using the ALLEGRO and MERLIN software programs. J. Med. Genet. 2005;42:e68. doi: 10.1136/jmg.2005.032029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoffmann K., Lindner T.H. easyLINKAGE-Plus–automated linkage analyses using large-scale SNP data. Bioinformatics. 2005;21:3565–3567. doi: 10.1093/bioinformatics/bti571. [DOI] [PubMed] [Google Scholar]
- 24.Thiele H., Nurnberg P. HaploPainter: a tool for drawing pedigrees with complex haplotypes. Bioinformatics. 2005;21:1730–1732. doi: 10.1093/bioinformatics/bth488. [DOI] [PubMed] [Google Scholar]
- 25.Inglis P.N., Boroevich K.A., Leroux M.R. Piecing together a ciliome. Trends Genet. 2006;22:491–500. doi: 10.1016/j.tig.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 26.Aerts S., Lambrechts D., Maity S., Van Loo P., Coessens B., De Smet F., Tranchevent L.C., De Moor B., Marynen P., Hassan B. Gene prioritization through genomic data fusion. Nat. Biotechnol. 2006;24:537–544. doi: 10.1038/nbt1203. [DOI] [PubMed] [Google Scholar]
- 27.Tranchevent L.C., Barriot R., Yu S., Van Vooren S., Van Loo P., Coessens B., De Moor B., Aerts S., Moreau Y. ENDEAVOUR update: a web resource for gene prioritization in multiple species. Nucleic Acids Res. 2008;36:W377–W384. doi: 10.1093/nar/gkn325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Janaswami P.M., Birkenmeier E.H., Cook S.A., Rowe L.B., Bronson R.T., Davisson M.T. Identification and genetic mapping of a new polycystic kidney disease on mouse chromosome 8. Genomics. 1997;40:101–107. doi: 10.1006/geno.1996.4567. [DOI] [PubMed] [Google Scholar]
- 29.Shalom O., Shalva N., Altschuler Y., Motro B. The mammalian Nek1 kinase is involved in primary cilium formation. FEBS Lett. 2008;582:1465–1470. doi: 10.1016/j.febslet.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 30.Vogler C., Homan S., Pung A., Thorpe C., Barker J., Birkenmeier E.H., Upadhya P. Clinical and pathologic findings in two new allelic murine models of polycystic kidney disease. J. Am. Soc. Nephrol. 1999;10:2534–2539. doi: 10.1681/ASN.V10122534. [DOI] [PubMed] [Google Scholar]
- 31.Hoefele J., Wolf M.T., O'Toole J.F., Otto E.A., Schultheiss U., Deschenes G., Attanasio M., Utsch B., Antignac C., Hildebrandt F. Evidence of oligogenic inheritance in nephronophthisis. J. Am. Soc. Nephrol. 2007;18:2789–2795. doi: 10.1681/ASN.2007020243. [DOI] [PubMed] [Google Scholar]
- 32.Katsanis N. The oligogenic properties of Bardet-Biedl syndrome. Hum. Mol. Genet. 2004;13(Spec No 1):R65–R71. doi: 10.1093/hmg/ddh092. [DOI] [PubMed] [Google Scholar]
- 33.Fauser S., Munz M., Besch D. Further support for digenic inheritance in Bardet-Biedl syndrome. J. Med. Genet. 2003;40:e104. doi: 10.1136/jmg.40.8.e104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huangfu D., Anderson K.V. Cilia and Hedgehog responsiveness in the mouse. Proc. Natl. Acad. Sci. USA. 2005;102:11325–11330. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.May S.R., Ashique A.M., Karlen M., Wang B., Shen Y., Zarbalis K., Reiter J., Ericson J., Peterson A.S. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev. Biol. 2005;287:378–389. doi: 10.1016/j.ydbio.2005.08.050. [DOI] [PubMed] [Google Scholar]
- 36.O'Regan L., Blot J., Fry A.M. Mitotic regulation by NIMA-related kinases. Cell Div. 2007;2:25. doi: 10.1186/1747-1028-2-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.White M.C., Quarmby L.M. The NIMA-family kinase, Nek1 affects the stability of centrosomes and ciliogenesis. BMC Cell Biol. 2008;9:29. doi: 10.1186/1471-2121-9-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Surpili M.J., Delben T.M., Kobarg J. Identification of proteins that interact with the central coiled-coil region of the human protein kinase NEK1. Biochemistry. 2003;42:15369–15376. doi: 10.1021/bi034575v. [DOI] [PubMed] [Google Scholar]
- 39.Hilton L.K., White M.C., Quarmby L.M. The NIMA-related kinase NEK1 cycles through the nucleus. Biochem. Biophys. Res. Commun. 2009;389:52–56. doi: 10.1016/j.bbrc.2009.08.086. [DOI] [PubMed] [Google Scholar]
- 40.Lanza D.C., Meirelles G.V., Alborghetti M.R., Abrile C.H., Lenz G., Kobarg J. FEZ1 interacts with CLASP2 and NEK1 through coiled-coil regions and their cellular colocalization suggests centrosomal functions and regulation by PKC. Mol Cell Biochem. 2010;338:35–45. doi: 10.1007/s11010-009-0317-9. [DOI] [PubMed] [Google Scholar]
- 41.Moser J.J., Fritzler M.J., Rattner J.B. Primary ciliogenesis defects are associated with human astrocytoma/glioblastoma cells. BMC Cancer. 2009;9:448. doi: 10.1186/1471-2407-9-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.