

Abstract
Hereditary sensory neuropathy type I (HSN I) is an axonal form of autosomal-dominant hereditary motor and sensory neuropathy distinguished by prominent sensory loss that leads to painless injuries. Unrecognized, these can result in delayed wound healing and osteomyelitis, necessitating distal amputations. To elucidate the genetic basis of an HSN I subtype in a family in which mutations in the few known HSN I genes had been excluded, we employed massive parallel exon sequencing of the 14.3 Mb disease interval on chromosome 14q. We detected a missense mutation (c.1065C>A, p.Asn355Lys) in atlastin-1 (ATL1), a gene that is known to be mutated in early-onset hereditary spastic paraplegia SPG3A and that encodes the large dynamin-related GTPase atlastin-1. The mutant protein exhibited reduced GTPase activity and prominently disrupted ER network morphology when expressed in COS7 cells, strongly supporting pathogenicity. An expanded screen in 115 additional HSN I patients identified two further dominant ATL1 mutations (c.196G>C [p.Glu66Gln] and c.976 delG [p.Val326TrpfsX8]). This study highlights an unexpected major role for atlastin-1 in the function of sensory neurons and identifies HSN I and SPG3A as allelic disorders.
Main Text
Recent technological developments in massive parallel sequencing have greatly facilitated the rapid and economical detection of the molecular basis for rare Mendelian disorders.1–3 To identify the genetic mutations responsible for distinct forms of inherited neuromuscular disorders in three families, we designed a 385k NimbleGen Sequence Capture array to selectively enrich and sequence the combined exonic information of disease-linked chromosomal loci. The first large family analyzed was diagnosed clinically with hereditary sensory neuropathy type I (HSN I [MIM 162400]), an axonal form of hereditary motor and sensory neuropathy distinguished by prominent early sensory loss and later positive sensory phenomena, including dysesthesia and shooting pains. In HSN I loss of sensation can lead to painless injuries, slow wound healing, and subsequent osteomyelitis, requiring distal amputations. Distal motor involvement is usually present in advanced cases and can be severe.4 The inheritance pattern in HSN I is autosomal dominant as a result of mutations in genes encoding serine palmitoyltransferases 1 and 2 (SPTLC1 [MIM 605712] and SPTLC2 [MIM 605713])5,6 and Ras-related GTPase 7 (RAB7 [MIM 602298]).7
The anonymized pedigree of the HSN I family studied here is shown in Figure 1. Affected and unaffected family members underwent a detailed clinical, neurological, and neurophysiological examination by experienced neurologists. After subjects received genetic counseling and gave written informed consent, a peripheral blood sample was taken for genetic analysis. The study was approved by the Ethics committees of the participating universities. Age at onset was in early adulthood, when most affected individuals exhibited trophic skin and nail changes and suffered from repeated foot ulcerations, leading to osteomyelitis and subsequent foot or toe amputations (Figure 2). Most patients presented with severe distal sensory loss and distal amyotrophy in the lower limbs but absent or minimal distal amyotrophy in the upper limbs. Patellar tendon reflexes ranged from normal to increased, and ankle reflexes ranged broadly from absent to increased, indicating upper motor-neuron involvement in some patients. A sensorimotor axonal neuropathy was almost consistently observed on electrophysiological testing. One 23-year-old patient (IV/2) with electrophysiologically prominent axonal nerve damage, but without foot ulcerations, was diagnosed with cerebral palsy in infancy because of pronounced early-onset lower-limb spasticity, but a detailed history revealed no indications of prenatal or perinatal hypoxia.
Figure 1.
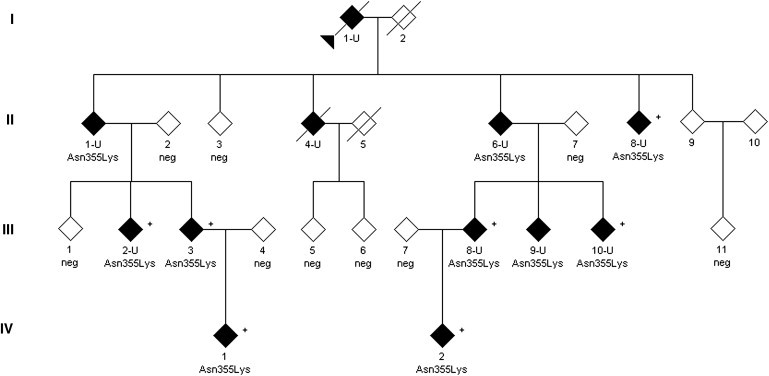
Partial Pedigree of a Family Affected by HSN I
The pedigree shows all individuals included in the linkage study (disease status at time of diagnosis is indicated). Additional unaffected family members and individuals not available for this study are not depicted for privacy. Individual II/3 was neurologically normal at the age of 69, but NCS could not be carried out. Also, the children of this individual did not have a history of gait disturbances or foot ulcerations. Medical history indicated that individuals II/9 and III/11 were unaffected, but they refused neurological and neurophysiological examination. However, individual III/11 agreed to participate in the genetic analysis. Filled symbols indicate affected individuals; empty symbols define unaffected individuals. U indicates patients with severe sensory neuropathy and foot ulcerations and/or amputations; patients with a + also presented with upper-motor-neuron signs. Neg indicates that an individual tested negative for the p.Asn355Lys mutation.
Figure 2.
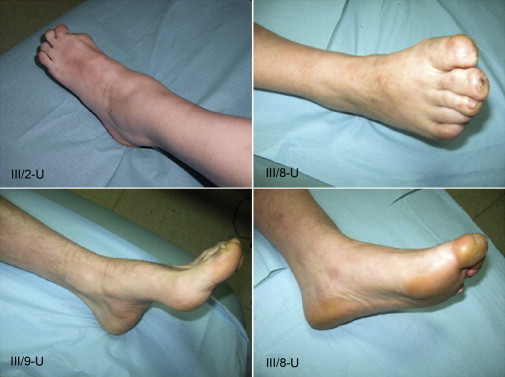
Clinical Findings in Patients Carrying the Asn355Lys Atlastin-1 Variant
Images of the feet of patients (III/2-U, III/8-U, and III/9-U) with prominent axonal sensory neuropathy, trophic skin and nail changes, distal muscle atrophy, mild pes cavus, and amputation of the great toe are shown.
After mutations in the genes known to be mutated in HSN I (SPTLC1, SPTLC2, and RAB7)5–7 were excluded, a genome-wide linkage scan performed with Affymetrix GeneChip® Human Mapping 10K arrays XbaI 142 2.0 (Affymetrix, Santa Clara, CA, USA) in ten affected and five unaffected individuals and three spouses from the HSN I family. Calculations of the parametric multipoint LOD score and haplotypes were obtained with the ALLEGRO program8 and an autosomal-dominant, fully penetrant model. A first evaluation exclusively considering individuals with foot ulcerations and/or axonal neuropathy to be affected localized the possible disease interval to either chromosome 5q or 14q (data not shown). However, when the two affected individuals—individual III/3, with marked nail changes and normal nerve conduction studies (NCS) but neuropathic changes on electromyography and pathologically brisk tendon reflexes, and individual IV/1, with mild spasticity but normal NCS—were included as well, the disease interval could be linked to a single locus on chromosome 14, bp 41,334,596–55,611,787 (Figure S1), with a maximum LOD score of 2.94. According to the ENSEMBL database (release 54 from May 2009, Homo sapiens Genes NCBI36), this interval contains 75 protein-encoding genes (Table S1) encompassing 924 exons.
To simultaneously analyze all 75 protein-coding genes within the disease region, we used array-based sequence capture followed by massive parallel resequencing. A Roche NimbleGen custom tiling 385K sequence capture array targeting the exonic sequences of all protein-encoding genes within the disease interval was designed with BioMart EnsEMBL release 54 May 2009 on the basis of the NCBI 36 assembly of the human genome (November 2005). Sequence capture was performed according to the manufactor's instructions (Roche NimbleGen). Twenty micrograms of genomic DNA from patient II/6-U was processed into a capture library compatible with GS FLX Titanium chemistry according to the NimbleGen Arrays User's Guide v.3.0, Dec. 2008 (Roche NimbleGen, Madison, WI). Four micrograms of the amplified capture library was processed into a sequencing library for the 454 GS-FLX according to the GS FLX Titanium General Library Preparation protocol (454 Life Sciences, Branford, CT) according to the manufacturer's recommended conditions (without the nebulization step). The captured sample library was sequenced with a quarter of a Titanium PicoTiterplate (70 × 75) on the GS-FLX platform with Titanium chemistry and standard settings. We generated ∼72.11Mb of sequence information (total of 222,522 reads), and 72.9% of uniquely mapped reads hit the target region. An effective >10-fold sequence coverage for ∼76% of the target sequence at the per base level (Figure S2) was achieved for the HSN I subtype locus on chromosome 14.
Applying high-confidence sequence-variant filtering with the standard GS Reference Mapper tool (reference: UCSC genome annotation database for the Mar. 2006 GenBank freeze assembled by NCBI hg18 build 36.1, dbSNP build 130) and manual curation, we identified 27 nonsynonymous sequence variants (Table S2), 25 of which were known single-nucleotide polymorphisms (dbSNPs). The two remaining sequence variants, both confirmed by Sanger sequencing, were not previously reported. Both segregated with the disease and affected the coding sequences of atlastin-1 (ATL1, NM_015915.4, c.1065C>A [p.Asn355Lys]; MIM 606439; Figure 3) and prostaglandin E receptor 2 (PTGER2, NM_000956.3, c.247T>G [p.Cys83Gly]; MIM 176804). Whereas detection of the PTGER2 sequence variant in 1/40 healthy control individuals points to a probable polymorphism, the missense variation p.Asn355Lys ATL1 at a highly conserved residue (Figure S3) was absent in 370 ethnically and age-matched control individuals, making disease causality highly likely.
Figure 3.
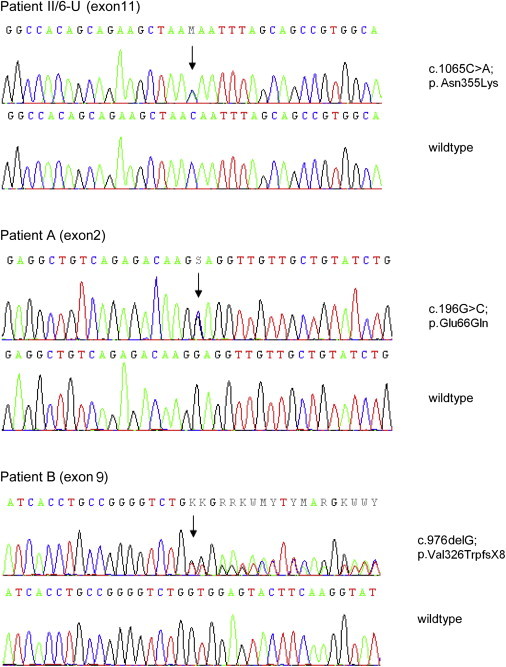
Detection of ATL1 Mutations by Direct Sequencing
Representative electropherograms derived from ATL1 sequencing of genomic DNA from patients (upper panel) compared to unaffected individuals (wild-type, lower panel). The positions of the heterozygous mutations are indicated with an arrow above the sequence. Effects of the mutations on amino acid sequence are shown on the right.
To evaluate further mutations in ATL1 as a potential cause of HSN I, we tested the entire ATL1 coding region in 115 additional patients classified as having familial or sporadic HSN I (for primer sequences, see Table S3); additional ATL1 mutations were found in two HSN patients for whom mutations in genes with known HSN mutations had been excluded previously (these genes include SPTLC1, SPTLC2, RAB7, WNK1 [MIM 605232], NTRK [MIM 191315], NGFB [MIM 162030], FAM134B [MIM 613114], CCT5 [MIM 610150], and the candidate genes SPTLC3 [MIM 611120], and NGFR [MIM 162010]). Patient A, aged 61 years, carried the missense change c.196G>C (p.Glu66Gln, Figure 3) at a highly conserved residue coded by exon 2 (Figure S3). He presented with progressive, ascending, severe sensory loss affecting all modalities in the lower legs but without foot ulcerations or paresis. Patellar tendon reflexes were reduced, and Achilles tendon reflexes were absent. Sural nerve biopsy revealed severe, chronic, slowly progressive neuropathy predominantly of the axonal type, and a moderate demyelinating component and minimal signs of regeneration were present. By report, the deceased mother had a similar severe sensory neuropathy of unknown etiology, but DNA was not available for genetic testing. Exon 9 of patient B contained a c.976delG nucleotide deletion predicted to cause a large C-terminal protein truncation of atlastin-1 (p.Val326TrpfsX8, Figure 3). This patient had adult-onset sensory neuropathy with ulcerations and lack of pain perception. There was neither muscle weakness nor autonomic symptoms. The patient had paresthesias in the fingers and occasional lancinating pains in his ankles. Patellar tendon reflexes were brisk, and Achilles tendon reflexes were absent. Electrophysiological testing was consistent with a sensorimotor axonal neuropathy. The father of patient B displayed a similar clinical phenotype and also carried this nucleotide deletion. According to his medical history, the index patient has one affected brother and two unaffected siblings; none of these individuals were available for testing or examination.
Subsequently, we investigated the functional consequences of these ATL1 mutations. COS7 cells were maintained, transfected with eukaryotic expression constructs, and analyzed by confocal immunofluorescence microscopy and immunoblotting as described previously.9–11 Site-directed mutagenesis was performed via the QuikChange method (Stratagene, La Jolla, CA). GTPase activity assays were performed with wild-type or mutant Myc-tagged atlastin-1 proteins immunopurified from COS7 cell extracts as described previously.12 In GTPase assays, the HSN I-associated Asn355Lys atlastin-1 mutant had partial but significant loss of GTPase activity in vitro when it was compared to wild-type atlastin-1 (Figure 4A). Expression of wild-type atlastin-1 in COS7 cells results in a more highly branched ER and punctate enrichment of atlastin-1 along the ER tubules, including at three-way junctions in the cell periphery and within aberrant ER sheets more centrally.11,13 Expression of Asn355Lys atlastin-1 in COS7 cells, at levels essentially identical to those of the wild-type protein, yielded prominent disruption of ER three-way junctions, similar to results seen upon expression of the Lys80Ala dominant-negative atlastin-1 mutant (equivalent to Lys44Ala in the dynamin GTPase) that lacks GTPase activity (Figures 4B–4D). However, the Glu66Gln atlastin-1 mutant exhibited no significant changes in GTPase activity compared to that of the wild-type protein. Also, no changes in ER morphology in COS7 cells were observed for the Glu66Gln atlastin-1 mutant when it was expressed at levels similar to those of the wild-type protein (Figure S4). Finally, the Val326TrpfsX8 atlastin-1 truncation mutant was localized diffusely throughout the cytoplasm in COS7 cells as a result of the lack of the C-terminal paired transmembrane domains11 that are necessary and sufficient for ER localization, but it did not significantly alter ER morphology, as assessed by coexpression with wild-type atlastin-1 and also by coexpression with RFP-tagged Sec61β, a resident ER protein (Figure S4). Importantly, immunoblot analysis (Figure S4) showed that expression levels of Val326TrpfsX8 atlastin-1 were markedly lower than those of wild-type atlastin-1 or the Glu66Gln and Asn355Lys mutants, probably as a result of decreased protein stability. These data indicate that disease pathogenesis, particularly in the case of Val326TrpfsX8 atlastin-1, might be related to loss of functional atlastin-1 protein, which has been shown to alter ER morphology.13,14 Even so, the absence of any clear pathogenic changes in heterologous cell expression studies, particularly for Glu66Gln atlastin-1, indicate that alterations in three-way junction formation of the tubular ER might not be a sensitive indicator for all types of atlastin-1 dysfunction. Consistent with this notion, a number of SPG3A mutant atlastin-1 proteins do not noticeably affect formation of three-way ER junctions, and this morphological change appears to correlate most closely with a given mutation's effects on atlastin-1 GTPase activity (P.-P.Z. and C.B, unpublished data). Furthermore, a number of SPG3A mutations have been demonstrated to disrupt other aspects of ER structure.15 Although we cannot completely rule out the possibility that the Glu66Gln and Val326TrpfsX8 atlastin-1 mutations are unrelated to HSN I pathogenesis, this appears to be unlikely given the positive family history for both patients A and B and the fact that these mutations were undetected in the large number of control subjects (150 for the Glu66Gln and 334 for the Val326TrpfsX8 mutation).
Figure 4.
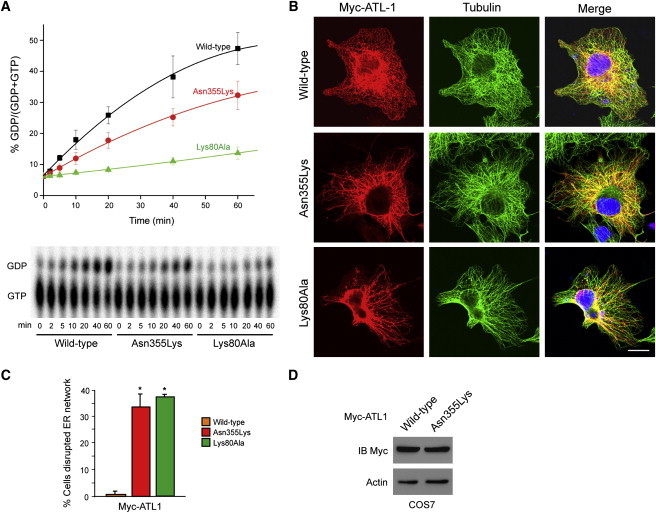
Asn355Lys Atlastin-1 Exhibits Altered GTPase Activity and Disrupts ER Morphology when It Is Expressed in Cells
(A) Myc-tagged wild-type atlastin-1 or the indicated missense mutants were immunopurified from COS7 cells, and GTP hydrolysis was plotted as a function of time (top). Representative thin-layer chromatography plates show conversion of GTP to GDP (bottom). Error bars represent means ± standard deviation.
(B) COS7 cells were transfected with Myc-tagged wild-type or mutant atlastin-1 and immunostained for Myc-epitope (red) and β-tubulin (green). Merged images are at the right. DAPI nuclear staining is in blue. The scale bar represents 20 μm.
(C) Quantification of ER disruption (three trials of n = 100 cells per condition, means ± SD); ∗p < 0.01.
(D) COS7 cells transfected with Myc-tagged wild-type or Asn355Lys atlastin-1 were immunoblotted for Myc-epitope. Actin levels were monitored as a control for protein loading.
Mutations in ATL1, a dynamin-related GTPase with orthologs in all eukaryotes,13 have been described previously for early-onset hereditary spastic paraplegia (SPG3A; MIM 182600), an inherited neurological disorder that affects axons of upper motor neurons in a length-dependent manner.16 Thus far, more than 40 sequence alterations in exons 4–10, 12, and 13 of ATL1, the vast majority missense changes (Figure 5), have been reported for SPG3A.16–18 In a small number of SPG3A patients, lower motor-neuron signs (e.g., distal amyotrophy of the lower limbs), abnormal motor and sensory NCS, and neuropathic findings on muscle and sural nerve biopsy indicative of a complicated ATL1-SPG3A phenotype have been noted.17 A recent study also described additional symptoms, including seizures, ataxia, and MRI hypertensities in some patients harboring ATL1 mutations.18 However, the prominent sensory disturbances and their complications produced by the ATL1 mutations reported here are highly unexpected and may shed additional light on the physiological role of atlastin-1. Structurally, most HSP-specific mutations in SPG3A are predicted to alter amino acids located on the surface of the globular, N-terminal region that contains the conserved GTPase domain (amino acid residues 30–337), but a majority do not disrupt GTPase motifs per se.11 Thus, these mutations might exert a pathogenic effect either by introducing an aberrant secondary structure that disturbs intramolecular associations or multimerization of atlastin-1 or by altering interactions of atlastin-1 with other proteins. Thus far, no clear functional distinction is evident between mutations causing SPG3A and those implicated in HSN I. High-resolution structural analysis of atlastin-1 might provide additional insights.
Figure 5.
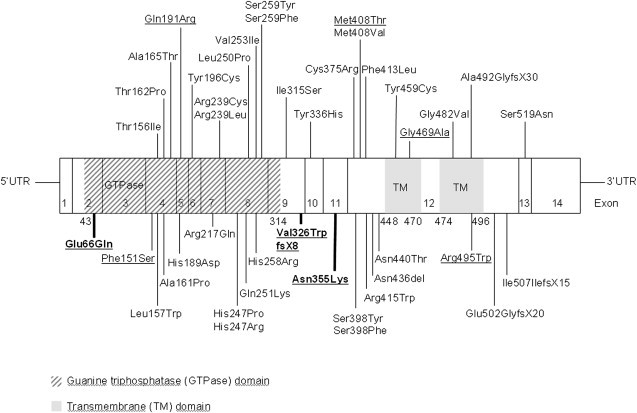
Schematic Model of Atlastin-1 Mutations in SPG3A and HSN I
The model shows the distribution of autosomal-dominant, SPG3A-associated mutations and their amino acid residue changes, as well as the novel ATL1 mutations (bold letters) associated with HSN I identified in this study. Mutations known to be associated with peripheral neuropathies are underlined. Exons 1–14 are indicated.
Atlastin-1 interacts with spastin (i.e., SPAST or SPG4) and the receptor expression enhancing protein-1 (REEP1, also known as SPG31), which together are mutated in about 50% of autosomal-dominant HSP cases,18,19 through predicted intramembrane hydrophobic hairpin domains.11 These proteins function in the generation of the tubular ER network in eukaryotes. This has led to the hypothesis that the most common HSPs are caused by ER network disruption, particularly via dysfunctional interactions with the microtubule cytoskeleton.11
How abnormal morphology of the ER might predispose to a long axonopathy remains unclear. The long axons of the corticospinal neurons, which are among the longest axons in the body, might be severely affected by the disruption of the cooperative regulation of polarized membrane and protein trafficking along microtubules.19 In cultured cortical neurons, atlastin-1 is enriched in axon growth cones, and shRNA-mediated depletion of atlastin-1 impaired axon elongation in neurons.9,11,14 Interestingly, a very recent study of atl1 in zebrafish demonstrated that atlastin-1 knock down results in abnormal architecture of spinal motor neurons, as well as an associated upregulation of the bone morphogenetic protein (BMP) signaling pathway.20 Because dysregulation of this pathway has been implicated in a number of other forms of HSPs, it will be important to study the interplay between regulation of ER morphology and BMP signaling in neurons.
When comparing the HSN I clinical phenotypes caused by mutations in SPTLC1, SPTLC2, RAB7, and ATL1, a persuasive overlap between HSN I caused by RAB7 mutations and that caused by ATL1 mutations is evident.7,21 In particular, the profound distal sensory loss affecting all sensory modalities to a similar degree from disease onset must be highlighted in both diseases. Also, in patients with RAB7 mutations, brisk tendon reflexes are common.21 Notably, both RAB7 and atlastin-1 are implicated in intracellular membrane trafficking and distribution, and RAB7 interacts with the SPG21 protein maspardin.22,23 Functional studies are needed for an investigation of any direct or indirect interactions between ATL1 and RAB7 and other HSN I genes. Furthermore, the variable occurence of upper and lower motor neuron signs and trophic complications in affected individuals could indicate the existence of modifier genes and/or compensatory molecular mechanisms. Uncovering these will lead to a better understanding of the diverse phenotypes elicited by ATL1 mutations.
Acknowledgments
We are grateful for the participation of the patients and families, and we thank C. Fischer, H. Knausz, J. Nagle, and T. Deconinck for expert technical assistance. This work was supported by the Austrian Science Fund (FWF, P19455-B05), the Oesterreichische Nationalbank (ÖNB, project 13010), and the Intramural Research Program of the National Institute of Neurological Disorders and Stroke, US National Institutes of Health. This project was in part funded by a Methusalem grant of the University of Antwerp, the Fund for Scientific Research (FWO-Flanders), the Medical Foundation Queen Elisabeth (GSKE), the Association Belge contre les Maladies Neuromusculaires (ABMM), and the Interuniversity Attraction Poles P6/43 Program of the Belgian Federal Science Policy Office (BELSPO). J.B. is supported by a PhD fellowship of the FWO-Flanders, and J.W. is supported by Deutsche Forschungsgemeinschaft grant WE 1406 13/1.
Supplemental Data
Web Resources
The URL for data presented herein is as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., Dent K.M., Huff C.D., Shannon P.T., Jabs E.W., Nickerson D.A. Exome sequencing identifies the cause of a Mendelian disorder. Nat. Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rehman A.U., Morell R.J., Belyantseva I.A., Khan S.Y., Boger E.T., Shahzad M., Ahmed Z.M., Riazuddin S., Khan S.N., Riazuddin S. Targeted capture and next-generation sequencing identifies C9orf75, encoding taperin, as the mutated gene in nonsyndromic deafness DFNB79. Am. J. Hum. Genet. 2010;86:378–388. doi: 10.1016/j.ajhg.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nikopoulos K., Gilissen C., Hoischen A., van Nouhuys C.E., Boonstra F.N., Blokland E.A.W., Arts P., Wieskamp N., Strom T.M., Ayuso C. Next-generation sequencing of a 40 Mb linkage interval reveals TSPAN12 mutations in patients with familial exudative vitreoretinopathy. Am. J. Hum. Genet. 2010;86:240–247. doi: 10.1016/j.ajhg.2009.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nicholson G.A. Hereditary sensory neuropathy I. In: Pagon R.A., Bird T.C., Stephens K., editors. GeneReviews. University of Washington; Seattle, WA: 2010. PMID: 20301564. [PubMed] [Google Scholar]
- 5.Dawkins J.L., Hulme D.J., Brahmbhatt S.B., Auer-Grumbach M., Nicholson G.A. Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat. Genet. 2001;27:309–312. doi: 10.1038/85879. [DOI] [PubMed] [Google Scholar]
- 6.Rotthier A., Auer-Grumbach M., Janssens K., Baets J., Penno A., Almeida-Souza L., Van Hoof K., Jacobs A., De Vriendt E., Schlotter-Weigel B. Mutations in the SPTLC2 subunit of serine palmitoyltransferase cause hereditary sensory and autonomic neuropathy type I. Am. J. Hum. Genet. 2010;87:513–522. doi: 10.1016/j.ajhg.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verhoeven K., De Jonghe P., Coen K., Verpoorten N., Auer-Grumbach M., Kwon J.M., FitzPatrick D., Schmedding E., De Vriendt E., Jacobs A. Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am. J. Hum. Genet. 2003;72:722–727. doi: 10.1086/367847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gudbjartsson D.F., Thorvaldsson T., Kong A., Gunnarsson G., Ingolfsdottir A. Allegro version 2. Nat. Genet. 2005;37:1015–1016. doi: 10.1038/ng1005-1015. [DOI] [PubMed] [Google Scholar]
- 9.Zhu P.-P., Soderblom C., Tao-Cheng J.-H., Stadler J., Blackstone C. SPG3A protein atlastin-1 is enriched in growth cones and promotes axon elongation during neuronal development. Hum. Mol. Genet. 2006;15:1343–1353. doi: 10.1093/hmg/ddl054. [DOI] [PubMed] [Google Scholar]
- 10.Rismanchi N., Soderblom C., Stadler J., Zhu P.-P., Blackstone C. Atlastin GTPases are required for Golgi apparatus and ER morphogenesis. Hum. Mol. Genet. 2008;17:1591–1604. doi: 10.1093/hmg/ddn046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park S.H., Zhu P.-P., Parker R.L., Blackstone C. Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin-1 coordinate microtubule interactions with the tubular ER network. J. Clin. Invest. 2010;120:1097–1110. doi: 10.1172/JCI40979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meijer I.A., Dion P., Laurent S., Dupré N., Brais B., Levert A., Puymirat J., Rioux M.F., Sylvain M., Zhu P.-P. Characterization of a novel SPG3A deletion in a French-Canadian family. Ann. Neurol. 2007;61:599–603. doi: 10.1002/ana.21114. [DOI] [PubMed] [Google Scholar]
- 13.Hu J., Shibata Y., Zhu P.-P., Voss C., Rismanchi N., Prinz W.A., Rapoport T.A., Blackstone C. A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell. 2009;138:549–561. doi: 10.1016/j.cell.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orso G., Pendin D., Liu S., Tosetto J., Moss T.J., Faust J.E., Micaroni M., Egorova A., Martinuzzi A., McNew J.A., Daga A. Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin. Nature. 2009;460:978–983. doi: 10.1038/nature08280. [DOI] [PubMed] [Google Scholar]
- 15.Namekawa M., Muriel M.-P., Janer A., Latouche M., Dauphin A., Debeir T., Martin E., Duyckaerts C., Prigent A., Depienne C. Mutations in the SPG3A gene encoding the GTPase atlastin interfere with vesicle trafficking in the ER/Golgi interface and Golgi morphogenesis. Mol. Cell. Neurosci. 2007;35:1–13. doi: 10.1016/j.mcn.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 16.Zhao X., Alvarado D., Rainier S., Lemons R., Hedera P., Weber C.H., Tukel T., Apak M., Heiman-Patterson T., Ming L. Mutations in a newly identified GTPase gene cause autosomal dominant hereditary spastic paraplegia. Nat. Genet. 2001;29:326–331. doi: 10.1038/ng758. [DOI] [PubMed] [Google Scholar]
- 17.Ivanova N., Claeys K.G., Deconinck T., Litvinenko I., Jordanova A., Auer-Grumbach M., Haberlova J., Löfgren A., Smeyers G., Nelis E. Hereditary spastic paraplegia 3A associated with axonal neuropathy. Arch. Neurol. 2007;64:706–713. doi: 10.1001/archneur.64.5.706. [DOI] [PubMed] [Google Scholar]
- 18.McCorquodale D.S., III, Ozomaro U., Huang J., Montenegro G., Kushman A., Citrigno L., Price J., Speziani F., Pericak-Vance M.A., Züchner S. Mutation screening of spastin, atlastin, and REEP1 in hereditary spastic paraplegia. Clin. Genet. 2010 doi: 10.1111/j.1399-0004.2010.01501.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blackstone C., O'Kane C.J., Reid E. Hereditary spastic paraplegias: Membrane traffic and the motor pathway. Nat. Rev. Neurosci. 2011;12:31–42. doi: 10.1038/nrn2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fassier C., Hutt J.A., Scholpp S., Lumsden A., Giros B., Nothias F., Schneider-Maunoury S., Houart C., Hazan L. Zebrafish atlastin controls motility and spinal motor axon architecture via inhibition of the BMP pathway. Nat. Neurosci. 2010;13:1380–1387. doi: 10.1038/nn.2662. [DOI] [PubMed] [Google Scholar]
- 21.Auer-Grumbach M., De Jonghe P., Wagner K., Verhoeven K., Hartung H.-P., Timmerman V. Phenotype-genotype correlations in a CMT2B family with refined 3q13-q22 locus. Neurology. 2000;55:1552–1557. doi: 10.1212/wnl.55.10.1552. [DOI] [PubMed] [Google Scholar]
- 22.Fukuda M. Regulation of secretory vesicle traffic by Rab small GTPases. Cell. Mol. Life Sci. 2008;65:2801–2813. doi: 10.1007/s00018-008-8351-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCray B.A., Skordalakes E., Taylor J.P. Disease mutations in Rab7 result in unregulated nucleotide exchange and inappropriate activation. Hum. Mol. Genet. 2010;19:1033–1047. doi: 10.1093/hmg/ddp567. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.