

Abstract
Microphthalmia with limb anomalies (MLA) is a rare autosomal-recessive disorder, presenting with anophthalmia or microphthalmia and hand and/or foot malformation. We mapped the MLA locus to 14q24 and successfully identified three homozygous (one nonsense and two splice site) mutations in the SPARC (secreted protein acidic and rich in cysteine)-related modular calcium binding 1 (SMOC1) in three families. Smoc1 is expressed in the developing optic stalk, ventral optic cup, and limbs of mouse embryos. Smoc1 null mice recapitulated MLA phenotypes, including aplasia or hypoplasia of optic nerves, hypoplastic fibula and bowed tibia, and syndactyly in limbs. A thinned and irregular ganglion cell layer and atrophy of the anteroventral part of the retina were also observed. Soft tissue syndactyly, resulting from inhibited apoptosis, was related to disturbed expression of genes involved in BMP signaling in the interdigital mesenchyme. Our findings indicate that SMOC1/Smoc1 is essential for ocular and limb development in both humans and mice.
Introduction
Microphthalmia with limb anomalies (MLA [MIM 206920]), also known as Waardenburg anophthalmia syndrome or ophthalmoacromelic syndrome, is a rare autosomal-recessive disorder first described by Waardenburg.1 It is characterized by ocular anomalies ranging from mild microphthalmia to true anophthalmia and by limb anomalies such as oligodactyly, syndactyly, and synostosis of the 4th and 5th metacarpals.2–4 The genetic cause for MLA has remained unknown.
It is widely known that secreted signaling molecules such as Sonic hedgehog (Shh), wingless-type MMTV integration site family (Wnt), transforming growth factor β (Tgf-β), bone morphogenetic proteins (Bmps), and fibroblast growth factor (Fgf) are involved in the development of many organs and tissues, including the eyes and limbs.5,6 In particular, mutations in BMP4 (MIM 112262) have resulted in anophthalmia with systemic manifestations, including polydactyly and/or syndactyly (also known as micropthalmia, syndromic 6, MCOPS6 [MIM 607932]),7 highlighting importance of BMP signaling in both the developing eye and limb.
SMOC1 (MIM 608488), which encodes SPARC (secreted protein acidic and rich in cysteine)-related modular calcium binding 1, is a member of the SPARC (also known as BM-40) matricellular protein family that modulates cell-matrix interaction by binding to many cell-surface receptors, the extracellular matrix, growth factors, and cytokines.8,9 SMOCs are extracellular glycoproteins with five domains: an N-terminal follistatin-like (FS) domain, two thyroglobulin-like (TY) domains, a domain unique to SMOC, and an extracellular calcium-binding (EC) domain.9 SMOC1 is widely expressed in various tissues with localization to basement membranes.9,10 Although the biological function of SMOC1 remains largely unknown, it has been recently reported that Xenopus smoc protein, the ortholog of human SMOC1, acts as a BMP antagonist,11 suggesting that human SMOC1 can also modulate BMP signaling.
Here, we demonstrate that SMOC1 mutations cause MLA. We also show that Smoc1 null mice recapitulated MLA phenotypes, indicating that SMOC1 plays essential roles in both eye and limb development in humans and mice.
Subjects and Methods
Subjects
A total of four families with one or two cases of MLA were analyzed in this study, including three previously reported families (A, B, and C).12,13 Family X from Turkey, which has been previously described,14 was newly recruited to this study. Detailed clinical information of all the patients is available in the literature,12,14 and phenotypes of patients with confirmed mutations are summarized in Table S1 (available online). A total of five affected and 16 unaffected members from the four families were analyzed in the linkage study. Genomic DNA was obtained from peripheral-blood leukocytes with the use of QuickGene 610-L (Fujifilm, Tokyo, Japan) after informed consent had been given. Experimental protocols were approved by the institutional review board of Yokohama City University School of Medicine.
SNP Genotyping, and Fine Mapping with Short Tandem Repeat Markers
Whole-genome SNP genotyping, with the use of GeneChip Human Mapping 50K Array XbaI (Affymetrix, Santa Clara, CA), and fine mapping of possible candidate regions, with the use of additional microsatellite markers, were performed as previously described.12,15 The list of primers used for fine mapping is presented in Table S2.
Linkage Analysis
Multipoint linkage analyses using aligned SNPs were performed with ALLEGRO software.16 Two-point linkage analyses of candidate regions were performed with the LINKAGE package MLINK (FASTLINK software, version 5.1). In each program, an autosomal-recessive model of inheritance with complete penetrance and a disease-allele frequency of 0.001 were applied.
Mutation Analysis of Candidate Genes
All coding exons and exon-intron boundaries of RAD51L1 (MIM 602948), ACTN1 (MIM 102575), ERH (MIM 601191), SRSF5 (MIM 600914), DCAF5 (MIM 603812), COX16, EXD2, GALNTL1, SLC39A9, KIAA0247, MED6 (MIM 602984), TTC9 (MIM 610488), MAP3K9 (MIM 600136), and SMOC1 (transcript variant 1, GenBank accession number NM_001034852.1) were analyzed in the probands of families A, C, and X. The transcript variant 2 of SMOC1 (GenBank accession number NM_022137.4) is 3 bp shorter than the variant 1, leading to an in-frame amino acid deletion at position 431. PCR was cycled 35 times at 94°C for 30 s, at 60°C for 30 s, and at 72°C for 30–90 s in a total volume of 20 μl containing 30 ng genomic DNA as a template, 0.5 μM forward and reverse primers, 200 μM each deoxyribonucleotide triphosphate (dNTP), 1 × ExTaq buffer, and 0.25 U ExTaq (Takara). All primers were designed with Primer3 software. Detailed information of primers is available upon request. PCR products were purified with ExoSAP (USB) and sequenced with BigDye Terminator 3.1 (Applied Biosystems) on a 3100 Genetic Analyzer. Sequences of patients were compared to reference genome sequences in the UCSC Genome Browser (February 2009 assembly) with Seqscape software, version 2.1 (Applied Biosystems).
Animals
Smoc1 mutant mice, created with the use of the Sleeping Beauty transposon system, have been previously described.17 Line PV384 was provided by the RIKEN BioResource Center through the National BioResource Project of MEXT, Japan. Three independent mouse lines (no. 1 to no. 3), each with a single insertion in intron 1 of Smoc1, were bred as heterozygotes. Lines 1 and 3 were backcrossed for at least four generations to a C57BL/6J background. Line 2 was maintained with a mixed background of C57BL/6J and ICR. We mainly analyzed line 1, but we confirmed similar phenotypes in lines 2 and 3. Animals were housed in accordance with protocols approved by the Institutional Animal Care and Use Committee at Yokohama City University, School of Medicine. PCR genotyping of mice was performed with the use of genomic DNA from yolk-sac, ear, or tail biopsies. The following primers were used: PV384-WF, 5′-AAAGGCTGGGAATTGTTTGA-3′; PV384-WR, 5′-TGCAGCTGAAACTGTCTCTCC-3′; PV384-MF, 5′-TGTCCTAACTGACTTGCCAAA-3′. The PV384-WF/PV384-WR primers amplified a 441 bp wild-type (WT) product, and the PV384-MF/PV384-WR primers amplified a 218 bp mutant product.
Southern Hybridization
Genomic DNA was extracted from livers or tail biopsies of PV384 heterozygous (Smoc1Tp/+) mice via standard protocols. The gene-trap insertions were analyzed by Southern hybridization with the use of 10 μg of SacI-, NdeI-, BglII-, and EcoRІ-digested DNA. The probe (451 bp), which hybridized to the internal ribosome entry site (IRES) in the gene-trap vector, was synthesized with the DIG PCR Probe Synthesis Kit (Roche) with the use of the following primers: 5′-CTAACGTTACTGGCCGAAGC-3′ and 5′- CCCAGATCAGATCCCATACAA-3′. Hybridization, washing, and detection of probes were performed according to the manufacturer's protocol. Images were captured with the FluorChem system (Alpha Innotech).
Cloning of Gene-Trap Insertion Sites
After identification of aberrant DNA fragments by Southern hybridization, NdeI-, SacI-, and EcoRІ-digested DNA from PV384 mice was fractionated by electrophoresis, and appropriately sized fragments containing Ol1 (other locus 1), Ol2, and Ol3 were isolated with a QIAEXII Gel Extraction Kit (QIAGEN). The isolated DNA was self-ligated by Ligation High ver.2 (Toyobo), precipitated with ethanol, and dissolved in 20 μl EB buffer (QIAGEN). Inverse PCR was performed in 25 μl reactions, containing 2 μl ligated DNA, 1 × PCR buffer for KOD FX, 0.4 mM each dNTP, 0.5 μM each primer, and 0.5 U KOD FX DNA polymerase (Toyobo). Primers common to Ol1, Ol2, and Ol3 were as follows: Inv-F, 5′- ATCGCCAGTTCTGTATGAACGGTCTGGTCTT-3′; Inv-R, 5′-CCCTCTTTACGTGCCAGCCATCTTAGAGATAC-3′. Confirmatory PCR of gene-trap insertion sites for Ol1, Ol2, and Ol3 loci was performed with the use of the following primers: Ol1-F, 5′-GAGTGGTATTCATTGGATTCTGCTGAT-3′; Ol2-F, 5′-AAATCCAGCTGGCCAACAGACTAAG-3′; Ol3-F, 5′-TTGCCGGGTAGACTCTATCAAGAACCA-3′; TBAL-R, 5′-CTTGTGTCATGCACAAAGTAGATGTCC-3′. Primer sets of Ol1-F/TBAL-R, Ol2-F/TBAL-R, and Ol3-F/TBAL-R could amplify 175 bp, 607 bp, and 767 bp products, respectively. These PCR primer pairs were also used for genotyping of mice harboring a single insertion at the Smoc1 locus.
Confirmation of Promoter- and Poly(A)-Trapped Transcripts
Whole embryos at embryonic day 10.5 (E10.5) and E11.5 were stored in RNAlater solution (QIAGEN). Total RNA was extracted from WT, Smoc1Tp/+, and Smoc1Tp/Tp embryos with the use of RNeasy Plus Mini (QIAGEN). One microgram total RNA was subjected to reverse transcription with the use of a PrimeScript 1st Strand Synthesis Kit with random hexamers (Takara). A control reaction with no reverse transcriptase was included in each experiment. PCR was performed in 20 μl reactions, containing 1 μl cDNA, 1 × PCR Buffer for KOD FX, 0.4 mM each dNTP, 0.3 μM each primer, and 0.4 U KOD FX (Toyobo). Primers used are listed below: Smoc1-F, 5′-GTCCCCACCTCCCCAAGTGCTTTGA-3′; LacZ-R, 5′-TGCCAAAAGACGGCAATATGGTGGAAA-3′; GFP-F, 5′-TCACATGGTCCTGCTGGAGTTCGTGAC-3′; Smoc1-R, 5′-ACACTTGCTCTGGCCAGCATCTTTGCAT-3′. Primer sets of Smoc1-F/Smoc1-R, Smoc1-F/LacZ-R, and GFP-F/Smoc1-R could amplify native Smoc1 (366 bp), promoter-trapped transcripts (Tp-LacZ, 500 bp) and poly(A)-trapped transcripts (Tp-GFP, 308 bp), respectively. The PCR conditions were 98°C for 10 s, 68°C for 1 min, for 30 cycles. Primers for ACTB18 were used as an internal control. PCR for ACTB was cycled 20 times at 94°C for 20 s, 60°C for 20 s, and 72°C for 30 s in a total volume of 10 μl containing 0.5 μl cDNA, 0.4 μM each primer, 0.2 mM each dNTP, 1 × ExTaq buffer, and 0.5 U ExTaq HS (Takara). All PCR products were electrophoresed on 2% agarose gels.
In Situ Hybridization
Embryos were collected between E9.5 and E13.5. Whole-mount in situ hybridization was carried out as previously described.19,20 Two fragments of Smoc1 cDNA were obtained as probes by RT-PCR, with the use of total RNA extracted from livers of E16.5 mouse embryos, and subcloned into pCR4-TOPO (Invitrogen). Primer sequences were as follows: probe 1-F, 5′-GTCTGCTCACGCCCCACT-3′; probe 1-R, 5′-CCTGAACCATGTCTGTGGTG-3′; probe P-F, 5′-CAGGAACAGGAAAGGGAAGA-3′; probe P-R, 5′-AAGGGAAAACCACACAGCAC-3′. PCR products were 1023 bp and 1578 bp, corresponding to nucleotide positions 275–1297 and 1849–3426 of the mouse Smoc1 cDNA (GenBank accession number NM_001146217.1), respectively. The cDNA fragment amplified with probe P-F and probe P-R primers was identical to the probe used in a previous report.21 Digoxigenin-labeled sense and antisense riboprobes were synthesized with the use of a digoxigenin RNA labeling kit (Roche). These two different antisense probes demonstrated identical staining patterns, and the control sense probes showed no staining. The expression pattern was confirmed with more than three embryos. In addition, the following probes were used: Bmp2 (gift from Y. Takahashi),22 Sox9 (gift from A. Yamada),22 Bmp7 (gift from E.J. Robertson), and Msx2 (gift from Dr. R.E. Maxson, Jr). The numbers of embryos examined were as follows (numerical quantity for WT, Smoc1Tp/+, and Smoc1Tp/Tp, respectively, shown in parentheses): Msx2 (2, 1, 3) at E11.5; Bmp2 (3, 0, 3), Bmp7 (3, 0, 3), Msx2 (3, 0, 3), and Sox9 (2, 1, 3) at E12.5; Bmp2 (1, 2, 3), Bmp7 (2, 1, 3), Msx2 (1, 2, 3), and Sox9 (1, 3, 4) at E13.5. Stained embryos were cleared in glycerol to enable images to be produced with a VHX-1000 digital microscope (Keyence).
Histology
Heads of embryos and newborns were fixed overnight in 4% paraformaldehyde in PBS at 4°C. These embryos were then washed in PBS. Frozen samples were serially sectioned at 16 μm (E14.5) and 20 μm (P0). The numbers of eyes examined (WT, Smoc1Tp/+, Smoc1Tp/Tp) were as follows: coronally sectioned at E14.5 (8, 10, 12), coronally sectioned at P0 (8, 10, 6), horizontally sectioned at P0 (2, 2, 4). For evaluation of ventral atrophy of the retina, only the coronally sectioned eyes were used. TB staining was performed according to standard protocols. Forelimbs of mice were fixed in 4% paraformaldehyde in PBS, decalcified in 10% EDTA, and embedded in paraffin. Forelimbs were serially sectioned at 4 μm and stained with hematoxylin and eosin.
Evaluation of Optic Nerve Diameter
The palatine and orbital bones were carefully removed to expose the optic chiasm and optic nerve. During the dissection process, 4% paraformaldehyde in PBS was frequently applied onto the gaps between the bone and optic nerve. Xylene cyanol was applied to enhance the outline of optic nerves at poastnatal day 0 (P0). Photographs of optic nerves were taken with a VHX-1000 digital microscope, and the diameter was measured for right and left optic nerves with the bundled software included with the VHX-1000 instrument.
Skeletal Staining
For skeletal preparations, mice were fixed in 99.5% ethanol after removal of the skin and viscera. Cartilage tissues were stained with 0.015% alcian blue and 20% acetic acid in 75% ethanol for three days at 37°C. After dehydration with 99.5% ethanol for three days, bones were stained with 0.002% alizarin red in 1% KOH. Then skeletons were cleared in 1% KOH for several weeks. For P14 mice, soft tissues were dissolved in 2% KOH before alizarin red staining.
Nile Blue Staining
For the study of apoptosis of hindlimbs at E13.5 and E14.5, Nile blue (NB) staining was performed on the basis of a previously described protocol,23 except that staining was performed at 37°C (not room temperature). Apoptosis was determined by NB-stained (deceased) cells. After rinsing in Tyrode solution, hindlimbs of control (WT and heterozygous littermates) and homozygous mice were evaluated. Photographs of dorsal aspects were taken with a VHX-1000 digital microscope. Experiments were repeated three times, and reproducible representative results are presented.
Statistical Analysis
Statistical analyses were performed with the use of non-repeated-measures ANOVA followed by Dunnett's post hoc test. The results are given as mean ± standard deviation, and the threshold p value for statistical significance was 0.01.
Results
Identification of Homozygous SMOC1 Mutations
We have previously mapped the MLA locus to a 422 kb region at 10p11.23 by analyzing three families (one Japanese family [A] and two Lebanese families [B and C]). This region contained only one gene, MPP7, in which no mutations were found.12 After a new Turkish family (X) was added to the analysis, the MLA locus was again searched by homozygosity mapping to the consanguineous families (X, B, and C) and haplotype mapping to family A for detection of compound-heterozygous mutations; however, we could not detect any common regions among the four families. We then focused on identifying common regions in any three of the four families to allow for locus heterogeneity (Table S3).
A locus at 14q24.1-q24.2, which showed the highest LOD score (3.936) among the candidate regions larger than 2.0 Mb, was highlighted among families A, C, and X. This locus was analyzed with the use of additional microsatellite markers, and a 3.0 Mb region containing 24 genes was identified (Figures 1A and 1B). A total of 14 genes were sequenced, and homozygous mutations were found in SMOC1: c.718C>T (p.Gln240X) in family A, c.664+1G>A in family C, and c.378+1G>A in family X (Figures 1C and 1D). All of these homozygous mutations were cosegregated with the disease phenotype, and the parents of the individuals with these mutations were heterozygous carriers (Figure 1C). We could not find any mutations in SMOC1 in family B, in which MLA is unlinked to the 14q24.1-q24.2 locus. Interestingly, in family A haplotypes of paternal and maternal alleles, each having the same mutation, are completely different (data not shown), suggesting that the same mutation may have occurred in separate events. The c.718C>T mutation was not detected in 289 healthy Japanese controls, including 100 Okinawa islanders. The other two mutations were not detected in ethnically matched controls (54 Lebanese and 99 Turkish subjects, respectively), nor in 289 Japanese controls. The two splice-donor-site mutations (c.664+1G>A and c.378+1G>A) are predicted to abolish a donor site, as predicted by ESEfinder, NetGene2, HSF2.4.1, SpliceView, and BDGP analysis (Table S4). Thus, the three mutations are likely to lead to a loss of functional SMOC1.
Figure 1.
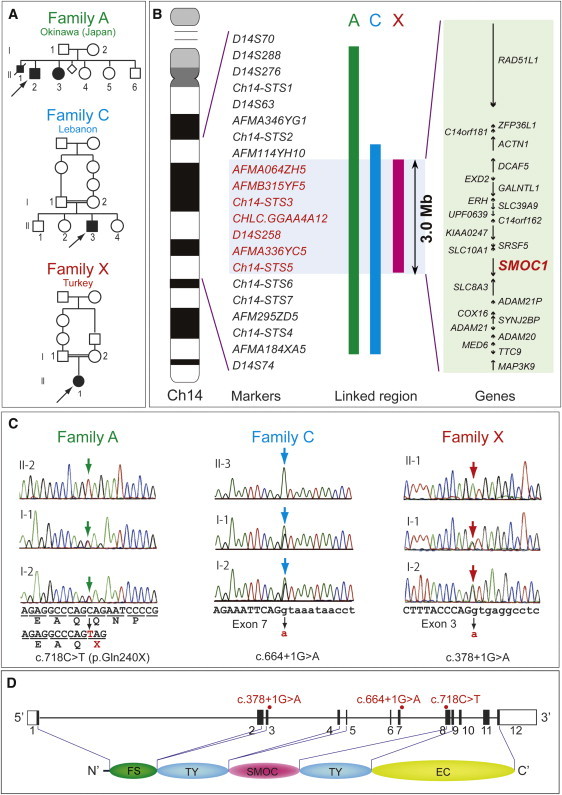
Genetic Analysis of Three Families with Members Affected by Microphthalmia with Limb Anomalies
(A) Pedigrees of the three families.
(B) Linkage analysis with SNPs and microsatellite markers on chromosome 14. From left to right: chromosome ideogram, genetic markers, linked regions of the three families, and genes mapped to the shortest overlapping linked region (between AFM114YH10 and Ch14-STS6 [UCSC coordinates, Feb. 2009: chromosome 14: 68,388,190–71,347,908 bp]).
(C) Sequences of mutations identified in each family. Affected patients in family A have a homozygous nonsense mutation (c.718C>T). Patients in families C and X have distinct homozygous splice-donor site mutations (c.664+1G>A and c.378+1G>A, respectively). For all mutations, parents of affected patients are heterozygous carriers, without exception. Sequences of the exon and intron are presented in upper and lower cases, respectively.
(D) At the top is a depiction of a schematic representation of SMOC1 consisting of 12 exons (UTR and coding exons are indicated by open and filled rectangles, respectively). The locations of three mutations are indicated by red dots. At the bottom, the functional domains of SMOC1 are depicted. Abbreviations are as follows: FS, the follistatin-like domain; TY, the thyroglobulin-like domain; SMOC, the domain unique to SMOC; and EC, the extracellular calcium-binding domain.
Smoc1 Expression in the Developing Eye and Limb in Mice
For the examination of Smoc1 expression in the developing eye and limb, whole-mount in situ hybridization of mouse embryos was performed. Smoc1 was expressed in the forebrain, midbrain, hindbrain, pharyngeal arch, somites, and forelimb buds at E9.5 (Figure 2A). At E10.5, Smoc1 expression was observed in the optic stalk (Figure 2B), and at E11.5, expression was localized to the closure site of the optic cup (Figure 2C). Expression of Smoc1 in developing limbs between E10.5 and E11.5 was observed in both dorsal and ventral regions, with a broader pattern of expression in dorsal regions, but expression was not detected in the most anterior, posterior, and distal parts of limb buds (Figures 2D and 2E). Expression coinciding with chondrogenic condensation was observed at E12.5 (Figure 2F), and expression then became restricted to future synovial joint regions at E13.5 (Figure 2G). This dynamic expression suggests that Smoc1 plays a critical role in ocular and limb development.
Figure 2.
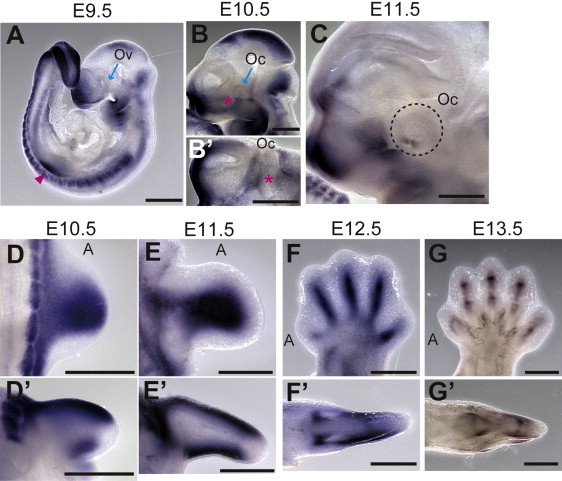
Smoc1 Expression in Mouse Embryos
Lateral views of embryos (A–C) and a ventral view of the left part of the head (B′, lateral view is shown at the top).
(A) At E9.5, Smoc1 was expressed in the forebrain, midbrain, hindbrain, pharyngeal arch, somites, and forelimb buds (magenta arrowhead), but not in the optic vesicle (Ov, blue arrow).
(B and B′) Expression in the optic stalk became evident at E10.5 (magenta asterisks), but was not evident in the optic cup (Oc, blue arrow).
(C) Expression was restricted to the closure site of the optic cup (dashed circle) at E11.5.
(D–G) Dorsal and (D′–G′) posterior view of the right hindlimbs (dorsal view is shown at the top in D′–G′). The anterior side is indicated by an A. (D and D′) At E10.5, Smoc1 was more widely expressed in the dorsal part of the limb bud than in the ventral part. Smoc1 expression is undetected in the most anterior, posterior, and distal parts of the limb bud. (E and E′) At E11.5, ventral expression was broader than that in the previous stage. (F and F′) At E12.5, expression was detected in areas consistent with chondrogenic condensation. (G and G′) At E13.5, Smoc1 expression became restricted to future joint regions. Scale bar represents 500 μm.
Ocular and Limb Anomalies in Smoc1 Null Mice
To investigate the pathological basis of MLA due to the loss of SMOC1 function, we obtained Smoc1 mutant mice, PV384.17 PV384 mice possess gene-trap insertions in the Smoc1 locus and in three other loci. After PV384 mice were bred with C57BL/6J or ICR mice, we obtained three independent lines (no. 1 to no. 3), each with a sole insertion in intron 1 of Smoc1 (Figure S1). We mainly analyzed line 1, but we confirmed similar phenotypes in lines 2 and 3. Heterozygous mutant mice (Smoc1Tp/+) were healthy and fertile. Homozygous mice (Smoc1TP/TP) were null mutants, as they showed no native transcript of Smoc1 (Figure S1E). Homozygous mice were viable at P0; however, they did not survive beyond the first 3 wks of life (Figure 3B). Their growth was retarded in comparison to WT and heterozygous littermates at P0 and P14 (Figures 3A and 3C). Developmental defects in eyes and optic nerves were evident at E14.5. Homozygous mice had relatively small eyes, and histological examinations revealed aplasia or hypoplasia of optic nerves (in 10 of 12 optic nerves), atrophy of the anteroventral part of the retina (in 11 of 12 eyes), and extension of the retinal pigmented epithelium (RPE) to the optic nerve (in 10 of 12 eyes) (Figures 3D–3I). These abnormalities were also observed at P0 (aplasia or hypoplasia of optic nerves [in 7 of 10 optic nerves], retinal atrophy [in 6 of 6 eyes], and RPE extension [in 3 of 6 eyes with identifiable optic nerves]) (Figures 3J–3M). WT or heterozygous littermates did not show any such abnormalities, except that a few eyes of heterozygous mice showed extension of the RPE at E14.5, but not at P0 (in 2 of 10 and 0 of 12 eyes, respectively). Toluidine blue (TB) staining showed ganglion cell layers that were thinned and irregular to varying degrees in homozygous mice, suggesting a reduced number of retinal ganglion cells (Figures 3J–3K′). Thus, Smoc1 is required for axon sprouting, elongation, or maintenance of retinal ganglion cells.24 Hypoplasia of optic nerves was further quantitatively confirmed by macroscopic examination: the average diameter of optic nerves of homozygous mice was significantly smaller than that of WT and heterozygous littermates at P0 and P14 (Figures 3L–3Q). These data clearly demonstrate that loss of Smoc1 in mice affects development of the body, retina, and optic nerves, in a manner similar to that seen in MLA patients.3,4
Figure 3.
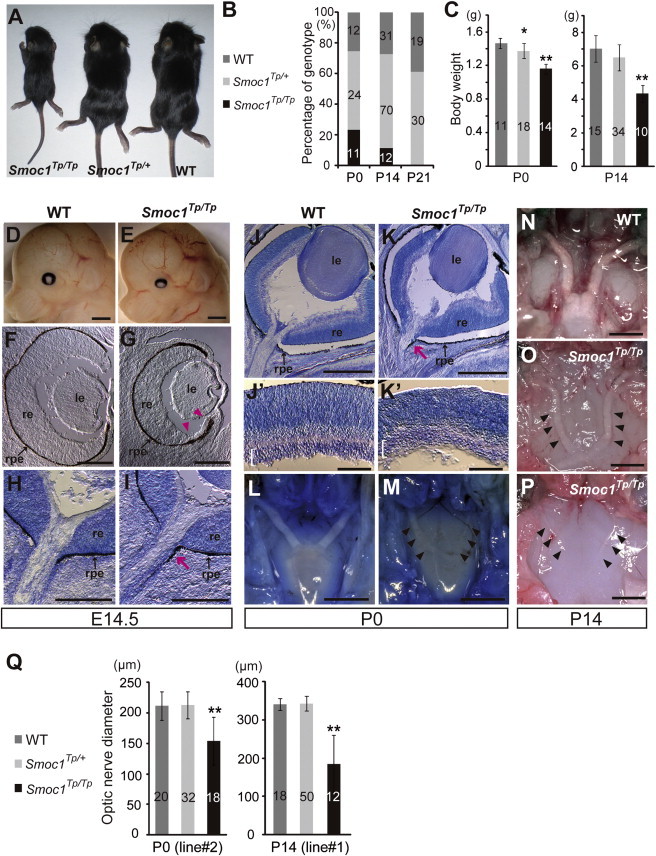
Growth and Ocular Phenotypes of Smoc1 Null Mice
(A) Representative Smoc1Tp/Tp mouse, showing a small body in comparison to Smoc1Tp/+ and WT littermates.
(B) Genotypes of living pups during the first 3 wk of life.
(C) Body weight of pups of each genotype at P0 (left panel) and P14 (right panel).
(D and E) Relatively small eyes were evident in Smoc1Tp/Tp mice in comparison to WT mice.
(F–K′) Coronal sections of eyes at E14.5 (F–I) and P0 (J–K′) with TB staining (H, I, and J–K′). (F-I) Atrophy of the anteroventral part of the retina (G, magenta arrowheads, dorsal view shown at the top), hypoplastic optic nerve, and extension of the RPE to the optic nerve (I, magenta arrow) in Smoc1Tp/Tp mice at E14.5. (J and K) Hypoplastic optic nerve and RPE extension in Smoc1Tp/Tp mice at P0 (K, magenta arrow). Note that sections in which optic nerves appeared most thick are presented in (H–K). (J′–K′) In higher-magnification views of (J and K), a thinned and irregular ganglion cell layer (white brackets) was observed in Smoc1Tp/Tp mice. Abbreviations are as follows: le, lens; re, retina; rpe, retinal pigmented epithelium.
(L–P) Ventral views of the brain showing optic nerves at P0 (L and M) and P14 (N–P), showing various degrees of optic nerve hypoplasia.
(Q) Optic nerve diameter. Optic nerves were significantly hypoplastic in Smoc1Tp/Tp mice in comparison to WT and Smoc1Tp/+ littermates.
The numbers of pups (B and C) or eyes (Q) corresponding to each genotype are indicated within bars. Error bars indicate standard deviation: ∗p < 0.01, compared with WT. ∗∗p < 0.01, compared with WT and Smoc1Tp/+. Scale bars represent 1 mm (D, E, and L–P), 200 μm (F–I), 500 μm (J and K), and 100 μm (J′ and K′).
Newborn homozygous mice could be readily identified by their hindlimb syndactyly and pes valgus, whereas no abnormalities were observed in WT and heterozygous pups (Figure 4 and Table 1). Interestingly, the severity of syndactyly varied between mouse lines: line 1 exclusively showed soft tissue syndactyly, whereas line 2 frequently showed four digits (Figures 4F and 4J). Skeletal preparations with alcian blue and alizarin red revealed that the foot with four digits had four phalanx and five metatarsals with fusion to each other (Figure 4K). Thus the Smoc1 null mutation resulted in a spectrum of phenotypes, from soft tissue syndactyly to four fused digits, probably due to different genetic backgrounds. Bowed tibiae and hypoplastic fibulae were also consistently observed in homozygous mice (Figures 4H and 4L). The articulation between tibia/fibula and calcanea of homozygous mice appeared malpositioned (Figures 4G and 4K), which might contribute to pes valgus. At P14, soft tissue syndactyly was also evident in most forelimbs of homozygous mice (Figures 4M–4O). Moreover, hindlimbs of homozygous mice showed synostosis between the 4th and 5th metatarsals (Figure 4T), which is observed in both the hands and the feet of MLA patients. Thus, many limb anomalies of MLA patients were recapitulated in Smoc1 null mice (Table S1).
Figure 4.
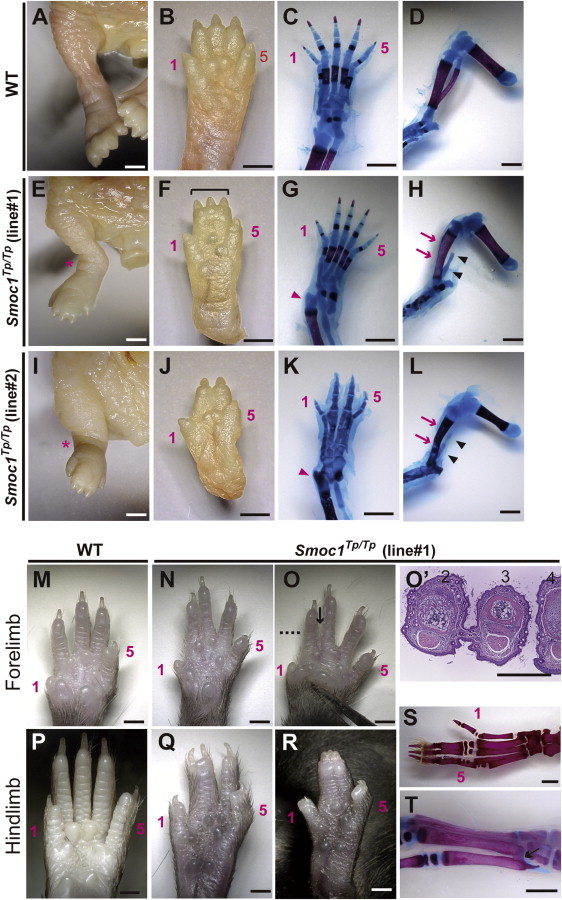
Limb Phenotypes of Smoc1 Null Mice
Limbs of WT (A–D, M, and P) and Smoc1Tp/Tp mice (E–L, N–O′, and Q–T) at P0 (A–L) and P14 (M–T). Digit identities are indicated by the numbers 1 (thumb, anterior) and 5 (little finger, posterior). Skeletal staining with alcian blue and alizarin red is presented (C, D, G, H, K, L, S, and T). Smoc1Tp/Tp mice showed pes valgus (E and I), soft tissue syndactyly (F and G), and four digits with metatarsal fusion (J and K). Malposition of the articulation between the tibia/fibula and the calcanea (G and K, magenta arrowheads), bowed tibia (magenta arrows), and hypoplastic fibula (arrowheads) of Smoc1Tp/Tp mice (H and L) were observed. 2/3 soft tissue syndactyly (N) and 2/3 webbing (O) were evident in forelimbs of Smoc1Tp/Tp mice. (O′) A transverse section taken at the level indicated by the dashed line in (O) showed 2/3 webbing. 2/3 syndactyly (Q), 2/3/4 syndactyly (R), synostosis between the 2nd and 3rd proximal phalanx and metatarsals (S), and synostosis between the 4th and 5th metatarsals (T, arrow), observed in the hindlimbs of Smoc1Tp/Tp mice. Scale bars represent 1 mm (A–O and P–T) or 500 μm (O′).
Table 1.
Limb Abnormalities in Smoc1Tp/Tp Mutants
| Genotype | Talipes Valgus (No. of Affected/Total No. of Pups) | Forelimb Abnormalities (No. of Limbs) |
Hindlimb Syndactyly (No. of Limbs) |
Other External Abnormalities (No. of Pups) | 4th and 5th Metatarsal Fusion (No. of Affected/Total No. of Limbs) | ||||
|---|---|---|---|---|---|---|---|---|---|
| None | 2/3a | 3/4b | 2/3/4c | 4 Digits | |||||
| Postnatal Day 0 | |||||||||
| Smoc1Tp/+ (line 1, C57BL/6J) | 0/42 | 0 | 84 | 0 | 0 | 0 | 0 | ||
| Smoc1Tp/+ (line 2, ICR mixed) | 0/38 | 0 | 76 | 0 | 0 | 0 | 0 | ||
| Smoc1Tp/Tp (line 1, C57BL/6J) | 10/10 | 0 | 3 | 0 | 3 | 12 | 2 | ||
| Smoc1Tp/Tp (line 2, ICR mixed) | 13/17 | 1d | 1 | 1 | 9 | 4 | 19 | cleft palate (3) | |
| Postnatal Day 14 | |||||||||
| Smoc1Tp/+ (line 1, C57BL/6J) | 0/70 | 0 | 140 | 0 | 0 | 0 | 0 | ||
| Smoc1Tp/Tp (line 1, C57BL/6J) | 11/11 | 18e | 2 | 7 | 3 | 8 | 2 | hypoplastic thumbs (5) | 9/10f |
Syndactyly between the 2nd and 3rd digits.
Syndactyly between the 3rd and 4th digits.
Syndactyly between the 2nd, 3rd, and 4th digits.
2/3 soft tissue syndactyly.
Eleven limbs showed 2/3 webbing, four limbs showed 2/3 soft tissue syndactyly, and one limb showed 3/4 syndactyly.
Based on examination of skeletal preparations.
Reduced Interdigital Apoptosis and Disturbed BMP Signaling
Among the various abnormalities caused by loss of Smoc1 function, we focused on soft tissue syndactyly, which was commonly observed in both fore- and hindlimbs of null mutants. It is possible that the syndactyly is caused by failed apoptotic regression of the interdigital mesenchyme. To examine this hypothesis, hindlimbs were stained with NB sulfate at E13.5 and E14.5, the time when interdigital apoptosis is most evident. In control embryos (WT and heterozygous littermates), NB-stained apoptotic cells were identified in the interdigital mesenchyme, where regression of the interdigital webbing occurs in the distal region (Figures 5A and 5C). By contrast, the number of apoptotic cells in the mesenchyme between digits 2 and 3 and digits 3 and 4 was dramatically reduced in homozygous mice at E13.5 and E14.5, along with persistent webbing in the distal region (Figures 5B and 5D, magenta asterisk). BMP signaling is involved in apoptosis of the interdigital mesenchyme.25,26 Bmp2, Bmp7, and Msx2, a direct target of BMP signaling, were strongly expressed in the interdigital mesenchyme of control hindlimbs at both E12.5 and E13.5. However, the expression of these three genes was profoundly reduced and perturbed in hindlimbs of homozygous mice (Figures 5E–5J). These data suggest that inhibition of apoptosis is spatiotemporally correlated to reduced and/or disturbed expression of genes involved in BMP signaling in the interdigital mesenchyme.
Figure 5.
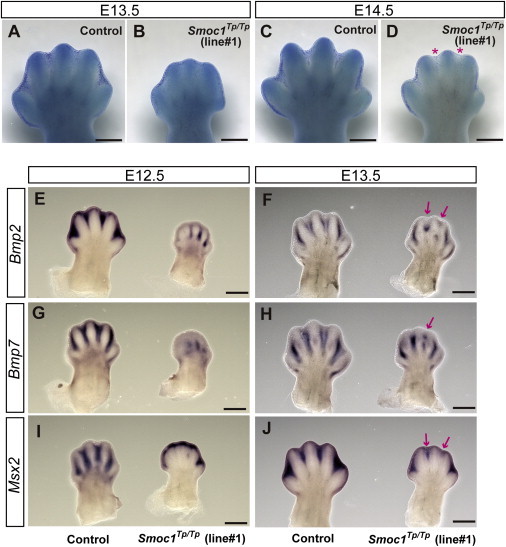
Reduced Apoptosis and Altered BMP Signaling in the Interdigital Mesenchyme of Smoc1 Null Mice
(A–D) NB staining of left hindlimbs at E13.5 (A and B) and E14.5 (C and D). In comparison to control embryos (WT and Smoc1Tp/+ littermates) (A and C), the number of NB-stained apoptotic cells in the interdigital mesenchyme of Smoc1Tp/Tp mice was dramatically reduced between digits 2 and 3 and digits 3 and 4 at both E13.5 and E14.5, and the webbing remained at a distal level (B and D, magenta asterisk).
(E–J) Whole-mount in situ hybridization of right hindlimbs at E12.5 (E, G, and I) and E13.5 (F, H, and J). At E12.5, interdigital expression of Bmp2, Bmp7, and Msx2 was profoundly delayed in the hindlimbs of Smoc1Tp/Tp mice, and their expression in the interdigital mesenchyme was apparently perturbed, even at E13.5 (magenta arrows). Scale bar represents 500 μm.
Discussion
In a previous report, we performed parametric linkage analysis with three families (families A, B, and C) and found 16 loci showing a LOD score (θ = 0.000) higher than 3.0. Additional microsatellite markers highlighted only one locus, 10p11.23.12 However, no mutations were found in the candidate gene MPP7.12 By recruiting a new family (family X) to this study, we successfully found homozygous mutations in SMOC1 in families A, C, and X. In family B, no SMOC1 mutations were found, indicating the genetic heterogeneity in MLA. Patients with SMOC1 mutations and Smoc1 null mice showed similar limb anomalies, such as oligodactyly, syndactyly, synostosis of 4th and 5th metacarpals, hypoplasia of fibula, and bowed tibia. Oligodactyly, syndactyly, and synostosis of 4th and 5th metacarpals are common in MLA patients.2–4 However, hypoplastic fibula and bowed tibia are less common in patients with MLA, as four out of 34 MLA patients showed these anomalies in the previous report.3 Although one patient with a SMOC1 mutation from family C did not show bowed tibia and hypoplastic fibula, these anomalies could be features specific to SMOC1 mutations. Further SMOC1 analysis of other MLA patients should delineate the phenotypic consequences caused by SMOC1 mutations.
Accumulating evidence suggests that BMP signaling plays crucial roles in early eye vesicle and limb patterning, skeletal formation, and apoptosis of the interdigital mesenchyme,25–29 and mutations involving BMP signaling cause human malformations including ocular, limb, and skeletal anomalies.7,30–33 Here, we present genetic evidence that SMOC1 is essential for ocular and limb development in humans and mice. Furthermore, Xenopus smoc can inhibit BMP signaling,11 suggesting that SMOC1/Smoc1 can also modulate BMP signaling in humans and mice. Indeed, we observed reduced and/or disturbed expression of genes involved in BMP signaling in the interdigital mesenchyme in Smoc1 null mice, and limb and ocular abnormalities associated with loss of Smoc1 function are consistent with phenotypic consequences of disturbed BMP signaling. Conditional inactivation of Bmp2 in the limb showed 3/4 syndactyly, and a similar deficiency of both Bmp2 and Bmp7 resulted in malformed fibulae in mice.25 Moreover, mice deficient in Fmn1, a repressor of BMP signaling, showed four digits, fused metatarsal bones, and an absence of fibulae in the hindlimbs,34 suggesting the importance of altered BMP signaling in these features. Concerning ocular phenotypes, haploinsufficiency of mouse Bmp4 resulted in a decreased number of ganglion layer cells and absence of the optic nerve similar to Smoc1 null mice,35 indicating that altered BMP signaling is also involved in the ocular phenotype. Interestingly, knockdown experiments of smoc by antisense morpholino in Xenopus showed absence or severe deformity of the eye and other anterior structures, which were accompanied by aberrant expression of otx2, tbx2 in the eye field.11 Mutations of OTX2 (MIM 600037) cause micropthalmia, syndromic 5 (MCOPS5 [MIM 610125]) in humans.36 Moreover, targeted disruption of Tbx2 resulted in a marked reduction in the size of the optic cup and a failure of optic nerve formation in mice.37 Thus, it is possible that loss of SMOC1 function could alter the expression of OTX2 and TBX2 (MIM 600747) by disturbing BMP signaling in human developing eyes.
It is unknown how the loss of functional SMOC1, a BMP antagonist, leads to reduced expression of genes involved in BMP signaling in the interdigital mesenchyme in Smoc1 null mice. In the case of Fmn1-deficient mice, the loss of the repressor of BMP signaling resulted in downregulation of Fgf4 and Shh and in upregulation of Gremlin expression at E10.5, and absence of apoptosis of the interdigital mesenchyme between the two middle digits at E13.5.34 Thus, there is a possibility that loss of SMOC1 could cause the imbalance among BMP, SHH, and FGF signaling, which would subsequently lead to reduced and/or disturbed expression of genes involved in BMP signaling in the interdigital mesenchyme. In fact, we observed reduced expression of Msx2 in the progressive zone of hindlimbs at E11.5 (Figure S2). Moreover, expression of Sox9, the initial cartilage condensation marker, showed abnormal limb patterning, suggesting that SMOC1 may affect BMP signaling even at early stages of limb development (Figure S3). Further examinations are required for understanding spatial and temporal actions of SMOC1/Smoc1 protein during limb development.
In conclusion, our data demonstrate that SMOC1/Smoc1 is an essential player in both ocular and limb development in humans and mice and give further support to the crucial roles of BMP signaling in these systems.
Acknowledgments
We would like to thank the patients and their families for their participation in this study. We thank Yoshiko Takahashi (Nara Institute of Science and Technology) and Atsushi Yamada (Showa University) for providing the Bmp2 and Sox9 probes; Elizabeth J. Robertson (University of Oxford) and Makoto Ishibashi (Kyoto University) for the Bmp7 probe; Robert E. Maxson, Jr. (University of Southern California Keck School of Medicine) for the Msx2 probe; Tomonori Hirose, Kazunori Akimoto, and Kazunori Sasaki (Yokohama City University) for providing useful information about mouse breeding, taking photos on a stereo microscope, and mRNA quantification; and Kohei Shiota and Sumiko Kimura (Kyoto University) for helpful comments about NB staining and limb anomalies. This work was supported by research grants from the Ministry of Health, Labour and Welfare (T. Furuichi, N. Miyake, N. Matsumoto, and H.S.) and the Japan Science and Technology Agency (N. Matsumoto), a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (T. Furuichi and N. Matsumoto), and a Grant-in-Aid for Young Scientist from the Japan Society for the Promotion of Science (K.N., H.D., N. Miyake, and H.S.). This work has been carried out at the Advanced Medical Research Center of Yokohama City University.
Contributor Information
Naomichi Matsumoto, Email: naomat@yokohama-cu.ac.jp.
Hirotomo Saitsu, Email: hsaitsu@yokohama-cu.ac.jp.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
BDGP, http://www.fruitfly.org/
ESEfinder 3.0, http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi?process=home
GenBank, http://www.ncbi.nlm.nih.gov/Genbank/
HSF2.4.1, http://www.umd.be/HSF/
NetGene2, http://www.cbs.dtu.dk/services/NetGene2/
Online Mendelian Inheritance in Man, http://www.ncbi.nlm.nih.gov/Omim
UCSC Genome Browser, http://genome.ucsc.edu/cgi-bin/hgGateway
SpliceView, http://zeus2.itb.cnr.it/∼webgene/wwwspliceview.html
References
- 1.Waardenburg P.J. Autosomally-recessive anophthalmia with malformations of the hands and feet. In: Waardenburg P.J., Franceschetti A., Klein D., editors. Genetics and Ophthalmology. Royal Van Gorcum; Assen, The Netherlands: 1961. p. 773. [Google Scholar]
- 2.Teiber M.L., Garrido J.A., Barreiro C.Z. Ophthalmo-acromelic syndrome: report of a case with vertebral anomalies. Am. J. Med. Genet. A. 2007;143A:2460–2462. doi: 10.1002/ajmg.a.31930. [DOI] [PubMed] [Google Scholar]
- 3.Garavelli L., Pedori S., Dal Zotto R., Franchi F., Marinelli M., Croci G.F., Bellato S., Ammenti A., Virdis R., Banchini G., Superti-Furga A. Anophthalmos with limb anomalies (Waardenburg opththalmo-acromelic syndrome): report of a new Italian case with renal anomaly and review. Genet. Couns. 2006;17:449–455. [PubMed] [Google Scholar]
- 4.Tekin M., Tutar E., Arsan S., Atay G., Bodurtha J. Ophthalmo-acromelic syndrome: report and review. Am. J. Med. Genet. 2000;90:150–154. doi: 10.1002/(sici)1096-8628(20000117)90:2<150::aid-ajmg12>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 5.Adler R., Canto-Soler M.V. Molecular mechanisms of optic vesicle development: complexities, ambiguities and controversies. Dev. Biol. 2007;305:1–13. doi: 10.1016/j.ydbio.2007.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeller R., López-Ríos J., Zuniga A. Vertebrate limb bud development: moving towards integrative analysis of organogenesis. Nat. Rev. Genet. 2009;10:845–858. doi: 10.1038/nrg2681. [DOI] [PubMed] [Google Scholar]
- 7.Bakrania P., Efthymiou M., Klein J.C., Salt A., Bunyan D.J., Wyatt A., Ponting C.P., Martin A., Williams S., Lindley V. Mutations in BMP4 cause eye, brain, and digit developmental anomalies: overlap between the BMP4 and hedgehog signaling pathways. Am. J. Hum. Genet. 2008;82:304–319. doi: 10.1016/j.ajhg.2007.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bornstein P., Sage E.H. Matricellular proteins: extracellular modulators of cell function. Curr. Opin. Cell Biol. 2002;14:608–616. doi: 10.1016/s0955-0674(02)00361-7. [DOI] [PubMed] [Google Scholar]
- 9.Vannahme C., Smyth N., Miosge N., Gösling S., Frie C., Paulsson M., Maurer P., Hartmann U. Characterization of SMOC-1, a novel modular calcium-binding protein in basement membranes. J. Biol. Chem. 2002;277:37977–37986. doi: 10.1074/jbc.M203830200. [DOI] [PubMed] [Google Scholar]
- 10.Gersdorff N., Müller M., Schall A., Miosge N. Secreted modular calcium-binding protein-1 localization during mouse embryogenesis. Histochem. Cell Biol. 2006;126:705–712. doi: 10.1007/s00418-006-0200-7. [DOI] [PubMed] [Google Scholar]
- 11.Thomas J.T., Canelos P., Luyten F.P., Moos M., Jr. Xenopus SMOC-1 inhibits BMP signaling downstream of receptor binding and is essential for post-gastrulation development in Xenopus. J. Biol. Chem. 2009;284:18994–19005. doi: 10.1074/jbc.M807759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamanoue H., Megarbane A., Tohma T., Nishimura A., Mizuguchi T., Saitsu H., Sakai H., Miura S., Toda T., Miyake N. A locus for ophthalmo-acromelic syndrome mapped to 10p11.23. Am. J. Med. Genet. A. 2009;149A:336–342. doi: 10.1002/ajmg.a.32656. [DOI] [PubMed] [Google Scholar]
- 13.Mégarbané A., Souraty N., Tamraz J. Ophthalmo-acromelic syndrome (Waardenburg) with split hand and polydactyly. Genet. Couns. 1998;9:195–199. [PubMed] [Google Scholar]
- 14.Cogulu O., Ozkinay F., Gündüz C., Sapmaz G., Ozkinay C. Waardenburg anophthalmia syndrome: report and review. Am. J. Med. Genet. 2000;90:173–174. [PubMed] [Google Scholar]
- 15.Miyake N., Kosho T., Mizumoto S., Furuichi T., Hatamochi A., Nagashima Y., Arai E., Takahashi K., Kawamura R., Wakui K. Loss-of-function mutations of CHST14 in a new type of Ehlers-Danlos syndrome. Hum. Mutat. 2010;31:966–974. doi: 10.1002/humu.21300. [DOI] [PubMed] [Google Scholar]
- 16.Gudbjartsson D.F., Thorvaldsson T., Kong A., Gunnarsson G., Ingolfsdottir A. Allegro version 2. Nat. Genet. 2005;37:1015–1016. doi: 10.1038/ng1005-1015. [DOI] [PubMed] [Google Scholar]
- 17.Keng V.W., Yae K., Hayakawa T., Mizuno S., Uno Y., Yusa K., Kokubu C., Kinoshita T., Akagi K., Jenkins N.A. Region-specific saturation germline mutagenesis in mice using the Sleeping Beauty transposon system. Nat. Methods. 2005;2:763–769. doi: 10.1038/nmeth795. [DOI] [PubMed] [Google Scholar]
- 18.Mamo S., Gal A.B., Bodo S., Dinnyes A. Quantitative evaluation and selection of reference genes in mouse oocytes and embryos cultured in vivo and in vitro. BMC Dev. Biol. 2007;7:14. doi: 10.1186/1471-213X-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parr B.A., Shea M.J., Vassileva G., McMahon A.P. Mouse Wnt genes exhibit discrete domains of expression in the early embryonic CNS and limb buds. Development. 1993;119:247–261. doi: 10.1242/dev.119.1.247. [DOI] [PubMed] [Google Scholar]
- 20.Saitsu H., Ishibashi M., Nakano H., Shiota K. Spatial and temporal expression of folate-binding protein 1 (Fbp1) is closely associated with anterior neural tube closure in mice. Dev. Dyn. 2003;226:112–117. doi: 10.1002/dvdy.10203. [DOI] [PubMed] [Google Scholar]
- 21.Tamplin O.J., Kinzel D., Cox B.J., Bell C.E., Rossant J., Lickert H. Microarray analysis of Foxa2 mutant mouse embryos reveals novel gene expression and inductive roles for the gastrula organizer and its derivatives. BMC Genomics. 2008;9:511. doi: 10.1186/1471-2164-9-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suzuki D., Yamada A., Amano T., Yasuhara R., Kimura A., Sakahara M., Tsumaki N., Takeda S., Tamura M., Nakamura M. Essential mesenchymal role of small GTPase Rac1 in interdigital programmed cell death during limb development. Dev. Biol. 2009;335:396–406. doi: 10.1016/j.ydbio.2009.09.014. [DOI] [PubMed] [Google Scholar]
- 23.Kimura S., Shiota K. Sequential changes of programmed cell death in developing fetal mouse limbs and its possible roles in limb morphogenesis. J. Morphol. 1996;229:337–346. doi: 10.1002/(SICI)1097-4687(199609)229:3<337::AID-JMOR8>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 24.Sernagor E., Eglen S.J., Wong R.O. Development of retinal ganglion cell structure and function. Prog. Retin. Eye Res. 2001;20:139–174. doi: 10.1016/s1350-9462(00)00024-0. [DOI] [PubMed] [Google Scholar]
- 25.Bandyopadhyay A., Tsuji K., Cox K., Harfe B.D., Rosen V., Tabin C.J. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2006;2:e216. doi: 10.1371/journal.pgen.0020216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robert B. Bone morphogenetic protein signaling in limb outgrowth and patterning. Dev. Growth Differ. 2007;49:455–468. doi: 10.1111/j.1440-169X.2007.00946.x. [DOI] [PubMed] [Google Scholar]
- 27.Dudley A.T., Lyons K.M., Robertson E.J. A requirement for bone morphogenetic protein-7 during development of the mammalian kidney and eye. Genes Dev. 1995;9:2795–2807. doi: 10.1101/gad.9.22.2795. [DOI] [PubMed] [Google Scholar]
- 28.Khokha M.K., Hsu D., Brunet L.J., Dionne M.S., Harland R.M. Gremlin is the BMP antagonist required for maintenance of Shh and Fgf signals during limb patterning. Nat. Genet. 2003;34:303–307. doi: 10.1038/ng1178. [DOI] [PubMed] [Google Scholar]
- 29.Furuta Y., Hogan B.L. BMP4 is essential for lens induction in the mouse embryo. Genes Dev. 1998;12:3764–3775. doi: 10.1101/gad.12.23.3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Asai-Coakwell M., French C.R., Berry K.M., Ye M., Koss R., Somerville M., Mueller R., van Heyningen V., Waskiewicz A.J., Lehmann O.J. GDF6, a novel locus for a spectrum of ocular developmental anomalies. Am. J. Hum. Genet. 2007;80:306–315. doi: 10.1086/511280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tassabehji M., Fang Z.M., Hilton E.N., McGaughran J., Zhao Z., de Bock C.E., Howard E., Malass M., Donnai D., Diwan A. Mutations in GDF6 are associated with vertebral segmentation defects in Klippel-Feil syndrome. Hum. Mutat. 2008;29:1017–1027. doi: 10.1002/humu.20741. [DOI] [PubMed] [Google Scholar]
- 32.Wyatt A.W., Osborne R.J., Stewart H., Ragge N.K. Bone morphogenetic protein 7 (BMP7) mutations are associated with variable ocular, brain, ear, palate, and skeletal anomalies. Hum. Mutat. 2010;31:781–787. doi: 10.1002/humu.21280. [DOI] [PubMed] [Google Scholar]
- 33.Ye M., Berry-Wynne K.M., Asai-Coakwell M., Sundaresan P., Footz T., French C.R., Abitbol M., Fleisch V.C., Corbett N., Allison W.T. Mutation of the bone morphogenetic protein GDF3 causes ocular and skeletal anomalies. Hum. Mol. Genet. 2010;19:287–298. doi: 10.1093/hmg/ddp496. [DOI] [PubMed] [Google Scholar]
- 34.Zhou F., Leder P., Zuniga A., Dettenhofer M. Formin1 disruption confers oligodactylism and alters Bmp signaling. Hum. Mol. Genet. 2009;18:2472–2482. doi: 10.1093/hmg/ddp185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang B., Smith R.S., Peters M., Savinova O.V., Hawes N.L., Zabaleta A., Nusinowitz S., Martin J.E., Davisson M.L., Cepko C.L. Haploinsufficient Bmp4 ocular phenotypes include anterior segment dysgenesis with elevated intraocular pressure. BMC Genet. 2001;2:18. doi: 10.1186/1471-2156-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ragge N.K., Brown A.G., Poloschek C.M., Lorenz B., Henderson R.A., Clarke M.P., Russell-Eggitt I., Fielder A., Gerrelli D., Martinez-Barbera J.P. Heterozygous mutations of OTX2 cause severe ocular malformations. Am. J. Hum. Genet. 2005;76:1008–1022. doi: 10.1086/430721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Behesti H., Papaioannou V.E., Sowden J.C. Loss of Tbx2 delays optic vesicle invagination leading to small optic cups. Dev. Biol. 2009;333:360–372. doi: 10.1016/j.ydbio.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.