

Abstract
Cowden syndrome (CS) and Bannayan-Riley-Ruvalcaba syndrome are allelic, defined by germline PTEN mutations, and collectively referred to as PTEN hamartoma tumor syndrome. To date, there are no existing criteria based on large prospective patient cohorts to select patients for PTEN mutation testing. To address these issues, we conducted a multicenter prospective study in which 3042 probands satisfying relaxed CS clinical criteria were accrued. PTEN mutation scanning, including promoter and large deletion analysis, was performed for all subjects. Pathogenic mutations were identified in 290 individuals (9.5%). To evaluate clinical phenotype and PTEN genotype against protein expression, we performed immunoblotting (PTEN, P-AKT1, P-MAPK1/2) for a patient subset (n = 423). In order to obtain an individualized estimation of pretest probability of germline PTEN mutation, we developed an optimized clinical practice model to identify adult and pediatric patients. For adults, a semiquantitative score—the Cleveland Clinic (CC) score—resulted in a well-calibrated estimation of pretest probability of PTEN status. Overall, decreased PTEN protein expression correlated with PTEN mutation status; decreasing PTEN protein expression correlated with increasing CC score (p < 0.001), but not with the National Comprehensive Cancer Network (NCCN) criteria (p = 0.11). For pediatric patients, we identified highly sensitive criteria to guide PTEN mutation testing, with phenotypic features distinct from the adult setting. Our model improved sensitivity and positive predictive value for germline PTEN mutation relative to the NCCN 2010 criteria in both cohorts. We present the first evidence-based clinical practice model to select patients for genetics referral and PTEN mutation testing, further supported biologically by protein correlation.
Introduction
Cowden syndrome (CS [MIM 158350]), presenting in adulthood, and Bannayan-Riley-Ruvalcaba syndrome (BRRS [MIM 153480]),1 a pediatric syndrome, show overlapping clinical features and may present with multisystem disease, including macrocephaly, various cancers, and skin, neurologic, and gastrointestinal manifestations.2,3 Because subsets of these two syndromes, together with other seemingly unrelated clinical syndromes,4 share a common etiology germline PTEN (MIM 601728) mutation,2 they are allelic and collectively referred to as PTEN hamartoma tumor syndrome (PHTS).5 Inheritance of this disorder is autosomal dominant, and penetrance is believed to be high (around 80%).5 The PTEN tumor suppressor gene, located on 10q23.3, encodes a dual-specificity phosphatase that can dephosphorylate both protein6 and phospholipid substrates.7
We formulated the International Cowden Consortium (ICC) operational diagnostic criteria8 14 years ago to select families and affected individuals for purposes of identifying the specific mutated gene. Over the last 10 years, we have continually revised this set of criteria for referral of patients for clinical germline PTEN mutation testing,9,10 with early expert opinion and clinical data derived from families studied from initial consortium studies suggesting that 85% of patients with CS had an identifiable PTEN mutation.2,11 The most recent National Comprehensive Cancer Network (NCCN) 2010 criteria,12 based primarily on these operational criteria, are useful, but they also have several disadvantages. These include the inability to quantitatively evaluate individual patients for their probabilities of testing positive for a PTEN mutation. Additionally, with the multisystem involvement of Cowden syndrome and rapid expansion of the clinical spectrum to include phenotypes such as autism13 and polyposis syndrome,14 the complexity of the current NCCN criteria involving multiple possible combinations of major and minor criteria render them challenging for use outside of a specialist community. Further, the current NCCN criteria is not refined for adult and pediatric populations, considering that CS and BRRS are clinically and epidemiologically distinct as a result of age-related penetrance and variable expression, even of the common underlying PTEN mutation.3 Finally, we note that there are no existing criteria based on large prospective patient cohorts for selection of patients for PTEN mutation testing.
Concurrently, advances in the understanding of the biology of PTEN in a patient-oriented setting have been inhibited by the lack of a common reference for measuring severity of phenotypes arising from PTEN protein deficiency or dysfunction. This is crucial, given insights supported by increasing laboratory evidence on the importance of tumor suppressor gene dosage. Small changes in the expression levels of tumor suppressor genes have been proposed to influence susceptibility to cancer,15 and there is experimental support from animal experiments to support a view that subtle variations of Pten dosage may also result in increased cancer susceptibility.16 Nonetheless, to date, there has been no evidence in the setting of a human population to support this view. From a molecular angle, as a lipid phosphatase that dephosphorylates phosphatidylinositol-3,4,5-triphosphate (PIP3) to phosphatidylinositol-4,5-phosphate (PIP2), PTEN is the key negative regulator of the phosphatidylinositol-3-kinase (PI3K) signal transduction cascade, inhibiting pathways of growth, proliferation, and survival.17
PTEN inactivation is associated with the activation of the AKT/mTOR signaling pathway when PTEN's lipid phosphatase is most relevant and with the mitogen-activated protein kinase (MAPK) signaling pathways with activated MAPK1/2 when PTEN's protein phosphatase pathway is more relevant.18–20
We thus conducted a prospective multicenter, multinational study, collecting phenotypic data on patients who met relaxed criteria for CS,21 in order to quantitatively assess both adult and pediatric patients for a priori risk of PTEN germline mutation in two large patient cohorts with a semiquantitative score. Subsequently, we evaluated whether there was correlation between PTEN protein dosage and the clinical score.
Subjects and Methods
Research Participants
For the two independent cohorts from the Cleveland Clinic (CC) and The Ohio State University (OSU; C.E., Principal Investigator), a total of 3042 patients was recruited into a protocol approved by the Institutional Review Boards for Protection of Human Subjects from both institutions (CC: 2005–2010; OSU: 2000–2006). Individuals who were probands and who met relaxed International Cowden Consortium operational criteria for CS (pathognomonic criteria, or at least two criteria, either major or minor)9,10 (Table 1) were recruited prospectively for these cohorts (Table 2). These patients were recruited from both community and academic medical centers throughout North America, Europe, and Asia. Upon providing informed consent, checklists to document presence or absence of specific features were completed by specialist genetic counselors or physicians concurrently with submission of samples. Specialist genetics staff reviewed all checklists and corresponded with the enrolling center; if necessary, further primary documentation of medical records was obtained for phenotype confirmation with patient consent.
Table 1.
Operational Criteria for Cowden Syndrome without Family History of Known PTEN Mutation
| Pathognomonic Criteria |
|---|
| Adult Lhermitte-Duclos disease (cerebellar tumors) |
| Mucocutaneous lesionsa |
| - Facial trichilemmomas, any numbera (at least two biopsy-proven trichilemmomasb) |
| - Acral keratoses |
| - Papillomatous papules |
| Mucosal lesions |
| Autism spectrum disorder and macrocephalyb |
| Major Criteria |
| Breast cancer |
| Thyroid cancer (nonmedullary) |
| Macrocephaly (megalocephaly) (i.e., 97th percentile and above) |
| Endometrial cancer |
| Mucocutaneous lesionsb |
| - One biopsy-proven trichilemmoma |
| - Multiple palmoplantar keratoses |
| - Multifocal cutaneous facial papules |
| - Macular pigmentation of glans penis |
| Multiple GI hamartomas or ganglioneuromasb |
| Minor Criteria |
| Other thyroid lesions (e.g., adenoma, multinodular goiter) |
| Mental retardation (i.e., IQ of 75 and below) |
| Gastrointestinal hamartomasa (single gastrointestinal hamartoma or ganglioneuromab) |
| Fibrocystic disease of the breast |
| Lipomas |
| Fibromas |
| Genitourinary tumors (especially renal cell carcinoma) |
| Genitourinary malformationsa |
| Uterine fibroids |
| Autism spectrum disorderb |
Present in this section as defined by ICC criteria only.
Present in this section as defined by NCCN 2010 criteria only.
Table 2.
Baseline Data for the Study Cohorts
| CC Adult Cohort | % | CC Adult Mutation Cohort | % | CC Pediatric Cohort | % | CC Pediatric Mutation Cohort | % | OSU Cohort | % | PTEN Mutation Cohort (OSU) | % | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number | 2007 | 105 | 92 | 28 | 943 | 157 | ||||||
| Age, median (range) | 55.9 (18.0–98.2) | 43.9 (3.0–78) | 8.2 (1.8–16.9) | 8.3 (3.0–16.9) | 48.5 (1.2–91.7) | 36.1 (1.7–73.1) | ||||||
| Gender, male | 117 | 5.8 | 34 | 32.4 | 59 | 64 | 21 | 75 | 164 | 17.4 | 70 | 44.6 |
| Gender, female | 1890 | 94.2 | 71 | 67.6 | 33 | 36 | 7 | 25 | 779 | 82.6 | 87 | 55.4 |
| Neurological | ||||||||||||
| Macrocephaly, presence | 538 | 26.8 | 79 | 75.2 | 87 | 95 | 28 | 100 | 385 | 40.8 | 135 | 86 |
| Extreme macrocephaly (male, ≥63 cm) | 15 | 13 | 11 | 32 | 2 | 3 | 2 | 10 | ||||
| Extreme macrocephaly (female, ≥60 cm) | 67 | 4 | 24 | 34 | 4 | 12 | 2 | 29 | ||||
| Lhermitte Duclos disease | 17 | 0.8 | 9 | 8.6 | 1 | 1 | 0 | 0 | 24 | 2.5 | 9 | 5.7 |
| Autism or developmental delay | 41 | 2 | 13 | 12.4 | 78 | 85 | 23 | 82 | 115 | 12.2 | 34 | 21.7 |
| Breast and Gynecological | ||||||||||||
| Invasive breast cancer | ||||||||||||
| <30 | 29 | 1.5 | 1 | 1.4 | 0 | 0 | 0 | 0 | 7 | 0.9 | 2 | 2.3 |
| 30–39 | 164 | 8.7 | 6 | 8.5 | 0 | 0 | 0 | 0 | 76 | 9.8 | 6 | 6.9 |
| 40–49 | 475 | 25.1 | 13 | 18.3 | 0 | 0 | 0 | 0 | 160 | 20.5 | 10 | 11.5 |
| ≥50 | 625 | 33.1 | 14 | 19.7 | 0 | 0 | 0 | 0 | 179 | 23 | 8 | 9.2 |
| Fibrocystic breast disease | 849 | 44.9 | 30 | 42.3 | 0 | 0 | 0 | 0 | 269 | 34.5 | 23 | 26.4 |
| Endometrial cancer | 74 | 9.5 | 12 | 13.8 | ||||||||
| <30 | 11 | 0.6 | 1 | 1.4 | 0 | 0 | 0 | 0 | 2 | 0.3 | 1 | 1.1 |
| 30–39 | 23 | 1.2 | 2 | 2.8 | 0 | 0 | 0 | 0 | 7 | 0.9 | 3 | 3.4 |
| 40–49 | 50 | 2.6 | 5 | 7 | 0 | 0 | 0 | 0 | 21 | 2.7 | 7 | 8 |
| ≥50 | 171 | 9 | 5 | 7 | 0 | 0 | 0 | 0 | 44 | 5.6 | 1 | 1.1 |
| Fibroids | 794 | 42 | 27 | 38 | 0 | 0 | 0 | 0 | 290 | 37.2 | 18 | 20.7 |
| Gastrointestinal | ||||||||||||
| Colorectal cancer | ||||||||||||
| <30 | 2 | 0.1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 30–39 | 3 | 0.1 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 0.1 | 0 | 0 |
| 40–49 | 15 | 0.7 | 3 | 2.9 | 0 | 0 | 0 | 0 | 5 | 0.5 | 2 | 1.3 |
| ≥50 | 33 | 1.6 | 2 | 1.9 | 0 | 0 | 0 | 0 | 10 | 1.1 | 0 | 0 |
| Polyposis syndrome (≥5) | 74 | 3.7 | 36 | 34.3 | 2 | 2 | 2 | 7 | 168 | 17.8 | 51 | 32.5 |
| Intestinal hamartoma, single | 23 | 1.1 | 16 | 15.2 | 5 | 5 | 3 | 11 | 11 | 1.2 | 10 | 6.4 |
| Intestinal hamartoma, multiplea | 11 | 0.5 | 8 | 7.6 | 0 | 0 | 0 | 0 | ||||
| Intestinal ganglioneuroma, single | 14 | 0.7 | 9 | 8.6 | 2 | 2 | 2 | 7 | 11 | 1.2 | 5 | 3.2 |
| Intestinal ganglioneuroma, multiplea | 10 | 0.5 | 7 | 6.7 | 0 | 0 | 0 | 0 | ||||
| Glycogenic acanthosis | 21 | 1 | 16 | 15.2 | 1 | 1 | 1 | 4 | 14 | 1.5 | 9 | 5.7 |
| Skin | ||||||||||||
| Trichilemmomas, biopsy proven | 13 | 0.6 | 6 | 5.7 | 0 | 0 | 0 | 0 | 112 | 11.9 | 39 | 24.8 |
| Oral papillomas | 160 | 8 | 16 | 15.2 | 3 | 3 | 1 | 4 | 210 | 22.3 | 72 | 45.9 |
| Penile freckling | 22 | 18.8 | 10 | 29.4 | 16 | 27 | 5 | 24 | 61 | 37.2 | 38 | 54.3 |
| Acral keratoses | 128 | 6.4 | 28 | 26.7 | 5 | 5 | 2 | 7 | 54 | 5.7 | 16 | 10.2 |
| Arteriovenous malformations | 19 | 0.9 | 12 | 11.4 | 4 | 4 | 2 | 7 | 14 | 1.5 | 10 | 6.4 |
| Skin lipomas | 528 | 26.3 | 45 | 42.9 | 25 | 27 | 13 | 46 | 336 | 35.6 | 89 | 56.7 |
| Fibromas | 121 | 6 | 17 | 16.2 | 0 | 0 | 0 | 0 | 2 | 0.2 | 1 | 0.6 |
| Endocrine | ||||||||||||
| Thyroid cancer | ||||||||||||
| <20 | 18 | 0.9 | 5 | 4.8 | 2 | 2 | 0 | 0 | 11 | 1.2 | 3 | 1.9 |
| 20–29 | 56 | 2.8 | 3 | 2.9 | 0 | 0 | 0 | 0 | 16 | 1.7 | 1 | 0.6 |
| 30–39 | 116 | 5.8 | 7 | 6.7 | 0 | 0 | 0 | 0 | 58 | 6.2 | 4 | 2.5 |
| 40–49 | 149 | 7.4 | 8 | 7.6 | 0 | 0 | 0 | 0 | 45 | 4.8 | 3 | 1.9 |
| ≥50 | 203 | 10.1 | 4 | 3.8 | 0 | 0 | 0 | 0 | 74 | 7.8 | 1 | 0.6 |
| Thyroid goiter, nodules, or adenomas | 710 | 35.4 | 72 | 68.6 | 4 | 4 | 3 | 11 | 182 | 19.3 | 48 | 30.6 |
| Hashimoto's thyroiditis | 151 | 7.5 | 22 | 21 | 0 | 0 | 0 | 0 | 19 | 2 | 5 | 3.2 |
| Genitourinary | ||||||||||||
| Renal cell carcinoma | 94 | 4.7 | 7 | 6.7 | 1 | 1 | 0 | 0 | 43 | 4.6 | 5 | 3.2 |
| Testicular germ cell tumor | 4 | 3.4 | 2 | 5.9 | 0 | 0 | 0 | 0 | 1 | 0.6 | 1 | 1.4 |
| Ovarian germ cell tumor | 5 | 0.3 | 2 | 2.8 | 0 | 0 | 0 | 0 | 1 | 0.1 | 1 | 1.1 |
| Congenital genitourinary malformations | 58 | 2.9 | 6 | 5.7 | 15 | 16 | 8 | 29 | 14 | 1.5 | 2 | 1.3 |
Data on specific head size measurement and exact polyp number are not available in the OSU cohort. For intestinal hamartoma and ganglioneuroma, OSU data are therefore presented without breakdown.
PTEN Mutation and Deletion Analysis
To understand the relationship between PTEN (NM_000314.4) mutations and the clinical phenotype, we had all subjects undergo PTEN mutation analysis, as described below. Genomic DNA was extracted from peripheral blood leukocytes via standard methods.22 Scanning of genomic DNA samples for PTEN mutations was performed as previously reported with a combination of denaturing gradient gel electrophoresis, high-resolution melting curve analysis (Idaho Technology), and direct Sanger sequencing (ABI 3730xl).23 Deletion analysis with the multiplex ligation-dependent probe amplification (MLPA) assay24 was performed with the P158 MLPA kit (MRC-Holland) according to manufacturer's protocol. All patients underwent resequencing of the PTEN promoter region as previously described.25 The primers used are available in Table S1 available online. Promoter mutations were defined as previously reported,11,25 except for −1084T>C; this variant has been reported in population controls of European descent (2/150),26 and further work is required to characterize it.
Analysis of PTEN and Other Downstream Proteins by Immunoblotting
Human immortalized lymphoblast-derived cell lines were obtained from each patient and cultured in RPMI-1640 supplemented with 20% fetal bovine serum. All cell lines were cultured at 37°C and 5% CO2.22 Whole-cell lysates were prepared with Mammalian Protein Extraction Reageant (Pierce) supplemented with protease inhibitor cocktail (Sigma-Aldrich). Lysates were separated by SDS-PAGE and transferred onto nitrocellulose. Antibodies used included anti-PTEN mouse monoclonal (Cascade Biosciences) at 1:5000, anti-phospho-AKT1 rabbit polyclonal (Cell Signaling) at 1:1000, anti-phospho-MAPK1/2 rabbit polyclonal (Cell Signaling) at 1:2000, anti-GAPDH rabbit monoclonal (Cell Signaling) at 1:20,000, and anti-actin mouse monoclonal at 1:20,000. The blots were scanned digitally with the Odyssey Imaging System (Li-Cor Biotechnology). Detected fluorescence intensities for protein bands were background adjusted and normalized between gels with the median expression of individual proteins on each blot.
Statistical Methods for Score Derivation and Validation
Risk data for the various phenotypes were obtained from a variety of sources. For breast, endometrial, and thyroid cancer, age-adjusted cumulative risk data were drawn from the Surveillance Epidemiology and End Results database. For other phenotypes, corresponding prevalence data were derived from the literature, with North American reports preferred: oral papillomas,27 arteriovenous malformations,28 autism/developmental delay,29 thyroid goiter,30 and fibroids.31 Where no standard population-based estimates were available, baseline prevalences were estimated based on best available expert opinion; where rarity of a feature precluded reliable estimates, an estimate of 0.01% baseline prevalence was used. Patients were classified into adult and pediatric age groups at a threshold of 18 years of age. Frequency data were generated from the CC data set, with corresponding relative risks (RR) calculated relative to baseline population risk. For the pediatric CC data set, a relatively high prevalence of PTEN mutations allowed ready derivation of highly predictive features from the relative frequencies of cases, and validation in the external OSU data set was directly performed. We refer to the criteria developed here in the pediatric setting as the CC criteria, which is distinct from the CC score for adults. For the adult data set, which was considerably larger, derivation of a score was more challenging. This may be due to a referral bias, which increased recruitment for patients with oncologic conditions, preventing the use of conventional regression modeling. Hence, we adopted a clinically driven modeling approach for data reduction and validation for weighing adult criteria with the CC data set, validating our approach in the separate OSU data set.32
We first selected the most predictive phenotypic features at initial clinical assessment, excluding certain characteristic dermatologic features of CS such as fibromas or skin (but not oral) papillomas due to loss of specificity in a community setting. For each feature, we considered the relative frequency against baseline, potential for referral bias (most evident in the cancers), and the prevalence of each feature. We weighted each feature with a prespecified approach: an RR relative to baseline of 1–5 corresponded to a weight of 1; RR of 5–10 with a weight of 2; RR of 10–25 with a weight of 4; RR of 25–50 with a weight of 6; RR of 50–100 with a weight of 8; RR > 100 with a weight of 10. High referral bias for individual features was recognized clinically, being additionally evident through relatively high RR when comparing prevalences in the study population relative to the community. We adjusted the weights for these features empirically through reduction by one risk tier, except where the weight was already at the lowest tier of 1, whereupon it was maintained. A score for each individual was obtained by the sum of weights of all positive features. We subsequently performed internal validation of this semiquantitative score in the CC and external validation in the OSU adult data set. All corresponding data fields were available in both data sets, with the exception of data on extreme macrocephaly and exact gastrointestinal polyp number in the OSU data set. Consequently, when evaluating the score performance in the OSU data set, we omitted extreme macrocephaly and matched polyp presence status with corresponding multiple polyp status. These changes would be expected to penalize the accuracy of the CC score relative to the NCCN criteria in the OSU data set; thus, our validation represents a conservative assessment of the CC score's performance. Performance of this score in both data sets was evaluated via measurements of sensitivity, specificity, accuracy, positive and negative predictive value, and receiver-operator characteristic (ROC) methodology. Likelihood-ratio analysis of each model nested in a summary model32,33 was performed to compare the performance of the CC score against the NCCN criteria. Calibration to obtain bias-corrected estimates of predicted versus observed values was performed with 200 bootstraps. All analyses were performed with R 2.11.1.34 For analysis correlating protein quantitation with PTEN variants, four sample groups were defined (wild-type, common variants of unknown significance [or SNPs; >1% prevalence], rare variants of unknown significance [<1%], and mutations), analysis of variance (ANOVA) testing was used for comparison between multiple groups, and t tests were used for two-group comparisons. Linear regression was used in evaluating the association between the CC score and PTEN protein. All tests were two-sided, and p < 0.05 was deemed significant.
Results
PTEN Mutation Spectrum in Patients
Of the 3042 individuals tested, 290 (9.5%) were found to have germline pathogenic PTEN mutations (Table S2). To be conservative, we excluded variants of unknown significance without proof of functionality from the mutation-positive group. In total, 85 (29%) missense mutations, 92 (32%) nonsense mutations, 42 (14%) small deletions, 24 (8%) small insertions, 3 (1%) indels, 8 (3%) large deletions, 19 (7%) splice-site donor mutations, 9 (3%) splice-site acceptor mutations, and 8 (3%) promoter mutations were found. The mutation spectrum for the 290 probands (Figure 1) reveals mutations that are distributed irregularly across all exons, with hot spots in exon 5 (n = 93, 32%), exon 7 (n = 37, 13%), and exon 8 (n = 45, 16%). A total of 45 mutations (16%) occurred within the exon 5 hot spot (aa 123–130), corresponding to the catalytic motif of the N-terminal phosphatase domain. A total of 101 mutations (35%) occurred within the C2 domain of the PTEN protein (aa 186–351).
Figure 1.
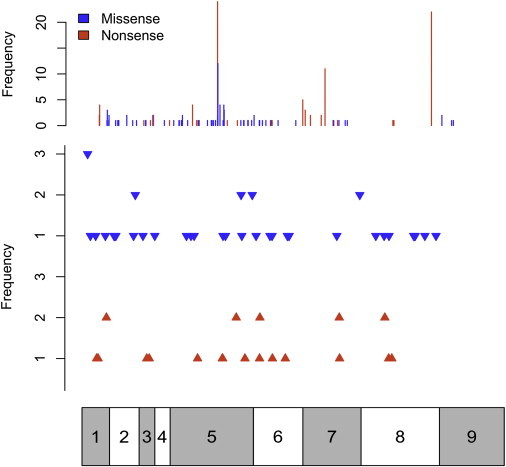
Consolidated PTEN Mutation Spectrum
Distribution and number of substitutions (missense and/or nonsense), small insertion mutations, and small deletion mutations across the gene. In the top panel, blue bars represent missense mutations and red bars represent nonsense mutations. In the second panel, the blue arrowheads represent small deletions and the red arrowheads represent small insertions along the gene. Complex mutations (indels, splice-site mutations, and large deletions) and promoter mutations are not depicted. For both panels, frequencies of both the substitution mutations and the insertion/deletion mutations are shown on the left. The bar below corresponds to the multiple exons of the PTEN cDNA molecule, with exon 1 on the left to exon 9 on the right, allowing for matching of mutation to exon. As evident, exons 5, 7, and 8 are sites of common mutations.
Pediatric Clinical Criteria Derivation
To derive appropriate clinical criteria to guide selection for PTEN testing, we analyzed genotype and phenotype data from pediatric (<18 years) and adult individuals separately, focusing on the CC data set (Table 2) to generate the criteria for validation testing in the OSU data set. For pediatric individuals, we determined that the presence of macrocephaly (occipitofrontal circumference [OFC] > 2 standard deviation [SD] over the population mean, or 97.5th percentile) was a necessary criterion for diagnosis, based on 100% prevalence at the point of diagnosis (Table 3). Neurologic (autism and developmental delay) and dermatologic (lipomas, oral papillomas) features represented extremely common secondary features; either or both systems were involved in 100% of patients with germline PTEN mutation. However, given that dermatologic features may often be overlooked, less-prevalent features in patients at first presentation in the pediatric setting are likely to be at least as important, such as vascular (such as arteriovenous) malformations, gastrointestinal polyps, thyroid goiter, and early-onset cancers (thyroid and germ cell).
Table 3.
Pediatric Clinical Criteria for PTEN Testinga
| Clinical Features | Percent Prevalence in CC Data Set of Pediatric Probands with PTEN Mutation |
|---|---|
| 1. Macrocephaly (≥2 SD) | 100% |
| 2. At least one of the following four additional criteria should be present: | |
| - Autism or developmental delay | 82% |
| - Dermatologic features, including lipomas, trichilemmomas, oral papillomas, penile freckling | 60% |
| - Vascular features, such as arteriovenous malformations or hemangiomas | 29% |
| - Gastrointestinal polyps | 14% |
In addition, pediatric-onset thyroid cancer and germ cell tumors (testicular cancer and dysgerminoma) are recognized associations of Cowden syndrome and should provoke consideration of PTEN testing.
Adult Clinical Score Derivation
For adults, additional weighing of the criteria was performed, resulting in the derivation of an appropriately weighted semiquantitative scoring system, referred to here as the CC score (Table 4). The generated score is useful for the calculation of point pretest probability of PTEN mutation. It is presented together with examples to illustrate its use (Figure 2). Increasing risk score was strongly associated with mutation-positive status (p < 0.001, Mann-Whitney test). ROC-based analysis demonstrates the variation of sensitivity and specificity in both data sets at multiple thresholds (Figures S1 and S2). At a threshold CC score of 10 (corresponding to a point pretest probability of approximately 3%; including and above the threshold score), sensitivity for diagnosis is 90%. Sensitivity falls accordingly as the threshold for CC score rises, so that when the threshold CC score is 15 (point pretest probability of 10%), the sensitivity is 72%. We report better performance for our overall approach relative to the NCCN 2010 criteria via conventional clinical and epidemiologic measures (Table 5). To evaluate for overfitting in the CC score, we used bootstrap validation on the resulting score within the CC data set (n = 200), demonstrating excellent calibration (Figure 3) and yielding an optimism-corrected concordance index of 0.91. Formal likelihood-ratio testing demonstrated considerably higher adequacy and strongly significant statistical benefit for the CC score relative to the NCCN criteria (Figure 4). The performance of the CC scoring system exceeded that of the NCCN criteria in cohorts from both centers, consistently showing superior predictive power, concordance indices, sensitivity, and specificity for the detection of PTEN mutations relative to the NCCN 2010 criteria.
Table 4.
CC Adult Score Derivation
| Population Risk | Relative Risk (Referral/Community) | Relative Risk (Mutants/Community) | Weight | |
|---|---|---|---|---|
| Neurological | ||||
| Macrocephaly, presence | 2 | 10.7 | 30.1 | 6 |
| Extreme (male, OFC ≥ 63 cm) | 0.2 | 130 | 160 | 10 |
| Extreme (female, OFC ≥ 60 cm) | 0.2 | 20 | 170 | 10 |
| Lhermitte Duclos disease | 0.01 | 80 | 860 | 10 |
| Autism or developmental delay | 5 | 0.2 | 1.2 | 1 |
| Breast and Gynecological | ||||
| Invasive breast cancer | ||||
| <30 | 0.06 | 23.3 | 23.3 | 4 |
| 30–39 | 0.43 | 20.2 | 19.8 | 4 |
| 40–49a | 1.45 | 17.3 | 12.6 | 2 |
| ≥50 | 10.93 | 3 | 1.8 | 1 |
| Fibrocystic breast disease | 10 | 4.5 | 4.2 | 1 |
| Endometrial cancer | ||||
| 20–29 | 0.01 | 50 | 140 | 10 |
| 30–39 | 0.06 | 20 | 46.7 | 6 |
| 40–49 | 0.19 | 13.7 | 36.8 | 6 |
| ≥50 | 2.43 | 3.7 | 2.9 | 1 |
| Fibroids | 13 | 3.2 | 2.9 | 1 |
| Gastrointestinal | ||||
| Polyposis syndrome (five or more, any type) | 1 | 3.7 | 34.3 | 6 |
| Intestinal hamartoma or ganglioneuroma, any number | 0.01 | 10 | ||
| Glycogenic acanthosis | 0.01 | 100 | 1520 | 10 |
| Skin | ||||
| Trichilemmomas, biopsy proven | 0.01 | 60 | 570 | 10 |
| Oral papillomas | 0.46 | 17.4 | 33 | 6 |
| Penile freckling | 1 | 18.8 | 29.4 | 6 |
| Acral keratoses | 10 | 0.6 | 2.7 | 1 |
| Arteriovenous malformations | 0.25 | 3.6 | 45.6 | 6 |
| Skin lipomas | 20 | 1.3 | 2.1 | 1 |
| Endocrine | ||||
| Thyroid cancer | ||||
| <20 | 0.01 | 90 | 480 | 10 |
| 20–29a | 0.06 | 46.7 | 48.3 | 4 |
| 30–39a | 0.13 | 44.6 | 51.5b | 4 |
| 40–49a | 0.17 | 43.5 | 44.7 | 4 |
| ≥50a | 0.57 | 17.7 | 6.7 | 1 |
| Thyroid goiter, nodules, adenomas, or Hashimoto's thyroiditis (one or more features) | 5 | 7.1 | 14.4 | 4 |
| Genitourinary | ||||
| Renal cell carcinoma | 1.49 | 3.2 | 4.5 | 1 |
For these specific features, referral bias was clinically recognized and accounted for by a standard downward modification of weight by a risk tier; this bias may also be recognized in a high RR (study population/community, relative to the RR in patients with PTEN mutations/community). Individual features for which high referral bias was evident, but where weight was already 1, were not further adjusted downward.
For this borderline RR just above the threshold of 50, given the relatively consistent RRs in adjacent groups for all populations, this weight was adjusted to 4 for consistency between the adjacent age groups.
Figure 2.
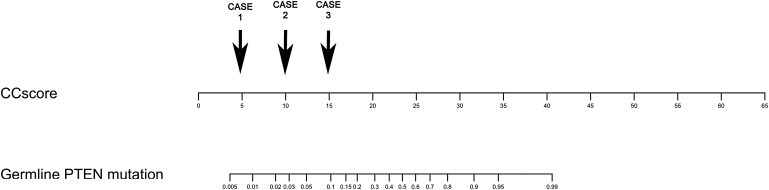
CC Score Nomogram for Obtaining a Corresponding Individual Point Pretest Probability of Germline PTEN Mutation
The CC score is first derived by a sum of the weights of positive features that is provided in Table 4. To illustrate, three hypothetical cases are presented, each corresponding to a CC score of 5 points, 10 points, and 15 points, respectively. Case 1 may present with breast cancer at age 55 (1 point), with background of thyroid cancer at age 44 (4 points), with a final score of 5 and corresponding point probability < 1%. Case 2 may present with breast cancer at age 38 (4 points) and concurrent macrocephaly (6 points), with a final score of 10 and corresponding point probability of 3%. Case 3 may present with a single hamartomatous gastrointestinal polyp (10 points) found on endoscopy, Hashimoto's thyroiditis (4 points), and lipomas (1 point), for a final score of 15 and corresponding point probability of 10%.
Table 5.
Comparison of the CC Pediatric Criteria and Adult Score Relative to the NCCN 2010 Criteria
|
Cleveland Clinic |
Ohio State University |
||||
|---|---|---|---|---|---|
| Nonmutant | Mutant | Nonmutant | Mutant | ||
| Adult | |||||
| CC scorea | Fails threshold | 1426 | 10 | 445 | 8 |
| Meets threshold | 476 | 95 | 267 | 102 | |
| Sensitivity/specificity | 90%/75% | 93%/62% | |||
| Concordance index | 0.83c | 0.83c | 0.78 | 0.78 | |
| NCCN criteriab | Fails criteria | 1310 | 27 | 474 | 17 |
| Meets criteria | 592 | 78 | 238 | 93 | |
| Sensitivity/specificity | 74%/69% | 85%/67% | |||
| Concordance index | 0.72 | 0.72 | 0.76 | 0.76 | |
| Pediatric | |||||
| CC criteriaa | Fails threshold | 6 | 0 | 7 | 1 |
| Meets threshold | 58 | 28 | 68 | 46 | |
| Sensitivity/specificity | 100%/10% | 98%/9% | |||
| NCCN criteriab | Fails criteria | 5 | 3 | 56 | 20 |
| Meets criteria | 59 | 25 | 19 | 27 | |
| Sensitivity/specificity | 89%/8% | 56%/74% | |||
| Overall | |||||
| CC scorea | Fails threshold | 1432 | 10 | 493 | 11 |
| Meets threshold | 533 | 124 | 294 | 146 | |
| Sensitivity/specificity | 93%/73% | 93%/63% | |||
| Concordance index | 0.83c | 0.83c | 0.76 | 0.76 | |
| NCCN criteriab | Fails criteria | 1315 | 30 | 530 | 37 |
| Meets criteria | 650 | 104 | 257 | 120 | |
| Sensitivity/specificity | 78%/67% | 76%/67% | |||
| Concordance index | 0.72 | 0.72 | 0.72 | 0.72 | |
For adults, using a threshold Cleveland Clinic (CC) score of 10 points and above for recommendation of PTEN testing.
National Comprehensive Cancer Network (NCCN) criteria, 2010.
Optimism corrected with 200 bootstraps.
Figure 3.
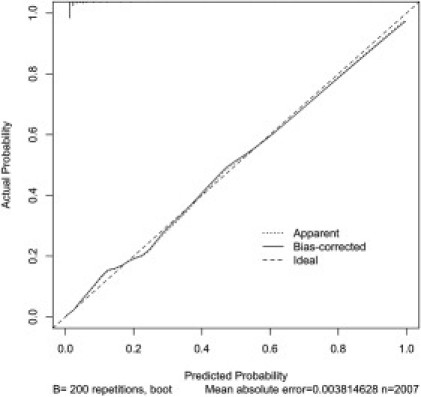
A Calibration Plot for Model-Based Predicted Probabilities of PTEN Mutation and Actual Outcomes
The calibration plot shows excellent bias-corrected correlation between observed and predicted values for the developed model within the CC data set, suggesting good internal calibration for the CC score model, that is, individual predicted and actual outcomes are similar. The dashed line at 45° (y = x) represents ideal agreement between observed and predicted probabilities of PTEN mutation.
Figure 4.
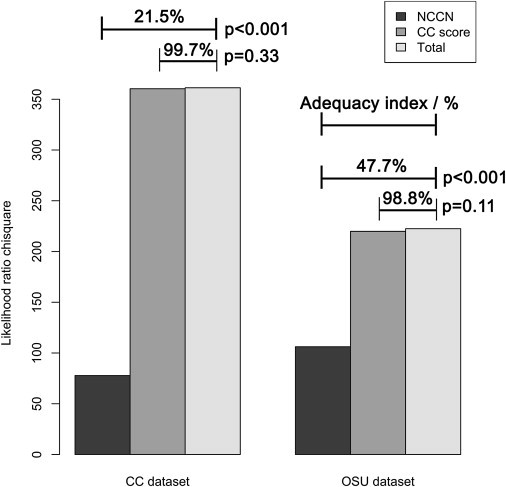
A Graph Showing that the CC Score Yields Superior Predictions Relative to the NCCN Criteria in Both the CC and the OSU Data Sets
Each data set shows corresponding likelihood ratio chi-squares for the NCCN criteria, the CC score, and a full model comprised of both criteria. The CC score confers statistically significant benefit to the NCCN criteria in PTEN mutation prediction, but the NCCN criteria do not confer benefit to the CC score. Higher adequacy indices are observed for the CC score relative to the NCCN criteria in both data sets.
PTEN Genotype, Clinical Score, and Downstream Pathway Proteins
We next sought to determine whether there was a correlation between PTEN mutation status and expression of proteins considered to be downstream readouts for PTEN signaling. For PTEN mutant patient-derived lymphoblasts (n = 24), PTEN protein was decreased and P-AKT1 was relatively increased (Figure 5) compared to wild-type PTEN, common variants (SNPs), and rare variants of unknown significance, whereas no significant overall difference was seen for P-MAPK1/P-MAPK2, actin, or GAPDH. It was noted on inspection that PTEN and P-AKT1 protein expression for the rare PTEN variants was intermediate between that of mutant and wild-type samples, although there was no statistical significance discerned between the protein profiles of rare variant lymphoblasts (n = 15) and the other groups of samples.
Figure 5.
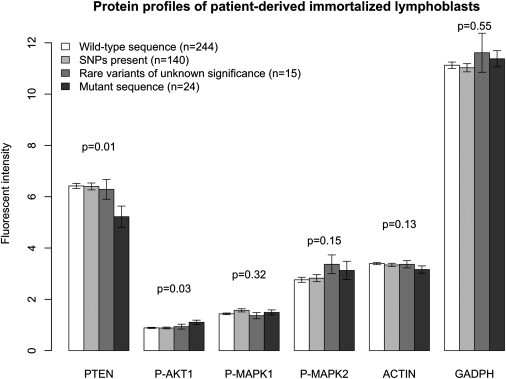
Bar Plot Showing Expression by Immunoblot of Downstream Readout Molecules of PTEN Function for Immortalized Lymphoblasts from 423 Patients
A statistically significant decrease in PTEN protein expression and increase in phospho-AKT1 expression are noted for samples with pathogenic mutations. Standard errors are shown in the error bar. P values are derived from ANOVA testing.
Finally, we evaluated whether an association existed between PTEN/P-AKT1 protein expression and CC score, which might serve as a potential index of phenotypic load of syndromic features. When analyzing the full data set, a strong inverse association was apparent between PTEN protein expression and CC score (p < 0.001; Figure 6). This association remained (p = 0.036) after excluding patients with pathogenic mutations (Figure 7). No correlation between P-AKT1 and CC score was noted in any subgroup of individuals.
Figure 6.
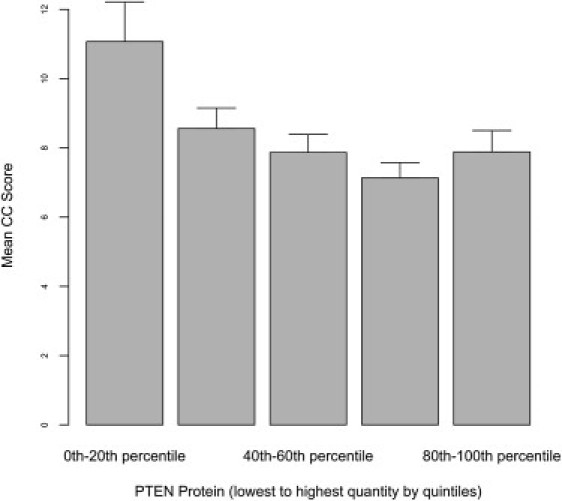
The Relationship between the CC Score and PTEN Protein Quantitation
This relationship between CC score and PTEN protein levels is classified by quintiles, lowest to highest from left to right of each graph, with an inverse association that is demonstrated between rising PTEN protein expression and decreasing mean CC score in the full data set on linear regression (p < 0.001). Standard errors are shown by error bar.
Figure 7.
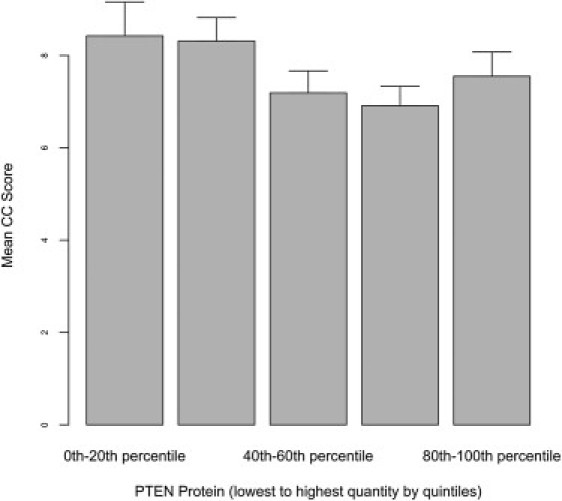
The Relationship between the CC Score and PTEN Protein Quantitation after Exclusion of Patients with PTEN Mutations
The relationship between CC score and PTEN expression levels is classified by quintiles, lowest to highest from left to right of each graph. An inverse association between rising PTEN protein expression and decreasing CC score is seen in samples derived from patients without pathogenic germline PTEN mutations (p = 0.036). Standard errors are shown by error bar.
Discussion
The criteria described here represent the collective experience derived from the largest prospective cohort of patients with germline pathogenic PTEN mutations to date (290 probands) with standard-of-care testing. This represents a core strength of this study. To our knowledge, outside of these two specialist centers, the largest single reported series from a single center includes 21 patients,35 and, to date, a total of 211 patients (both probands and affected family members) have been reported in the entire published medical literature.35 Our study is limited to probands only, this being critical for an accurate evaluation of the relative importance of each feature in initial screening through the reduction of ascertainment bias. In terms of the mutation spectra, our summary study confirms our previously reported profiles from smaller data sets.36
Pediatric Criteria
The classification of our recommendations for PTEN mutation testing into distinct adult and pediatric criteria has demonstrated considerable clinical utility in our cohorts. This success is likely multifactorial in nature, with reasons including age-specific phenotypic penetrance and, importantly, the different health services utilized in the distinct age groups: for example, macrocephaly and delayed development are well-established clinical problems for pediatricians who use head circumference measurements routinely, whereas cancers and polyps are common presenting problems in adult medicine. Indeed, it is possible that patients who present in childhood are more readily identified than adult patients, given that features such as macrocephaly and autism are likely to result in early consults with pediatricians, who are often familiar with issues of genetics. At the same time, patients presenting and being diagnosed during adulthood with PTEN mutations tend to have fewer overt disease manifestations because of many reasons, including a selection bias, in which more severely affected individuals may already have been diagnosed during childhood, true age-related penetrance, and less physician familiarity. In terms of the phenotypic spectrum, the particularly high prevalence of neurologic features such as autism and developmental delay (>80%) validate early observations by our group,13 demonstrating previously unrecognized PTEN mutations in patients with autism spectrum disorder and macrocephaly. The inclusion of this feature into our clinical criteria is particularly noteworthy, because autism was not hitherto recognized in pediatric patients with BRRS, and germline PTEN mutation is now recognized as one of the most common single gene causes of autism.26,37 Thus, the pediatric CC criteria that we have developed de novo here, maximizing sensitivity, represents an important guideline for pediatricians to select children for referral to genetic professionals for PTEN mutation testing. Such an approach is aided by the routine use of head circumference measurements, and this macrocephaly is present in all patients diagnosed in childhood. Indeed, because macrocephaly is similarly present in almost all adult patients, it is likely that active evaluation of pediatric patients with macrocephaly may result in earlier diagnosis.
Adult Clinical Scoring
For adult patients, our study establishes useful clinical thresholds that can be readily translated to community practice for referral of patients for specialist evaluation. We recommend a threshold CC score of 10 (or a point pretest probability of 3%) and above for referral to medical genetics for specialist evaluation, this permitting a sensitivity of at least 90%. We present a range of scenarios that would satisfy this threshold. It is clear that age of presentation for cancer profoundly influences risk: patients presenting with endometrial, thyroid, or breast cancer below the age of 40 must be evaluated for other suggestive features of germline PTEN mutation, particularly macrocephaly. For patients presenting with first onset of cancer above 40, combinations of the above cancers, together with other suggestive nonmalignant features, are more common. Similarly, gastroenterologists or surgeons who encounter patients with polyposis syndromes (at least five polyps) should also assess the past medical history of these individuals for the presence of other types of cancer, particularly breast, endometrial, or thyroid cancer, or relevant combinations. These scenarios are useful, given that PTEN mutation will be considered in the majority of adult patients only after the diagnosis of cancer or polyposis syndrome. Similarly, based on our results, we would recommend consideration of PTEN mutation analysis in patients with documented vascular malformations as defined by ultrasound evaluation38 as an important feature of adult PHTS. This feature is recognized in BRRS as an important component of the syndrome, but it is not part of the NCCN diagnostic criteria, even though vascular malformations have been reported in association with CS since the 1970s.39
For adult patients seen by front-line clinicians, genetic etiologies are most often considered if a patient presents with a serious disorder under the age of 40. This common clinical approach for referral of such patients for genetics evaluation is supported by our results, which show that PTEN mutation should be considered particularly in patients with breast or endometrial cancer with onset below the age of 40, as well as thyroid cancer with onset below age of 50. However, a not-insignificant proportion of our patients did present with cancer with first onset above age 40, as seen in Table 2. For such patients with late onset of cancer, a majority has distinct phenotypic features or cancer combinations such as breast and thyroid cancers or thyroid and endometrial cancers, which certainly should mandate clinical evaluation. For such individuals, we have demonstrated previously that head circumference measurement alone in the setting of a high-risk breast cancer clinic population was useful in identifying patients with germline PTEN mutation.40 For these patients who are often first recognized by gastroenterologists or surgeons, we also recommend evaluation by genetics professionals, which may also be useful for consideration of other polyposis syndromes. Indeed, our criteria here underline our recent report that PTEN mutation may also underpin a distinct gastrointestinal polyposis phenotype in adults,14 to be considered alongside other polyposis syndromes such as familial adenomatous polyposis and Peutz-Jeghers syndrome. We have demonstrated that the measurement of head circumference is a useful clinical maneuver to facilitate diagnosis in these patients.14 Although colorectal cancer is recognized in association with PTEN mutation,14 it does not confer sufficient additional diagnostic value in the community setting.
Advantages of Clinical Scoring over Existing Criteria
The semiquantitative CC score has several advantages over the NCCN criteria. First, the CC score is more accurate and can provide individualized estimates of probability. In particular, the ability to quantify these probabilities is critical for an educated discussion between patients and healthcare providers. Further, adoption of quantitative risk assessment by health management organizations in genetic screening policies underlines their importance. The CC score permits a corresponding estimate for the use of specialist genetics staff and represents a tool for individualized counseling and testing of patients with the wide variety of phenotypes that CS represents. Second, because of the complex and multisystem nature of CS, the current NCCN criteria involve complex rules involving multiplicity of combinations between major and minor criteria inaccessible to most clinicians. A clinical score, represented by a single number as a sum of weights, can be calculated relatively simply. Third, we have found that the age of onset for cancer in patients is crucial for clarifying the diagnosis, and this has been readily incorporated. Finally, accrued clinical experience, together with our active recruitment of patients from the community, demonstrated that certain dermatologic features of CS patients, although characteristic to the eye of the trained specialist, were much more challenging to apply to the community setting.41
Performance
Although it was clear that the CC score outperformed the NCCN criteria, it is of interest that the fraction of adult patients with PTEN mutations in the CC and OSU cohorts is relatively low (7.6%). Certainly the relaxed recruitment criteria (any two features) for recruitment with the goal of detecting more subtle phenotypes were likely to be the main reason. The strictly applied ICC/NCCN criteria, which we first developed on early consortium data based on a retrospective and cross-sectional series, were previously reported to result in a high PTEN mutation frequency (85%). Based on this study, it is now clear that the ICC/NCCN criteria have a lower positive predictive value of 15%–30% in a prospective setting, where patients are accrued in a setting closer to the community. That germline alterations of other genes may contribute to this phenotype is of considerable interest, and we have previously identified germline SDHB and SDHD mutations in 10% of research participants with a similar clinical phenotype and without germline PTEN mutations.22 Furthermore, we have recently identified ∼35% of individuals, who have similar clinical phenotypes but without PTEN mutations, with germline hypermethylation of the KILLIN-PTEN bidirectional promoter resulting in downregulation of KILLIN.42
Molecular Correlates of Clinical Score
In addition to the clinical dimension of a disease, useful clinical scoring should also reflect biological and molecular aspects of the disease. Experimental data from nonhuman models has accrued, supporting the molecular concept of subtle variations of Pten protein dosage deficiency as a key influence on carcinogenesis, with a Pten dose reduction of 20% associated with manifestation of murine breast cancer and altered steady-state biology of mammary tissue.16 Our study provides direct support from human clinical data for the idea that PTEN protein dosage may influence a multisystem phenotype, showing that the molecular phenotype of PTEN protein deficiency is correlated with increased CC score and consequently increased CS phenotypic load. Thus, even though the average decrease of PTEN protein is relatively subtle, its correlation with PTEN mutation and elevated CC score is highly provocative in light of these insights. Although the mechanisms of this dose reduction remain unclear, possible explanations include hereditary variants in other genes that regulate PTEN expression or its localization.43–45 It is also possible that extended upstream PTEN promoter variants may affect this expression. Either way, our results imply that phenotypes comprising a complex multisystem syndrome may be underpinned by more genetic variation than would be expected, manifesting in a common PTEN protein deficiency pathway with a genetic basis. The provocative idea that PTEN protein deficiency may underpin a pathogenic clinical phenotype, however, requires additional validation. When evaluating other proteins that are downstream readouts of the PTEN signaling pathway, the increased expression of phospho-AKT1 in PTEN-mutated samples is expected.46 The absence of association between phospho-MAPK1/2 protein expression and PTEN protein expression or mutant status, as would be expected,19 requires additional investigation of the relative impairments of the lipid and protein phosphatase activities of the mutations. Overall, our results highlight that the CC score, in correlation with protein quantitation, represents a novel resource for the interrogation of PTEN function in the clinical setting, consistent with the “phenomic” approach we have previously advocated.21
In terms of limitations, our data are derived from two referral cohorts representing patients recruited at two major cancer genetics centers, with consequent possible referral bias. It is also likely that referral bias for certain clinical features (particularly adult cancers) may result in overrepresentation of certain patient groups in our patient cohort. We have described our method to adjust for this referral bias through the reduction of score weightage for these features. The most important test of this model and adjustment approach for referral bias is whether it may be validated externally. We were able to demonstrate excellent performance through both calibration and external validation, demonstrating that the approach we have undertaken performs well in a prospective real-world setting. Until a population-based sampling approach to screen community patients for CS is performed, our approach represents the best available evidence-based clinical approach to identifying patients with germline PTEN mutations, over the current NCCN criteria. Most importantly, its excellent performance in the real-world setting that we have validated here implies that it is of practice-changing importance.
Overall, we have developed a useful semiquantitative scoring system to evaluate patients for the prior probability of PTEN germline mutations, validated in two large separate prospective cohorts, representing an evidence-based advance on existing NCCN criteria. We make practice recommendations with regard to the evaluation of patients for germline PTEN mutations, guided by the data accrued above. Additionally, in demonstrating correlation between this score and PTEN protein expression in immortalized lymphocytes, we provide direct support from clinical studies for the concept of gene dosage for PTEN, highlighting the contribution of PTEN protein deficiency to the complex phenotypic features recognized in CS.
Acknowledgments
We would like to thank the Genomic Medicine Biorepository of the Cleveland Clinic Genomic Medicine Institute, as well as the Eng laboratory personnel over the last 14 years who have contributed technical assistance, technical advice, and helpful discussions. We would also like to express our gratitude to all the patients and clinical collaborators from all the centers around the world who have contributed their time and specimens over the last 10 years. This study is funded, in part, by the National Cancer Institute (P01CA124570 and R01CA118989), American Cancer Society (RPG-02-151-01-CCE), William Randolph Hearst Foundations, and Doris Duke Distinguished Clinical Scientist Award (all to C.E.). M.-H.T. is the Lee Foundation (Singapore) Fellow and an Ambrose Monell Foundation Cancer Genomic Medicine Clinical Fellow at the Cleveland Clinic Genomic Medicine Institute. C.E. is the Sondra J. and Stephen R. Hardis Chair of Cancer Genomic Medicine at the Cleveland Clinic. C.E. is the recipient of an American Cancer Society Clinical Research Professorship, generously funded, in part, by the F.M. Kirby Foundation.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Cleveland Clinic Genomic Medicine Institute: adult and pediatric criteria for individualized risk estimation, http://www.lerner.ccf.org/gmi/ccscore/
National Comprehensive Cancer Network (NCCN), http://www.nccn.org/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
References
- 1.Gorlin R.J., Cohen M.M., Jr., Condon L.M., Burke B.A. Bannayan-Riley-Ruvalcaba syndrome. Am. J. Med. Genet. 1992;44:307–314. doi: 10.1002/ajmg.1320440309. [DOI] [PubMed] [Google Scholar]
- 2.Marsh D.J., Coulon V., Lunetta K.L., Rocca-Serra P., Dahia P.L., Zheng Z., Liaw D., Caron S., Duboué B., Lin A.Y. Mutation spectrum and genotype-phenotype analyses in Cowden disease and Bannayan-Zonana syndrome, two hamartoma syndromes with germline PTEN mutation. Hum. Mol. Genet. 1998;7:507–515. doi: 10.1093/hmg/7.3.507. [DOI] [PubMed] [Google Scholar]
- 3.Orloff M.S., Eng C. Genetic and phenotypic heterogeneity in the PTEN hamartoma tumour syndrome. Oncogene. 2008;27:5387–5397. doi: 10.1038/onc.2008.237. [DOI] [PubMed] [Google Scholar]
- 4.Eng C., Thiele H., Zhou X.P., Gorlin R.J., Hennekam R.C., Winter R.M. PTEN mutations and proteus syndrome. Lancet. 2001;358:2079–2080. doi: 10.1016/S0140-6736(01)07110-0. [DOI] [PubMed] [Google Scholar]
- 5.Hobert J.A., Eng C. PTEN hamartoma tumor syndrome: An overview. Genet. Med. 2009;11:687–694. doi: 10.1097/GIM.0b013e3181ac9aea. [DOI] [PubMed] [Google Scholar]
- 6.Li D.M., Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 1997;57:2124–2129. [PubMed] [Google Scholar]
- 7.Myers M.P., Pass I., Batty I.H., Van der Kaay J., Stolarov J.P., Hemmings B.A., Wigler M.H., Downes C.P., Tonks N.K. The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc. Natl. Acad. Sci. USA. 1998;95:13513–13518. doi: 10.1073/pnas.95.23.13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nelen M.R., Padberg G.W., Peeters E.A., Lin A.Y., van den Helm B., Frants R.R., Coulon V., Goldstein A.M., van Reen M.M., Easton D.F. Localization of the gene for Cowden disease to chromosome 10q22-23. Nat. Genet. 1996;13:114–116. doi: 10.1038/ng0596-114. [DOI] [PubMed] [Google Scholar]
- 9.Eng C. Will the real Cowden syndrome please stand up: Revised diagnostic criteria. J. Med. Genet. 2000;37:828–830. doi: 10.1136/jmg.37.11.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pilarski R., Eng C. Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome. J. Med. Genet. 2004;41:323–326. doi: 10.1136/jmg.2004.018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou X.P., Waite K.A., Pilarski R., Hampel H., Fernandez M.J., Bos C., Dasouki M., Feldman G.L., Greenberg L.A., Ivanovich J. Germline PTEN promoter mutations and deletions in Cowden/Bannayan-Riley-Ruvalcaba syndrome result in aberrant PTEN protein and dysregulation of the phosphoinositol-3-kinase/Akt pathway. Am. J. Hum. Genet. 2003;73:404–411. doi: 10.1086/377109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.The National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology (2010). Genetic/Familial High-Risk Assessment: Breast and Ovarian Cancer (Version 1.2010). http://www.nccn.org.
- 13.Butler M.G., Dasouki M.J., Zhou X.P., Talebizadeh Z., Brown M., Takahashi T.N., Miles J.H., Wang C.H., Stratton R., Pilarski R., Eng C. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J. Med. Genet. 2005;42:318–321. doi: 10.1136/jmg.2004.024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heald B., Mester J., Rybicki L., Orloff M.S., Burke C.A., Eng C. Frequent gastrointestinal polyps and colorectal adenocarcinomas in a prospective series of PTEN mutation carriers. Gastroenterology. 2010;139:1927–1933. doi: 10.1053/j.gastro.2010.06.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan H., Dobbie Z., Gruber S.B., Markowitz S., Romans K., Giardiello F.M., Kinzler K.W., Vogelstein B. Small changes in expression affect predisposition to tumorigenesis. Nat. Genet. 2002;30:25–26. doi: 10.1038/ng799. [DOI] [PubMed] [Google Scholar]
- 16.Alimonti A., Carracedo A., Clohessy J.G., Trotman L.C., Nardella C., Egia A., Salmena L., Sampieri K., Haveman W.J., Brogi E. Subtle variations in Pten dose determine cancer susceptibility. Nat. Genet. 2010;42:454–458. doi: 10.1038/ng.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stambolic V., Suzuki A., de la Pompa J.L., Brothers G.M., Mirtsos C., Sasaki T., Ruland J., Penninger J.M., Siderovski D.P., Mak T.W. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 18.Wu X., Senechal K., Neshat M.S., Whang Y.E., Sawyers C.L. The PTEN/MMAC1 tumor suppressor phosphatase functions as a negative regulator of the phosphoinositide 3-kinase/Akt pathway. Proc. Natl. Acad. Sci. USA. 1998;95:15587–15591. doi: 10.1073/pnas.95.26.15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weng L.P., Smith W.M., Brown J.L., Eng C. PTEN inhibits insulin-stimulated MEK/MAPK activation and cell growth by blocking IRS-1 phosphorylation and IRS-1/Grb-2/Sos complex formation in a breast cancer model. Hum. Mol. Genet. 2001;10:605–616. doi: 10.1093/hmg/10.6.605. [DOI] [PubMed] [Google Scholar]
- 20.Weng L.P., Brown J.L., Eng C. PTEN coordinates G(1) arrest by down-regulating cyclin D1 via its protein phosphatase activity and up-regulating p27 via its lipid phosphatase activity in a breast cancer model. Hum. Mol. Genet. 2001;10:599–604. doi: 10.1093/hmg/10.6.599. [DOI] [PubMed] [Google Scholar]
- 21.Zbuk K.M., Eng C. Cancer phenomics: RET and PTEN as illustrative models. Nat. Rev. Cancer. 2007;7:35–45. doi: 10.1038/nrc2037. [DOI] [PubMed] [Google Scholar]
- 22.Ni Y., Zbuk K.M., Sadler T., Patocs A., Lobo G., Edelman E., Platzer P., Orloff M.S., Waite K.A., Eng C. Germline mutations and variants in the succinate dehydrogenase genes in Cowden and Cowden-like syndromes. Am. J. Hum. Genet. 2008;83:261–268. doi: 10.1016/j.ajhg.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van der Stoep N., van Paridon C.D., Janssens T., Krenkova P., Stambergova A., Macek M., Matthijs G., Bakker E. Diagnostic guidelines for high-resolution melting curve (HRM) analysis: An interlaboratory validation of BRCA1 mutation scanning using the 96-well LightScanner. Hum. Mutat. 2009;30:899–909. doi: 10.1002/humu.21004. [DOI] [PubMed] [Google Scholar]
- 24.Schouten J.P., McElgunn C.J., Waaijer R., Zwijnenburg D., Diepvens F., Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Teresi R.E., Zbuk K.M., Pezzolesi M.G., Waite K.A., Eng C. Cowden syndrome-affected patients with PTEN promoter mutations demonstrate abnormal protein translation. Am. J. Hum. Genet. 2007;81:756–767. doi: 10.1086/521051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Buxbaum J.D., Cai G., Chaste P., Nygren G., Goldsmith J., Reichert J., Anckarsäter H., Rastam M., Smith C.J., Silverman J.M. Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2007;144B:484–491. doi: 10.1002/ajmg.b.30493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bouquot J.E., Gundlach K.K. Oral exophytic lesions in 23,616 white Americans over 35 years of age. Oral Surg. Oral Med. Oral Pathol. 1986;62:284–291. doi: 10.1016/0030-4220(86)90010-1. [DOI] [PubMed] [Google Scholar]
- 28.Al-Shahi R., Warlow C. A systematic review of the frequency and prognosis of arteriovenous malformations of the brain in adults. Brain. 2001;124:1900–1926. doi: 10.1093/brain/124.10.1900. [DOI] [PubMed] [Google Scholar]
- 29.Rosenberg S.A., Zhang D., Robinson C.C. Prevalence of developmental delays and participation in early intervention services for young children. Pediatrics. 2008;121:e1503–e1509. doi: 10.1542/peds.2007-1680. [DOI] [PubMed] [Google Scholar]
- 30.Matovinovic J. Endemic goiter and cretinism at the dawn of the third millennium. Annu. Rev. Nutr. 1983;3:341–412. doi: 10.1146/annurev.nu.03.070183.002013. [DOI] [PubMed] [Google Scholar]
- 31.DeWaay D.J., Syrop C.H., Nygaard I.E., Davis W.A., Van Voorhis B.J. Natural history of uterine polyps and leiomyomata. Obstet. Gynecol. 2002;100:3–7. doi: 10.1016/s0029-7844(02)02007-0. [DOI] [PubMed] [Google Scholar]
- 32.Harrell F.E. Springer-Verlag; New York: 2001. Regression Modelling Strategies: With Applications to Linear Models, Logistic Regression and Survival Analysis. [Google Scholar]
- 33.Tan M.H., Kanesvaran R., Li H., Tan H.L., Tan P.H., Wong C.F., Chia K.S., Teh B.T., Yuen J., Chong T.W. Comparison of the UCLA Integrated Staging System and the Leibovich score in survival prediction for patients with nonmetastatic clear cell renal cell carcinoma. Urology. 2010;75:1365–1370. doi: 10.1016/j.urology.2009.07.1289. [DOI] [PubMed] [Google Scholar]
- 34.Ihaka R., Gentleman R. R: A language for data analysis and graphics. J. Comput. Graph. Statist. 1996;5:299–314. [Google Scholar]
- 35.Riegert-Johnson D.L., Gleeson F.C., Roberts M., Tholen K., Youngborg L., Bullock M., Boardman L.A. Cancer and Lhermitte-Duclos disease are common in Cowden syndrome patients. Hered. Cancer Clin. Pract. 2010;8:6. doi: 10.1186/1897-4287-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marsh D.J., Kum J.B., Lunetta K.L., Bennett M.J., Gorlin R.J., Ahmed S.F., Bodurtha J., Crowe C., Curtis M.A., Dasouki M. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum. Mol. Genet. 1999;8:1461–1472. doi: 10.1093/hmg/8.8.1461. [DOI] [PubMed] [Google Scholar]
- 37.McBride K.L., Varga E.A., Pastore M.T., Prior T.W., Manickam K., Atkin J.F., Herman G.E. Confirmation study of PTEN mutations among individuals with autism or developmental delays/mental retardation and macrocephaly. Autism Res. 2010;3:137–141. doi: 10.1002/aur.132. [DOI] [PubMed] [Google Scholar]
- 38.Paltiel H.J., Burrows P.E., Kozakewich H.P., Zurakowski D., Mulliken J.B. Soft-tissue vascular anomalies: Utility of US for diagnosis. Radiology. 2000;214:747–754. doi: 10.1148/radiology.214.3.r00mr21747. [DOI] [PubMed] [Google Scholar]
- 39.Turnbull M.M., Humeniuk V., Stein B., Suthers G.K. Arteriovenous malformations in Cowden syndrome. J. Med. Genet. 2005;42:e50. doi: 10.1136/jmg.2004.030569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shiovitz S., Everett J., Huang S.C., Orloff M.S., Eng C., Gruber S.B. Head circumference in the clinical detection of PTEN hamartoma tumor syndrome in a clinic population at high-risk of breast cancer. Breast Cancer Res. Treat. 2010;124:459–465. doi: 10.1007/s10549-010-0839-6. [DOI] [PubMed] [Google Scholar]
- 41.Pilarski R. Cowden syndrome: A critical review of the clinical literature. J. Genet. Couns. 2009;18:13–27. doi: 10.1007/s10897-008-9187-7. [DOI] [PubMed] [Google Scholar]
- 42.Bennett K.L., Mester J., Eng C. Germline epigenetic regulation of KILLIN in Cowden and Cowden-like syndromes. JAMA. 2010;304:2724–2731. doi: 10.1001/jama.2010.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baker S.J. PTEN enters the nuclear age. Cell. 2007;128:25–28. doi: 10.1016/j.cell.2006.12.023. [DOI] [PubMed] [Google Scholar]
- 44.Shen W.H., Balajee A.S., Wang J., Wu H., Eng C., Pandolfi P.P., Yin Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128:157–170. doi: 10.1016/j.cell.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 45.Trotman L.C., Wang X., Alimonti A., Chen Z., Teruya-Feldstein J., Yang H., Pavletich N.P., Carver B.S., Cordon-Cardo C., Erdjument-Bromage H. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. 2007;128:141–156. doi: 10.1016/j.cell.2006.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Puc J., Keniry M., Li H.S., Pandita T.K., Choudhury A.D., Memeo L., Mansukhani M., Murty V.V., Gaciong Z., Meek S.E. Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer Cell. 2005;7:193–204. doi: 10.1016/j.ccr.2005.01.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.