

Abstract
Electrophysiological studies have implicated voltage-gated proton channels in several specific cellular contexts. In neutrophils, they mediate charge compensation associated with the oxidative burst of phagocytosis. Molecular characterization of the hydrogen voltage-gated channel 1 (HVCN1) has enabled identification of unanticipated and diverse functions: HVCN1 not only modulates signaling from the B-cell receptor following B-cell activation and histamine release from basophils, but also mediates pH-mediated activation of spermatozoa, as well as acid secretion by tracheal epithelium. The importance of HVCN1 in pH regulation during phagocytosis was established by surprising evidence indicating its first-responder role. In this review, we examine recent findings from a functional perspective, and discuss the potential of HVCN1 as a therapeutic target for autoimmune and other diseases.
Introduction
The voltage-gated proton channel (HVCN1, also called HV1 and VSOP) was a mysterious protein for a number of years. Proton currents were identified in snail neurons and amphibian oocytes in the 1980s [1–3], in mammalian cells in 1991 [4], and in human cells in 1993 [5–7]. However, the gene encoding the channel was not discovered until 2006 when two groups cloned human and murine HVCN1 genes [8, 9], prompting a wave of new studies on voltage-gated proton channel function and regulation. Historically, proton currents have been associated with the NADPH oxidase in phagocytic cells, where they mediate rebalancing of charges across the plasma membrane to allow optimal NADPH oxidase-mediated reactive oxygen species (ROS) production [10–13]. Availability of an HVCN1-deficient mouse line has facilitated further functional characterization of HVCN1, opening new avenues for the investigation of its roles in other cell types. We now know that the proton channel is expressed in such diverse cell types as neutrophils, basophils, B cells, spermatozoa and airway epithelial cells and that it performs specific functions in each of them. This review will highlight the significance of such functions and the emerging potential to exploit HVCN1 as a therapeutic target in applications such as allergy, autoimmune diseases and contraception.
Key electrophysiological properties of the proton channel
Consideration of the electrophysiological properties of voltage-gated proton channels leads to the conclusion that their fundamental function is acid extrusion from cells [14]. Proton channels are perfectly selective for protons. Furthermore, they are ion channels, not carriers or exchangers, and acid extrusion occurs independently of other ionic concentrations [1, 2, 14]. Voltage-gated proton channels open upon membrane depolarization, with a sigmoid time course. However, their open probability depends strongly on pH on both sides of the membrane. The threshold voltage at which proton channels begin to open was determined empirically to be 40(pHo-pHi) +20 mV [15]. In practical terms, this means that at symmetrical pH, the channel opens 20 mV positive to the Nernst potential for protons, EH, that increasing pHo or decreasing pHi by one unit shifts the entire gH-V (proton conductance-voltage) relationship by −40 mV, and that at all physiologically achievable pH, the channel opens only when the electrochemical proton gradient is outwards. Thus, proton channels are designed to open only when this will result in acid extrusion from cells.
Proton channels have been found in a multitude of cell types within two dozen species, based on both direct voltage-clamp evidence and gene homology [16]. In electrophysiological studies, widely used to provide unequivocal evidence of expression, proton channel function is characterized in the plasma membrane. However, indirect evidence supports the presence of proton channels in phagosomes [17] and in the Golgi complex [18]. When overexpressed in HeLa cells, HVCN1 protein appears mainly in intracellular compartments [19]; whether this reflects a catabolic pathway or a function in organelles is unclear.
The current that flows through a single proton channel is quite small, compared to other ion channels. For a moderate driving force, a Na+ or K+ channel might conduct a few picoamperes (10−12 A) of current (~107 ions/s), but a proton channel conducts only a few femtoamperes (10−15 A) of current (~104 H+/s) [20]. The smaller conductance reflects the much lower permeant ion concentration; the K+ concentration in cells is 106 times larger than the H+ concentration. Nevertheless, macroscopic (collective) proton currents in many cells are as large or larger than K+ currents [21], because nature is clever enough to express large numbers of H+ channels. Having a tiny conductance is useful for proton channels in small organelles like phagosomes, because this enables a finely controlled response.
Discovery of the gene encoding the proton channel was hampered by a lack of selective inhibitors. The most potent inhibitor is Zn2+, which although rather promiscuous, inhibits H+ currents more effectively than Ca2+ currents, for example [22]. Unlike traditional ion channel blockers, Zn2+ does not occlude the channel, but instead binds to the external surface of the molecule where it slows channel opening and shifts the voltage dependence positively [23]. The effects of Zn2+ are strongly attenuated at low pHo, reflecting competition between protons and Zn2+ for binding site(s). Modeling this competition suggested that Zn2+ binding is coordinated between 2 to 3 Histidine (His) residues [23]. Subsequently, mutation of the human proton channel confirmed that His140 and His193 (Figure 1) both contribute to Zn2+ effects [8]. Zn2+ appears to bind at the interface between monomers in the HVCN1 dimer (see below), where it prevents channel opening [24, 25].
Figure 1. Sequence of human HVCN1 showing its arrangement in the membrane.

Two sites on the intracellular amino terminus have been shown to influence channel opening: phosphorylation of Thr29 strongly enhances opening in leukocytes [45], whereas the missense mutation M91T reduces it in airway epithelial cells [27]. Histidines constituting Zn2+ binding site(s) [8, 24, 25] are indicated, together with transmembrane domains (four rectangular boxes).
The HVCN1 protein and structure-function relationships
In 2006, human HVCN1 was cloned [5], as were homologous genes in mice and Ciona intestinalis [9]. The human HVCN1 (Figure 1) is expressed in two isoforms, the full-length protein (273 amino acids), and a short form, which lacks the first 20 amino acids, thus far documented only in B cells [26]. The latter protein isoform derives from translation from an alternative initiation site. One naturally occurring mutation has been identified, M91T, which reduces the probability of channel opening [27].
The protein encoded by the HVCN1 gene has two remarkable features. First, no explicit aqueous pore that might provide a conduction pathway similar to other ion channels is evident [8, 9]. This feature was not altogether unexpected, because several properties of proton channels suggested the lack of a conventional aqueous pathway. That the deuterium conductance is only 50% that of H+ [28], conduction is strongly temperature-sensitive [29, 30], and the channel is perfectly selective [31, 32] suggested that protons permeate proton channels by a hydrogen-bonded chain mechanism [33], which does not require a continuous aqueous pathway. A recent study concludes that protons hop across frozen or non-exchangeable waters without displacing them [34]. Larger ions, in contrast, permeate ordinary ion channels by moving in single file along with water molecules. The second remarkable feature of HVCN1 is its astounding similarity to the voltage-sensing domain (VSD) of voltage-gated K+, Na+, and Ca2+ channels [8, 9]. Most voltage-gated ion channels are tetramers of subunits that span the membrane 6 times (Figure 2). The first four domains (S1–S4) comprise the voltage sensing apparatus (VSD), and the remaining two domains (S5–S6) from each of the four monomers collectively form a single aqueous pathway across the membrane. Although using 24 membrane-spanning domains to form a single pore might seem needlessly baroque, the result is a four-fold amplification of the voltage sensitivity of a single VSD. For ion channels that mediate nerve impulses or trigger contraction of muscle fibers, extreme sensitivity to small changes in membrane potential is essential.
Figure 2. Architectural distinction between K+ channels and proton channels.
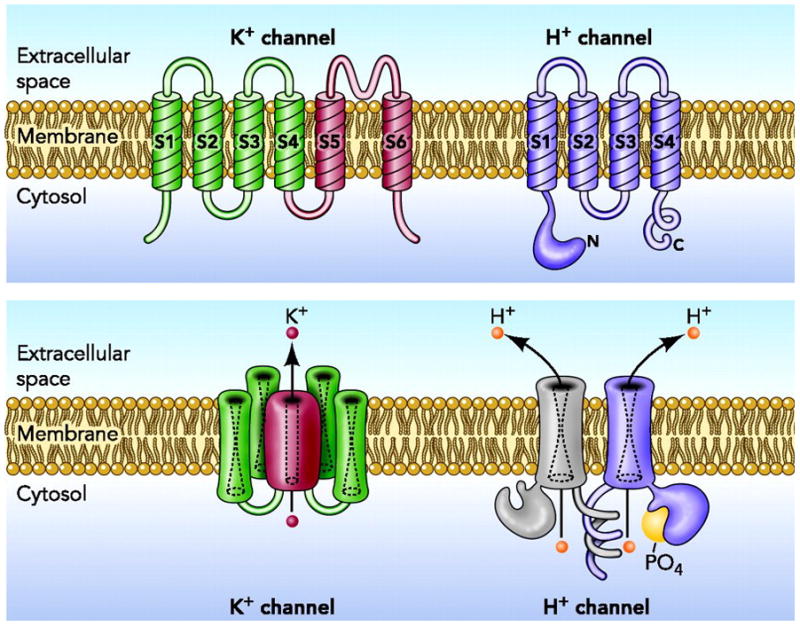
Voltage-gated K+ channels are tetramers of the subunit shown in the top left panel. The four S4-S5 domains combine to create a single central pore in the assembled channel (lower panel). The voltage-gated H+ channel (right, upper and lower panel) is a dimer, but each protomer contains its own conduction pathway and can function as a monomer. Proton channel activity is greatly enhanced by phosphorylation at Thr29 (Figure 1). Figure modified from [21] with permission of the American Physiological Society.
The discovery of the HVCN1 gene, and its homology with the VSDs of other, more widely studied ion channels, opened floodgates of interest in this molecule, with the result that new discoveries are being made rapidly. Diverse evidence indicates that the proton channel exists as a dimer, with a separate conduction pathway in each protomer [35–37]. Although most voltage-gated ion channels are tetramers with a single conduction pathway (Figure 2), the ClC (Cl− channel) family of Cl− channels and Cl− /H+ antiporters also are dimers with permeation pathways in each subunit [38–40]. Truncation of the HVCN1 carboxy terminus results in monomeric channels that retain most functions of the dimer [24, 36, 37]. The two protomers do not function independently in the dimer, however, but interact during gating [24, 25, 41, 42]. One consequence of this cooperativity is that the dimer has twice the voltage sensitivity of the monomer [41], despite channel opening being slower [24, 25, 37]. Teleologically, strong voltage sensitivity may be more important to the cell than rapid opening kinetics [24]. For example, in phagocytes, proton channels counterbalance the effects of NADPH oxidase, which remains active for minutes (Box 1). Strong voltage sensitivity activates proton channels before excessive depolarization can occur, precluding self-inhibition of this electrogenic enzyme [10], which both produces and is inhibited by membrane depolarization [13].
Box 1. NADPH oxidase and reactive oxygen species (ROS).
A schematic representation of NADPH oxidase and HVCN1 function in phagocytes (figure I). The NADPH oxidase enzymatic complex assembles on the plasma (or phagosome) membrane of granulocytes, macrophages and B cells upon stimulation. The membrane bound components, gp91phox (phox refers to phagocyte oxidase) and p22phox, bind the cytosolic components (p47phox, p67phox, p40phox and Rac) that are recruited to the membrane, which in phagocytic cells invaginates to produce a phagosome. NADPH oxidase produces superoxide anion, O2 •−, a precursor to other reactive oxygen species. Most O2 •− dismutates to hydrogen peroxide, H2O2, which is converted to HOCl by myeloperoxidase, MPO. Gp91phox, also called Nox2, contains an electron transport chain comprising NADPH, FAD and two heme groups that pass electrons sequentially to O2 at an external binding site, to produce O2 •− The enzyme is electrogenic, because electrons extracted from cytoplasmic NADPH are translocated to extracellular or intraphagosomal O2 that is thereby reduced to O2 •− [10]. Without charge compensation, the membrane would depolarize to extreme positive voltages at which NADPH oxidase ceases to function [13]. Proton currents provide most of this charge compensation [49], and alleviate the cytosolic acidification that results from NADPH utilization and reconstitution by the hexose monophosphate shunt (HMS), which also inhibits NADPH oxidase [51]. Other transporters that may contribute to pH regulation include ClC-3 (a Cl−/H+ antiporter), H-ATPase, and the Na+/H+ antiporter. ROS enable clearance of engulfed bacteria, as shown by impaired immune responses in chronic granulomatous disease (CGD) patients [81]. CGD occurs when mutations to components of NADPH oxidase prevent its function. The disease affects phagocytes and, to a lesser extent, B cells [82]. The primary goal of the high levels of ROS produced by phagocytes is clearance of bacteria. In B cells, where ROS production is about 10 times smaller than in phagocytes, ROS have been implicated in BCR signaling [66]. ROS temporarily inhibit protein-tyrosine phosphatases to allow initiation and amplification of BCR downstream signaling pathways. ROS oxidize the cysteine residue in tyrosine phosphatases catalytic domain to a sulfenic acid (-SOH). The reaction is reversible and the active thiolate anion (-S−) can be quickly reformed by reduction, mediated by the many reducing agents present in the cytosol. In recent years the number of signaling pathways and phosphatases shown to be regulated by oxidation has increased considerably [83, 84]. Figure reprinted from [21] with permission of the American Physiological Society.
Proton channels in phagocytes regulate NADPH oxidase activity
The best-established function of proton channels is in leukocytes that phagocytose and kill bacteria (Box 1). Proton channel properties change dramatically in activated leukocytes [43, 44]. In the enhanced gating mode, they open faster and at less positive voltages and close more slowly, substantially increasing the proton current. Enhanced gating results primarily from phosphorylation of HVCN1 by PKC (protein kinase C) on Thr29 of the intracellular amino terminus [45], and is prevented or reversed by PKC inhibitors [46, 47]. Part of this response may involve arachidonic acid (AA) [6, 48], produced locally by activated phagocytes, although it is unclear whether the effect is direct or via AA-mediated stimulation of PKC [47]. Without enhanced gating, proton channels would still open during NADPH oxidase activity, triggered by membrane depolarization, decreased intracellular pH (pHi), and increased pH in the extracellular milieu or phagosome. However, in the enhanced gating mode, more proton channels open with smaller depolarization. This, in turn, improves the efficiency of NADPH oxidase by 15–20% by minimizing depolarization-induced self-inhibition [49].
At the same time that proton flux compensates charge, it also prevents large changes in both cytoplasmic and phagosomal pH. Regulation of pHi is essential, because NADPH oxidase activity is optimal at pHi 7.5 and decreases drastically at more acidic and more basic pH [50]. The importance of proton channels in minimizing pHi changes during phagocytosis was demonstrated recently in a study using the pH-sensitive dye, SNARF (seminaphtorhodafluor) [51]. When a neutrophil first engulfed an opsonized zymosan particle, there was a precipitous drop in pHi, followed by an almost equally rapid recovery. This acidification reflected H+ generation as a consequence of NADPH oxidase activity, since it was abolished by the oxidase inhibitor, diphenylene iodonium (DPI). Inhibiting either Na+/H+ antiport or proton channel activity prevented pHi recovery. Acidification was more rapid in the presence of Zn2+ (but not inhibitors of other transporters) and in the HVCN1-deficient mouse [51], indicating that the proton channel is the first transporter to respond during phagocytosis. When proton channels were inhibited or absent, pHi dropped to levels known to inhibit NADPH oxidase [50]. This raises the philosophical question of whether charge compensation or pHi regulation is more important to the cell. Fortunately, proton channels perform both functions simultaneously and inseparably.
Using proton channels to compensate charge in phagocytes has two additional benefits [49]. Proton efflux minimizes osmotic effects that would occur if other ions performed this function. If all charge were compensated by K+, the phagosome would swell to ~20 times its original volume [49, 52]. Finally, H+ is required inside the phagosome as a substrate to form H2O2 and HOCl, the main products of NADPH oxidase, both of which are generated at a high rate [53].
The downstream effects of HVCN1 deficiency in neutrophils were investigated both in terms of ROS production and neutrophil function [17, 51, 54, 55]. All studies support the concept that HVCN1 is required for optimal ROS production. Furthermore, NADPH oxidase-dependent bacterial killing was significantly reduced in HVCN1-deficient neutrophils in vitro [54]. Bacterial clearance in HVCN1-deficient mice in vivo after peritoneal injection of Staphylococcus aureus was slightly but not significantly impaired. Clearance of Pseudomonas aeurginosa and Burkholderia cepacia was not impaired. The residual ROS production in HVCN1-deficient cells may be sufficient to ensure bacterial clearance to a point where the infection can be resolved. Patients with variant forms of chronic granulomatous disease (CGD, Box 1) whose NADPH oxidase activity is reduced to 3–30% normal may have milder symptoms than CGD patients with complete absence of NADPH oxidase activity [56–60]. However, defects in HVCN1 activity might become more relevant in different settings of inflammation and/or infection. Further work may clarify this point. HVCN1-deficient neutrophils also had diminished chemotactic responses to the bacterial peptide N-formyl-Met-Ile-Val-Ile-Leu (fMIVIL) in vitro [55]. Ca2+ entry was reduced, which in turn impaired migration and actin depolymerization. It would be interesting to see whether in vivo defects in HVCN1-deficient mice are more apparent in neutrophil responses in which migration is critical.
HVCN1 in basophils facilitates histamine release
Another cell with abundant HVCN1 expression is the basophil [61], not surprisingly given its lineage relationship with eosinophils and neutrophils. On the other hand, basophils lack NADPH oxidase, which seems to be the raison d’être for proton channels in phagocytes. Agents that stimulate histamine release by basophils, including anti-IgE, the phorbol ester PMA (phorbol 12-myristate 13-acetate), and the chemotactic peptide fMLF (formyl-Met-Leu-Phe), all enhanced the gating of proton channels in human basophils [62]. Inhibiting proton channels with Zn2+ abolished histamine release, suggesting that proton channels in basophils might compensate charge or regulate pH. Measurement of pHi during anti-IgE stimulation revealed acidification, which was exacerbated by Zn2+. Thus, stabilization of pHi is a likely function of HVCN1 in basophils. Histamine release is a clear example of a cellular process that is unrelated to NADPH oxidase activity, but is mediated by proton channels.
HVCN1 sustains optimal B cell receptor (BCR) signaling
Proton currents were discovered in human B cells in 2002 [63] and the HVCN1 protein was identified in a proteomic screen of plasma membrane proteins expressed by mantle cell lymphoma cells in the peripheral blood [64]. Protein expression levels in resting naïve and memory B cells were comparable to granulocytes; however, HVCN1 was down-regulated in proliferating B cells, such as cells in the germinal centre or primary cells stimulated in vitro via CD40 (a tumor necrosis factor receptor family member) in the presence of IL-4 (interleukin-4) [26]. Down-regulation may be mediated by BCL6 (B-cell lymphoma 6), since HVCN1 appears to be a direct target for BCL6-mediated transcriptional repression [65]. This pattern of expression suggests HVCN1 involvement in the initial phase of B-cell activation, which indeed was found impaired during in vivo and in vitro stimulation of HVCN1-deficient B cells. The impairment was mediated by diminished ROS production following BCR stimulation. ROS had been postulated to ensure correct BCR signal propagation by inhibiting protein tyrosine phosphatases, such as SHP-1 (Figure 3 and Box 1). This has been somewhat controversial [66] due to the possibility that reducing conditions in the cytosol would scavenge any ROS as soon as they were formed. Recent evidence, however, suggests that there are mechanisms to temporarily block the reducing action of cytosolic scavenging proteins such as peroxiredoxin [67]. The HVCN1-deficient model therefore provides a new way to illustrate how attenuation of ROS production in B cells has a detrimental effect on BCR downstream signaling. Diminished ROS correlates with diminished oxidation of the protein tyrosine phosphatase SHP-1, which dephosphorylates and therefore inhibits a crucial kinase in B-cell activation, spleen tyrosine kinase (Syk) [68]. Syk controls many downstream pathways such as MAP kinase activation, Ca2+ mobilization (from ER-stores as well as entry from the extracellular milieu) and PI3 kinase activation [69, 70]. Syk activation was indeed impaired in BCR-stimulated HVCN1-deficient cells, but not in the initial phases of activation, only at later time-points. The defect could be “rescued” by treating the cells with low doses of a SHP-1 inhibitor, sodium stibogluconate [71]. Surprisingly, not all Syk downstream pathways were affected equally as neither ERK activation nor Ca2+ mobilization was impaired. This suggests that these events are controlled by the very early activation of Syk and are not affected in the absence of sustained Syk activation. On the other hand, the protein kinase Akt, which is downstream of PI3K [72], was impaired in HVCN1-deficient cells and this resulted in decreased cell metabolism, as both mitochondrial respiration and glycolysis were diminished following BCR stimulation (Figure 3). The defects in HVCN1-deficient B cells were specific for BCR stimulation as downstream signaling from TLR4 (Toll-like receptor 4) and CD40 were unimpaired.
Figure 3. Schematic representation of HVCN1 in the context of B cell receptor (BCR) signaling.

Antigen (Ag) binding to the BCR results in phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) in the Igα/β heterodimer by Lyn, a src-family tyrosine kinase, creating docking sites for Syk [85]. This serves to amplify BCR signaling by further recruitment and activation of Syk, which leads to PI3K activation, activation of Akt and increased glucose uptake and metabolism. Amplification of signaling is negatively regulated by CD22, which is also phosphorylated by Lyn, providing a docking site for protein tyrosine phosphatase (PTP) SHP-1 [86]. SHP-1 dephosphorylates Syk, counterbalancing ITAM-Syk mediated signal amplification. SHP-1 is inhibited by ROS, which oxidize a cysteine residue in the catalytic site of the enzyme. BCR stimulation results in ROS generated by the NADPH oxidase enzymatic complex, which transfers electrons across the plasma/endosome membrane to molecules of oxygen. The transfer of one electron results in the production of O2 that combines with protons to form H2O2 and O2, which freely diffuse through the membrane (2O2 + 2H+ H2O2 + O2). ROS generate a localized oxidizing environment causing inhibition of SHP-1, which results in amplification of BCR signal. HVCN1 sustains NADPH oxidase activity through charge compensation and intracellular pH regulation; therefore, in the absence of HVCN1, the oxidizing environment cannot be maintained and this results in SHP-1 remaining more active, diminishing BCR-signal strength. Figure adapted from [26].
Consistent with the BCR-specific defect in signaling, HVCN1 was found to be associated with the BCR complex and to colocalize with the receptor upon stimulation. This raises the possibility that close proximity of H+ transport to the BCR might be important. Whether proximity is necessary to support NADPH oxidase activity or for other reasons is unknown and requires further investigation. Intriguingly, Ca2+ entry is impaired in the absence of HVCN1 in neutrophils [55] but not in B cells [26], which might reflect different signal regulation of Ca2+ mobilization in neutrophils and B cells or differences in membrane depolarization. Ca2+ entry is impaired by the large depolarization attained by HVCN1-deficient neutrophils, which attenuates entry of positive ions [55]. In contrast with neutrophils, B cells have substantial K+ conductances [73] that would abrogate any depolarization due to NADPH oxidase activity. Consequently, the driving force for Ca2+ entry should be preserved.
HVCN1 regulates human spermatozoa activation
Gene expression analysis (http://biogps.gnf.org/#goto=genereport&id=84329 and [9]) revealed HVCN1 expression in human testis and recently a role for HVCN1 in human spermatozoa was proposed [74]. Spermatozoa reside in the testis in a quiescent state that is maintained by low cytosolic pH, below 6.5. The abundant proton channels are thought to be inhibited by a high concentration of Zn2+ present in seminal fluid. Once the sperm enter the female reproductive tract, Zn2+ might be buffered by albumin and other proteins, relieving the inhibition. The resulting efflux of acid through proton channels increases pHi, triggering capacitation (maturation) that is necessary for egg fertilization.
Other species may lack this mechanism. Zn2+ enhances sperm motility in the Japanese eel [75]. Mouse spermatozoa lack proton currents, suggesting a different mechanism of alkalinization [74] although the murine cells investigated were less mature than the human counterparts [76]. Notably, HVCN1-deficient mice do not exhibit obvious fertility defects.
HVCN1 regulates pH in airway mucosa
A recent study by Iovannisci et al. [27] demonstrated HVCN1 involvement in establishing the pH in airway epithelia. Low pH is associated with diseases such as asthma and cystic fibrosis while alkaline pH is associated with rhinitis [77, 78], which emphasizes that pH regulation of airway surface liquid (ASL) needs to be finely tuned. The pH in ASL is controlled by acid secretion from airway epithelial cells. As in most cells, multiple mechanisms contribute to acid secretion. In human nasal mucosa, proton channels secrete over half the acid measured in ASL; this fraction changes in chronic rhinosinusitis [78]. Iovannisci et al. [27] found that HVCN1 opens when extracellular pH exceeds 7. They emphasize that in airway epithelia, proton channel gating is modulated by the difference in pH across the membrane rather than depolarization, because the epithelial membrane potential is stable.
Interestingly, this group identified a human subject with a missense mutation that produced a substitution of Met→Thr at position 91. Mutation M91T inhibited channel opening, with the mutant requiring +20 mV additional depolarization or ~0.5 unit more alkaline mucosal pH to open the same number of channels. The mutation is likely to be heterozygous, which implies two possible scenarios: 1) the mutant has dominant expression, in which case the observed effect is mediated by M91T/M91T dimers; or 2) dimers with the altered phenotype are composed of one mutant and one wild-type copy, suggesting that M91T/M91T dimers might have an even stronger phenotype. Surprisingly, Met91 is not well conserved among species. Macaca mulatta, Canis familiaris and Bos taurus have Thr at position 91 and mice have Arg. It is not clear if this produces different characteristics for HVCN1 in different species.
Therapeutic implications of targeting HVCN1
As descriptions of HVCN1 involvement in diverse cellular processes proliferate, the potential of HVCN1 as a therapeutic target expands. Beyond the archetypal role of HVCN1 in supporting bactericidal ROS production by phagocytes, recent studies describe a role for HVCN1 in regulating production of ROS intended for signaling. Significantly, in basophils, spermatozoa and airway epithelia, HVCN1 serves functions altogether unrelated to ROS production. Its regulation of histamine release by basophils and the threshold of activation of autoreactive B cells make HVCN1 a desirable therapeutic target in allergic reactions and autoimmune diseases characterized by hyperreactive B cells, such as rheumatoid arthritis and systemic lupus erythematosus. For B cells in particular, the potential for HVCN1 inhibitors to impair optimal B-cell activation without complete abrogation of B-cell responses should provide a useful therapeutic window, avoiding the increased susceptibility to infections risked by strong immunosuppression. Some B-cell malignancies have been shown to rely on BCR signaling [79, 80]; thus, inhibiting HVCN1 in HVCN1-expressing tumors could help reduce the survival signal that cells derive via engagement of their BCR. An HVCN1 inhibitor introduced into the female reproductive system might block capacitation (activation) of spermatozoa and thus act as a contraceptive. In asthma and cystic fibrosis, inhibiting HVCN1 should ameliorate mucosal acidification, preventing epithelial injury that may contribute to asthma pathogenesis. But can HVCN1 inhibition be achieved? Its structural similarities to other voltage-gated cation channels raise the possibility that some side-targeting might occur. And even if side-targeting could be avoided, lack of a crystal structure makes it difficult to predict which residues mediate proton transport and how the conformation could be altered to achieve proton transport inhibition. Identifying an effective inhibitor might be a difficult task, but surely the therapeutic possibilities will be abundant.
Concluding remarks
Interest in voltage-gated proton channels mushroomed after identification of the gene in 2006. The molecule has its own unique charm, with perfect selectivity, tiny conductance, strong modulation by pH and unusual dimeric architecture. Its homology to the VSD of other voltage-gated ion channels makes it a unique model system in which voltage-gating mechanisms may be studied. Many questions remain unanswered regarding the structure of the channel (Box 2); surely a great boost to HVCN1 research would come from unveiling the crystal structure. In addition to helping to unravel the molecular mechanisms underlying proton conductance, structural information would likely prove invaluable in the design of HVCN1 inhibitors. These inhibitors, in turn, would help further understanding of HVCN1 function in cells.
Box 2. Major questions for future research.
What is the crystal structure?
What interactions take place between monomers during gating of the dimeric proton channel complex?
Where is the permeation pathway? How is selectivity achieved? What is the difference between the open and closed states?
Which cells express proton channels, and for what purposes?
Are there other naturally-occurring mutations in the HVCN1 sequence that have consequences for its function?
Can selective inhibitors HVCN1 be identified? Can inhibitors be targeted to specific tissues? Will these inhibitors ameliorate diseases that might benefit from HVCN1 inhibition?
So far HVCN1 has been shown to regulate neutrophil, basophil and B-cell activation, human sperm capacitation and pH in mucosal airway epithelial cells. As expression in more cells is discovered, new functions will undoubtedly be identified. Furthermore, additional mutations or polymorphisms that alter channel properties might emerge. Exploration of what are now only putative proton channels in other species will provide clues to understanding differences in HVCN1 properties in diverse species. Beyond its intrinsic interest to evolutionary biologists, comparison of a wide range of proteins will illuminate our understanding of how these extraordinary molecules work.
Figure I.
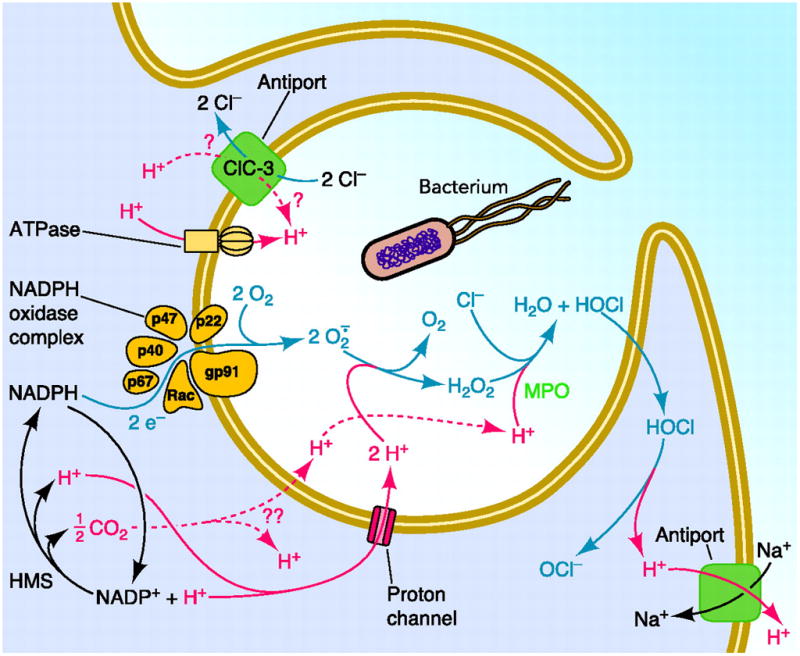
Schematic representation of NADPH oxidase and HVCN1 function in phagocytes.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Melania Capasso, Institute of Cancer, Barts and The London School of Medicine & Dentistry, Queen Mary University of London, Charterhouse Square, London, EC1M 6BQ, UK.
Thomas E. DeCoursey, Department of Molecular Biophysics and Physiology, Rush University Medical Center, 1750 West Harrison Street, Chicago, IL 60612-3824, USA
Martin J. S. Dyer, MRC Toxicology Unit, University of Leicester, Hodgkin Building, Lancaster Road, Leicester, LE1 9HN, UK
References
- 1.Thomas RC, Meech RW. Hydrogen ion currents and intracellular pH in depolarized voltage-clamped snail neurones. Nature. 1982;299:826–828. doi: 10.1038/299826a0. [DOI] [PubMed] [Google Scholar]
- 2.Byerly L, et al. Rapidly activating hydrogen ion currents in perfused neurones of the snail, Lymnaea stagnalis. J Physiol. 1984;351:199–216. doi: 10.1113/jphysiol.1984.sp015241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barish ME, Baud C. A voltage-gated hydrogen ion current in the oocyte membrane of the axolotl, Ambystoma. J Physiol. 1984;352:243–263. doi: 10.1113/jphysiol.1984.sp015289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeCoursey TE. Hydrogen ion currents in rat alveolar epithelial cells. Biophys J. 1991;60:1243–1253. doi: 10.1016/S0006-3495(91)82158-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernheim L, et al. A voltage-dependent proton current in cultured human skeletal muscle myotubes. J Physiol. 1993;470:313–333. doi: 10.1113/jphysiol.1993.sp019860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeCoursey TE, Cherny VV. Potential, pH, and arachidonate gate hydrogen ion currents in human neutrophils. Biophys J. 1993;65:1590–1598. doi: 10.1016/S0006-3495(93)81198-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Demaurex N, et al. Proton currents in human granulocytes: regulation by membrane potential and intracellular pH. J Physiol. 1993;466:329–344. [PMC free article] [PubMed] [Google Scholar]
- 8.Ramsey IS, et al. A voltage-gated proton-selective channel lacking the pore domain. Nature. 2006;440:1213–1216. doi: 10.1038/nature04700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sasaki M, et al. A voltage sensor-domain protein is a voltage-gated proton channel. Science. 2006;312:589–592. doi: 10.1126/science.1122352. [DOI] [PubMed] [Google Scholar]
- 10.Henderson LM, et al. The superoxide-generating NADPH oxidase of human neutrophils is electrogenic and associated with an H+ channel. Biochem J. 1987;246:325–329. doi: 10.1042/bj2460325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henderson LM, et al. Superoxide generation by the electrogenic NADPH oxidase of human neutrophils is limited by the movement of a compensating charge. Biochem J. 1988;255:285–290. [PMC free article] [PubMed] [Google Scholar]
- 12.Henderson LM, et al. Internal pH changes associated with the activity of NADPH oxidase of human neutrophils. Further evidence for the presence of an H+ conducting channel. Biochem J. 1988;251:563–567. doi: 10.1042/bj2510563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeCoursey TE, et al. The voltage dependence of NADPH oxidase reveals why phagocytes need proton channels. Nature. 2003;422:531–534. doi: 10.1038/nature01523. [DOI] [PubMed] [Google Scholar]
- 14.DeCoursey TE. Voltage-gated proton channels and other proton transfer pathways. Physiol Rev. 2003;83:475–579. doi: 10.1152/physrev.00028.2002. [DOI] [PubMed] [Google Scholar]
- 15.Cherny VV, et al. The voltage-activated hydrogen ion conductance in rat alveolar epithelial cells is determined by the pH gradient. J Gen Physiol. 1995;105:861–896. doi: 10.1085/jgp.105.6.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeCoursey TE. Voltage-gated proton channels. Cell Mol Life Sci. 2008;65:2554–2573. doi: 10.1007/s00018-008-8056-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Okochi Y, et al. Voltage-gated proton channel is expressed on phagosomes. Biochem Biophys Res Commun. 2009;382:274–279. doi: 10.1016/j.bbrc.2009.03.036. [DOI] [PubMed] [Google Scholar]
- 18.Schapiro FB, Grinstein S. Determinants of the pH of the Golgi complex. J Biol Chem. 2000;275:21025–21032. doi: 10.1074/jbc.M002386200. [DOI] [PubMed] [Google Scholar]
- 19.Li SJ, et al. The role and structure of the carboxyl-terminal domain of the human voltage-gated proton channel Hv1. J Biol Chem. 2010;285:12047–12054. doi: 10.1074/jbc.M109.040360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cherny VV, et al. Properties of single voltage-gated proton channels in human eosinophils estimated by noise analysis and by direct measurement. J Gen Physiol. 2003;121:615–628. doi: 10.1085/jgp.200308813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeCoursey TE. Voltage-gated proton channels find their dream job managing the respiratory burst in phagocytes. Physiology (Bethesda) 2010;25:27–40. doi: 10.1152/physiol.00039.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahaut-Smith MP. The effect of zinc on calcium and hydrogen ion currents in intact snail neurones. J Exp Biol. 1989;145:455–464. doi: 10.1242/jeb.145.1.455. [DOI] [PubMed] [Google Scholar]
- 23.Cherny VV, DeCoursey TE. pH-dependent inhibition of voltage-gated H+ currents in rat alveolar epithelial cells by Zn2+ and other divalent cations. J Gen Physiol. 1999;114:819–838. doi: 10.1085/jgp.114.6.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Musset B, et al. Zinc inhibition of monomeric and dimeric proton channels suggests cooperative gating. J Physiol. 2010;588:1435–1449. doi: 10.1113/jphysiol.2010.188318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Musset B, et al. Oligomerization of the voltage gated proton channel. Channels (Austin) 2010;4:260–265. doi: 10.4161/chan.4.4.12789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Capasso M, et al. HVCN1 modulates BCR signal strength via regulation of BCR-dependent generation of reactive oxygen species. Nat Immunol. 2010;11:265–272. doi: 10.1038/ni.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iovannisci D, et al. Function of the HVCN1 proton channel in airway epithelia and a naturally occurring mutation, M91T. J Gen Physiol. 2010;136:35–46. doi: 10.1085/jgp.200910379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DeCoursey TE, Cherny VV. Deuterium isotope effects on permeation and gating of proton channels in rat alveolar epithelium. J Gen Physiol. 1997;109:415–434. doi: 10.1085/jgp.109.4.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeCoursey TE, Cherny VV. Temperature dependence of voltage-gated H+ currents in human neutrophils, rat alveolar epithelial cells, and mammalian phagocytes. J Gen Physiol. 1998;112:503–522. doi: 10.1085/jgp.112.4.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuno M, et al. Temperature dependence of proton permeation through a voltage-gated proton channel. J Gen Physiol. 2009;134:191–205. doi: 10.1085/jgp.200910213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeCoursey TE, Cherny VV. Voltage-activated hydrogen ion currents. J Membr Biol. 1994;141:203–223. doi: 10.1007/BF00235130. [DOI] [PubMed] [Google Scholar]
- 32.Lukacs GL, et al. Proton conductance of the plasma membrane: properties, regulation, and functional role. Am J Physiol. 1993;265:C3–14. doi: 10.1152/ajpcell.1993.265.1.C3. [DOI] [PubMed] [Google Scholar]
- 33.Nagle JF, Morowitz HJ. Molecular mechanisms for proton transport in membranes. Proc Natl Acad Sci U S A. 1978;75:298–302. doi: 10.1073/pnas.75.1.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramsey IS, et al. An aqueous H+ permeation pathway in the voltage-gated proton channel Hv1. Nat Struct Mol Biol. 2010;17:869–875. doi: 10.1038/nsmb.1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee SY, et al. Dimeric subunit stoichiometry of the human voltage-dependent proton channel Hv1. Proc Natl Acad Sci U S A. 2008;105:7692–7695. doi: 10.1073/pnas.0803277105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tombola F, et al. The voltage-gated proton channel Hv1 has two pores, each controlled by one voltage sensor. Neuron. 2008;58:546–556. doi: 10.1016/j.neuron.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koch HP, et al. Multimeric nature of voltage-gated proton channels. Proc Natl Acad Sci U S A. 2008;105:9111–9116. doi: 10.1073/pnas.0801553105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller C, White MM. A voltage-dependent chloride conductance channel from Torpedo electroplax membrane. Ann N Y Acad Sci. 1980;341:534–551. doi: 10.1111/j.1749-6632.1980.tb47197.x. [DOI] [PubMed] [Google Scholar]
- 39.Middleton RE, et al. Homodimeric architecture of a ClC-type chloride ion channel. Nature. 1996;383:337–340. doi: 10.1038/383337a0. [DOI] [PubMed] [Google Scholar]
- 40.Ludewig U, et al. Two physically distinct pores in the dimeric ClC-0 chloride channel. Nature. 1996;383:340–343. doi: 10.1038/383340a0. [DOI] [PubMed] [Google Scholar]
- 41.Gonzalez C, et al. Strong cooperativity between subunits in voltage-gated proton channels. Nat Struct Mol Biol. 2010;17:51–56. doi: 10.1038/nsmb.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tombola F, et al. The opening of the two pores of the Hv1 voltage-gated proton channel is tuned by cooperativity. Nat Struct Mol Biol. 2010;17:44–50. doi: 10.1038/nsmb.1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bánfi B, et al. A novel H+ conductance in eosinophils: unique characteristics and absence in chronic granulomatous disease. J Exp Med. 1999;190:183–194. doi: 10.1084/jem.190.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DeCoursey TE, et al. Simultaneous activation of NADPH oxidase-related proton and electron currents in human neutrophils. Proc Natl Acad Sci U S A. 2000;97:6885–6889. doi: 10.1073/pnas.100047297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Musset B, et al. Identification of Thr29 as a critical phosphorylation site that activates the human proton channel Hvcn1 in leukocytes. J Biol Chem. 2010;285:5117–5121. doi: 10.1074/jbc.C109.082727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bankers-Fulbright JL, et al. Regulation of human eosinophil NADPH oxidase activity: a central role for PKC. J Cell Physiol. 2001;189:306–315. doi: 10.1002/jcp.10022. [DOI] [PubMed] [Google Scholar]
- 47.Morgan D, et al. Sustained activation of proton channels and NADPH oxidase in human eosinophils and murine granulocytes requires PKC but not cPLA2 activity. J Physiol. 2007;579:327–344. doi: 10.1113/jphysiol.2006.124248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Henderson LM, Chappell JB. The NADPH-oxidase-associated H+ channel is opened by arachidonate. Biochem J. 1992;283:171–175. doi: 10.1042/bj2830171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murphy R, DeCoursey TE. Charge compensation during the phagocyte respiratory burst. Biochim Biophys Acta. 2006;1757:996–1011. doi: 10.1016/j.bbabio.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 50.Morgan D, et al. The pH dependence of NADPH oxidase in human eosinophils. J Physiol. 2005;569:419–431. doi: 10.1113/jphysiol.2005.094748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morgan D, et al. Voltage-gated proton channels maintain pH in human neutrophils during phagocytosis. Proc Natl Acad Sci U S A. 2009;106:18022–18027. doi: 10.1073/pnas.0905565106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reeves EP, et al. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. 2002;416:291–297. doi: 10.1038/416291a. [DOI] [PubMed] [Google Scholar]
- 53.Winterbourn CC, et al. Modeling the reactions of superoxide and myeloperoxidase in the neutrophil phagosome: implications for microbial killing. J Biol Chem. 2006;281:39860–39869. doi: 10.1074/jbc.M605898200. [DOI] [PubMed] [Google Scholar]
- 54.Ramsey IS, et al. Hv1 proton channels are required for high-level NADPH oxidase-dependent superoxide production during the phagocyte respiratory burst. Proc Natl Acad Sci U S A. 2009;106:7642–7647. doi: 10.1073/pnas.0902761106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.El Chemaly A, et al. VSOP/Hv1 proton channels sustain calcium entry, neutrophil migration, and superoxide production by limiting cell depolarization and acidification. J Exp Med. 2010;207:129–139. doi: 10.1084/jem.20091837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lew PD, et al. A variant of chronic granulomatous disease: deficient oxidative metabolism due to a low-affinity NADPH oxidase. N Engl J Med. 1981;305:1329–1333. doi: 10.1056/NEJM198111263052207. [DOI] [PubMed] [Google Scholar]
- 57.Shurin SB, et al. Impaired granulocyte superoxide production and prolongation of the respiratory burst due to a low-affinity NADPH-dependent oxidase. Blood. 1983;62:564–571. [PubMed] [Google Scholar]
- 58.Roos D, et al. Chronic granulomatous disease with partial deficiency of cytochrome b558 and incomplete respiratory burst: variants of the X-linked, cytochrome b558-negative form of the disease. J Leukoc Biol. 1992;51:164–171. doi: 10.1002/jlb.51.2.164. [DOI] [PubMed] [Google Scholar]
- 59.Curnutte JT. Chronic granulomatous disease: the solving of a clinical riddle at the molecular level. Clin Immunol Immunopathol. 1993;67:S2–15. doi: 10.1006/clin.1993.1078. [DOI] [PubMed] [Google Scholar]
- 60.Bu-Ghanim HN, et al. Molecular analysis in three cases of X91- variant chronic granulomatous disease. Blood. 1995;86:3575–3582. [PubMed] [Google Scholar]
- 61.Cherny VV, Thomas LL, DeCoursey TE. Voltage-gated proton currents in human basophils. Biologicheskie Membrany. 2001;18:458–465. [Google Scholar]
- 62.Musset B, et al. A pH-stabilizing role of voltage-gated proton channels in IgE-mediated activation of human basophils. Proc Natl Acad Sci U S A. 2008;105:11020–11025. doi: 10.1073/pnas.0800886105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schilling T, et al. Voltage-activated proton currents in human lymphocytes. J Physiol. 2002;545:93–105. doi: 10.1113/jphysiol.2002.028878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Boyd RS, et al. Protein profiling of plasma membranes defines aberrant signaling pathways in mantle cell lymphoma. Mol Cell Proteomics. 2009;8:1501–1515. doi: 10.1074/mcp.M800515-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Basso K, et al. Integrated biochemical and computational approach identifies BCL6 direct target genes controlling multiple pathways in normal germinal center B cells. Blood. 2010;115:975–984. doi: 10.1182/blood-2009-06-227017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reth M. Hydrogen peroxide as second messenger in lymphocyte activation. Nat Immunol. 2002;3:1129–1134. doi: 10.1038/ni1202-1129. [DOI] [PubMed] [Google Scholar]
- 67.Woo HA, et al. Inactivation of peroxiredoxin I by phosphorylation allows localized H2O2 accumulation for cell signaling. Cell. 2010;140:517–528. doi: 10.1016/j.cell.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 68.Dustin LB, et al. Expression of dominant-negative src-homology domain 2-containing protein tyrosine phosphatase-1 results in increased Syk tyrosine kinase activity and B cell activation. J Immunol. 1999;162:2717–2724. [PubMed] [Google Scholar]
- 69.Kurosaki T. Molecular mechanisms in B cell antigen receptor signaling. Curr Opin Immunol. 1997;9:309–318. doi: 10.1016/s0952-7915(97)80075-1. [DOI] [PubMed] [Google Scholar]
- 70.Beitz LO, et al. SYK is upstream of phosphoinositide 3-kinase in B cell receptor signaling. J Biol Chem. 1999;274:32662–32666. doi: 10.1074/jbc.274.46.32662. [DOI] [PubMed] [Google Scholar]
- 71.Pathak MK, Yi T. Sodium stibogluconate is a potent inhibitor of protein tyrosine phosphatases and augments cytokine responses in hemopoietic cell lines. J Immunol. 2001;167:3391–3397. doi: 10.4049/jimmunol.167.6.3391. [DOI] [PubMed] [Google Scholar]
- 72.Franke TF, et al. PI3K: downstream AKTion blocks apoptosis. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 73.Wulff H, et al. K+ channel expression during B cell differentiation: implications for immunomodulation and autoimmunity. J Immunol. 2004;173:776–786. doi: 10.4049/jimmunol.173.2.776. [DOI] [PubMed] [Google Scholar]
- 74.Lishko PV, et al. Acid extrusion from human spermatozoa is mediated by flagellar voltage-gated proton channel. Cell. 2010;140:327–337. doi: 10.1016/j.cell.2009.12.053. [DOI] [PubMed] [Google Scholar]
- 75.Yamaguchi S, et al. Zinc is an essential trace element for spermatogenesis. Proc Natl Acad Sci U S A. 2009;106:10859–10864. doi: 10.1073/pnas.0900602106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Florman HM, et al. Shedding light on sperm pHertility. Cell. 2010;140:310–312. doi: 10.1016/j.cell.2010.01.035. [DOI] [PubMed] [Google Scholar]
- 77.Fischer H, Widdicombe JH. Mechanisms of acid and base secretion by the airway epithelium. J Membr Biol. 2006;211:139–150. doi: 10.1007/s00232-006-0861-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cho DY, et al. Proton secretion in freshly excised sinonasal mucosa from asthma and sinusitis patients. Am J Rhinol Allergy. 2009;23:e10–13. doi: 10.2500/ajra.2009.23.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen L, et al. SYK-dependent tonic B-cell receptor signaling is a rational treatment target in diffuse large B-cell lymphoma. Blood. 2008;111:2230–2237. doi: 10.1182/blood-2007-07-100115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Davis RE, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463:88–92. doi: 10.1038/nature08638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Holland SM. Chronic granulomatous disease. Clin Rev Allergy Immunol. 2010;38:3–10. doi: 10.1007/s12016-009-8136-z. [DOI] [PubMed] [Google Scholar]
- 82.Volkman DJ, et al. B cell lines as models for inherited phagocytic diseases: abnormal superoxide generation in chronic granulomatous disease and giant granules in Chediak-Higashi syndrome. J Immunol. 1984;133:3006–3009. [PubMed] [Google Scholar]
- 83.Meng TC, et al. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 84.Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15:247–254. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- 85.Reth M, Brummer T. Feedback regulation of lymphocyte signalling. Nat Rev Immunol. 2004;4:269–277. doi: 10.1038/nri1335. [DOI] [PubMed] [Google Scholar]
- 86.Cornall RJ, et al. Polygenic autoimmune traits: Lyn, CD22, and SHP-1 are limiting elements of a biochemical pathway regulating BCR signaling and selection. Immunity. 1998;8:497–508. doi: 10.1016/s1074-7613(00)80554-3. [DOI] [PubMed] [Google Scholar]