

Abstract
Verapamil has been shown to be neuroprotective in several acute neurotoxicity models due to blockade of calcium entry into neurons. However, the potential use of verapamil to treat chronic neurodegenerative diseases has not been reported. Using rat primary mesencephalic neuron/glia cultures, we report that verapamil significantly inhibited LPS-induced dopaminergic neurotoxicity in both pre- and post-treatment experiments. Reconstituted culture studies revealed that the presence of microglia was essential in verapamil-elicited neuroprotection. Mechanistic studies showed that decreased production of inflammatory mediators from LPS-stimulated microglia underlay neuroprotective property of verapamil. Further studies demonstrated that microglial NADPH oxidase (PHOX), the key superoxide-producing enzyme, but not calcium channel in neurons, is the site of action for the neuroprotective effect of verapamil. This conclusion was supported by the following two observations: 1) Verapamil failed to show protective effect on LPS-induced dopaminergic neurotoxicity in PHOX-deficient (deficient in the catalytic subunit of gp91phox) neuron/glia cultures; 2) Ligand binding studies showed that the binding of [3H]Verapamil onto gp91phox transfected COS-7 cell membranes was higher than the non-transfected control. The calcium channel-independent neuroprotective property of verapamil was further supported by the finding that R(+)-verapamil, a less active form in blocking calcium channel, showed the same potency in neuroprotection, inhibition of pro-inflammatory factors production and binding capacity to gp91phox membranes as R(-)-verapamil, the active isomer of calcium channel blocker. In conclusion, our results demonstrate a new indication of verapamil-mediated neuroprotection through a calcium channel-independent pathway and provide a valuable avenue for the development of therapy for inflammation-related neurodegenerative diseases.
Keywords: calcium channel blocker, oxidative stress, neuroinflammation
1. Introduction
Calcium channel blockers (CCB) were developed to reduce smooth and cardiac muscle excitability and have proven to be beneficial for hypertension, ischemic heart disease, and cardiac arrhythmia (Triggle, 1991) by blocking voltage-dependent calcium channels (Fleckenstein, 1983). In addition, CCB have been shown experimentally to affect a wide range of disorders outside the cardiovascular system; for example, CCBs attenuate acute hepato-cellular injury (Deakin et al., 1991; Mustafa and Olson, 1999) and prolong survival time in various animal models of endotoxic shock (Bosson et al., 1985). Furthermore, reports have also found neuroprotective effects of CCB in cerebral ischemia (Ginsberg, 2008; Kobayashi and Mori, 1998; Li et al., 2007; Schurr, 2004). Previous studies have also found that verapamil inhibits activation of superoxide production in neutrophils (Irita et al., 1986) and macrophages (Hotchkiss et al., 1997). These findings prompted us to investigate possible roles of superoxide and NADPH oxidase, a key superoxide-producing enzyme in microglia, (PHOX, phagocyte oxidase), in neuroprotective effects of verapamil.
Verapamil belongs to the dihydropyridine family of the CCBs and is used clinically to treat high blood pressure, angina and cardiac arrhythmias. It is generally believed that neuroprotection exhibited by verapamil is due to acute blockade of calcium entry into neurons. However, neurodegenerative diseases are associated with prolonged progressive degeneration, oxidative stress and microglial activation and the potential use of verapamil in chronic neurodegenerative diseases, such as Parkinson’s disease, has not been studied.
While the clinical effects of verapamil mentioned above are believed to be mediated through blockade of the calcium channel, the molecular mechanism underlying verapamil-elicited neuroprotection is unclear. Using two optical isomers of verapamil (S-(-) verapamil, an effective CCB, and R-(+)-verapamil, which exhibits lower affinity in binding to the calcium channel) to investigate the underlying molecular mechanism of verapmil’s protective effect on dorpaminergic (DA) neurons in in vitro models of PD. Here, we report that both isomers of verapamil are equi-potent in down-regulating LPS-induced microglia activation and show similar efficacy in neuroprotection, suggesting that verapamil-mediated neuroprotection is not mediated through its blockade of calcium channel. Instead, our studies show that verapamil-elicited neuroprotection is through inhibition of microglial PHOX activity via binding to its catalytic subunit gp91. These results suggest a potential new therapeutic use of verapamil with a novel mechanism for the treatment of inflammation-related neurodegenerative diseases.
2. Materials and Methods
2.1 Animals
Timed-pregnant (gestational day 14) adult female Fisher 344 rats were purchased from Charles River Laboratories (Kingston MA, USA). Eight-wk-old (25–30 g) male and female B6.129S6-Cybbtm1Din (PHOX−/−) and C57BL/6J (PHOX+/+) mice were purchased from Jackson Laboratories (Bar Harbor, Maine, USA) and maintained in a strict pathogen free environment. Housing and breeding of the animals were performed in strict accordance with the National Institutes of Health guidelines.
2.2 Reagents
Lipopolysaccharide (LPS) (strain O111:B4) was purchased from Calbiochem (San Diego, CA, USA). Geneticin was purchased from Gibco (Rockville, MD). Puromycin was purchased from Invitrogen (Carlsbad, CA). Cell culture ingredients were obtained from Life Technologies (Grand Island, NY, USA). [3H]Dopamine (DA, 34.8 Ci·mmol-1) were purchased from PerkinElmer (Boston, MA, USA). [3H]Verapamil (80 Ci·mmol-1) were purchased from American Radiolabled Chemicals Inc. The polyclonal antibody against tyrosine hydroxylase was purchased from Millipore Corporation Marlboro, MA, USA). The biotinylated horse anti-mouse and goat anti-rabbit secondary antibodies were purchased from Vector Laboratories (Burlingame, CA, USA). WST-1 was purchased from Dojindo Laboratories (Gaithersburg, MD, USA). TNF-α enzyme-linked immunosorbent assay (ELISA) kits were purchased from R&D Systems Inc. (Minneapolis, MN, USA). All other reagents came from Sigma Aldrich Chemical Co. (St. Louis, MO, USA).
2.3 Primary cultures
2.3.1 Mesencephalic neuron-glia cultures
Rat and mouse ventral mesencephalic neuron-glia cultures were prepared using a described protocol (Liu et al., 2000a). Briefly, midbrain tissues were dissected from day 14 Fisher 344 rat embryos or day 13 mouse embryos (PHOX+/+ or PHOX−/−). Cells were dissociated via gentle mechanical trituration in minimum essential medium (MEM) and immediately seeded (5×105/well) in poly D-lysine (20 μg·mL-1) precoated 24-well plates. Cells were seeded in maintenance media and treated with the treatment media described previously. Three days after seeding, the cells were replenished with 500 μL of fresh maintenance media. Cultures were exposed 7 days after seeding.
2.3.2 Microglia-enriched cultures
Primary enriched-microglia cultures were prepared from whole brains of 1-day-old Fisher 344 rat pups, using the previously described procedure (Liu et al., 2000a). Briefly, after removing meninges and blood vessels, the brain tissue was gently triturated and seeded (5×107) in 175 cm2 flasks. One week after seeding, the media was replaced. Two weeks after seeding, cells reached confluence and microglia were shaken off and either replated at 1 × 105 in a 96-well plate precoated with poly d-Lysine, or reseeded on top of a neuron-enriched culture in a 24-well plate at 1 × 105 (20 %) for a microglia add-back culture. Cells were treated 24 h after seeding enriched microglia.
2.3.3 Neuron-enriched cultures
Mesencephalic neuron-glia cultures were seeded (5×105/well) in 24 well plates precoated with poly d-lysine. Twenty-four hours postseeding, 5-10 μmol·L-1 cytosine ß-d-arabinofuranoside was added to the culture. After 2 days, the cytosine ß-d-arabinofuranoside was removed and replaced with fresh media. Neuron-enriched cultures are 98 % pure, as indicated by ICC staining with OX-42 and GFAP antibodies. Neuron-enriched cultures were treated 7 days post-seeding. For microglia or astrocyte add-back cultures, microglia or astrocyte were plated on top of the neuron-enriched culture at 6 days postseeding. The amount of microglia or astrocyte added represents 20 % of the total cell number in the cultures. Cells were treated 7 days after the initial seeding of the neuron-enriched cultures.
2.3.4 Astrocyte-enriched cultures
Primary astrocytes were prepared as described previously (Liu et al. 2001). Briefly, the mixed glial cultures, after the separation of microglia, were detached with trypsin–EDTA and seeded in the same culture medium as that used for microglia. After at least five consecutive passages, cells were added to neuron–enriched cultures in 24- well plates for DA uptake experiments. Immunocytochemical staining of the astrocyte-enriched cultures with either OX-42 or anti-GFAP antibody indicated purity of 98%.
2.3.5 Microglia-depleted cultures
Primary microglia-depleted cultures were prepared as described previously (Wang et al., 2006) by adding 1 μmol·L-1 leucine methyl ester into primary neuron-glia cultures 24 h after initial seeding. The medium was changed 6 days later. Seven-day-old cultures were used for treatment. At the time of treatment, ICC analysis indicated that the microglial composition was <0.1%.
2.4 COS7 cell lines cultures
COS-7 cells were maintained at 37°C in DMEM (Dulbecco’s modified Eagle’s medium; Sigma) supplemented with 10 % (v/v) fetal calf serum, 50 units·mL-1 penicillin and 50 μg·mL-1 streptomycin. For COS7-gp91 cell line, geneticin (Gibco) 0.8 mg·mL-1 and puromycin (Inviogen) 2μg·mL-1 were added in a humidified incubator with 5% CO2 and 95% air. The cells were split or harvest every 3-5 days.
2.5 [3H]Dopamine uptake assay
[3H]-DA uptake assays were performed as described (Liu et al., 2000b). Briefly, cells were incubated for 20 min at 37°C with 1 μM [3H]-DA in Krebs-Ringer buffer (16 mmol·L-1 sodium phosphate, 119 mmol·L-1 NaCl, 4.7 mmol·L-1 KCl, 1.8 mmol·L-1 CaCl2, 1.2 mmol·L-1 MgSO4, 1.3 mmol·L-1 EDTA, and 5.6 mmol·L-1 glucose; pH 7.4). Cells were washed with ice-cold Krebs-Ringer buffer three times, after which the cells were collected in 1N NaOH. Radioactivity was determined by liquid scintillation counting. Nonspecific DA uptake observed in the presence of mazindol (10 μ mol·L-1) was subtracted.
2.6 Membrane preparation and binding assay
Cells were lysed and homogenized with lysis buffer (20 nmol·L-1 Tris, pH=7.4, 2 mmol·L-1 EDTA, 10μg·mL-1 CLAP, a mixture of protease inhibitors: chymostatin, leupeptin, antipain and pepstatin A). Lysate was transferred to a 50 mL conical tube and centrifuged at 250 g for 10 min at 4°C. The supernatant was transferred and spun at 100,000 g for 90 min. Pellets were resuspended in 2 mL lysis buffer. Protein content was determined by Bradford’s method. 500μg membrane extracts or BSA was incubated with [3H]Verapamil for 120 min at 4°C in hanks buffer which containing 1% BSA, making a final volume of 500 μL. Incubation was terminated by adding 1 mL of ice-cold assay buffer followed by immediate filtration through glass fiber filters (Sigma-Aldrich LOT NoZK022) supported on a 12-port filter manifold. Filters were immediately washed three times with 5 mL of ice-cold assay buffer and dried in an oven at 60°C for 2 h before adding 15mL of Triton-toluene based scintillation fluid. Membrane bound radio-ligand trapped in the filters were counted in a Packard liquid scintillation analyzer (TRI-CARB 2300TR, Packard BioScience, USA). In each experiment, nonspecifically bound radioligand agents were measured by incubating BSA protein. Specific binding was then obtained by deducting this value from the total binding of radio-ligand agents for each sample.
2.7 Immunocytochemistry
Immunostaining was performed as previously described (Gao et al., 2002). Briefly, cultures were fixed for 20min with 3.7% formaldehyde in phosphate buffered saline (PBS). After two PBS washes (10 minutes), cultures were treated with a 1% solution of hydrogen peroxide 10 minutes, followed by three PBS washes, then incubated 40 minutes in blocking solution containing PBS, 1% bovine serum albumin, 0.4% Triton X-100 and 4% normal goat serum. Cultures were incubated overnight at 4°C with a primary antibody against TH (1:20,000) in DAKO antibody diluent. The next day cultures were washed three times with PBS; incubated 1hr with goat anti-rabbit biotinylated secondary antibody in PBS containing 0.3% Triton X-100 (1:277); followed by three PBS washes.
Subsequently, cultures were incubated 1hr with Vectastain ABC reagents diluted in PBS containing 0.3% Triton X-100, followed by three PBS washes. Color was developed with 3,3’-diaminobenzidine/urea-hydrogen peroxide tablets from Sigma dissolved in water for 3 minutes. Images were recorded, for morphological analysis, with an inverted microscope (Nikon. Tokyo, Japan) connected to a charge-coupled device camera (DAGE-MTI, Michigan City, IN) operated with MetaMorph software (Universal Imaging Corporation, Downingtown, PA). Nine representative areas per well of a 24-well plate were counted under the microscope at 100 × magnification by three individuals. The average of these scores was reported.
2.8 Superoxide assay
Extracellular superoxide (O2−) production from microglia was determined by measuring the superoxide dismutase (SOD) inhibitable reduction of 2-(4-lodophenyl)-3-(4-nitrophenyl)-5-(2, 4,-disulfophenyl)-2H-tetrazolium, monosodium salt, WST-1. Briefly, 200 μL of primary enriched-microglia were seeded (1×105/well) in 96-well plates. Cells were then incubated for 24 h at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Immediately before treatment, cells were washed twice with Hanks balanced salt solution (HBSS). To each well, 100 μL of HBSS with or without SOD (600 U·mL-1), 50 μL of vehicle or LPS, and 50 μL of WST-1 (1 mmol·L-1) in HBSS were added. The cultures were incubated for 30 min at 37°C and 5% CO2 and 95% air. The absorbance at 450 nm was read with a Spectra Max Plus microtiter plate spectrophotometer (Molecular Devices, Sunnyvale, CA, USA). The amount of SOD-inhibitable superoxide was calculated and expressed as percent of vehicle-treated control cultures.
2.9 TNF-α assay
The production of TNF-α was measured with a commercial ELISA kit from R&D Systems.
2.10 Nitrite assay
As an indicator of nitric oxide production, the amount of nitrite accumulated in culture supernatant was determined with a colorimetric assay using Griess reagent [1% sulfanilamide, 2.5% H3PO4, 0.1% N-(1-naphthyl) ethylenediamine dihydrochloride]. Briefly, 50 μL of Griess reagent and 50 μL of culture supernatant were incubated in the dark at room temperature for 10 min. After incubation, the absorbance at 540 nm was determined with the Spectra Max Plus microplate spectrophotometer. The sample nitrite concentration was determined from a sodium nitrite standard curve.
2.11 Statistical analysis
The data are expressed as the mean ± SEM and statistical significance was assessed with an ANOVA followed by Bonferroni’s t test. A value of P < 0.05 was considered statistically significant.
3. Results
3.1 Verapamil protects dopaminergic neurons from LPS-induced toxicity
We first examined whether LPS-induced damage of dopaminergic neurons could be prevented by verapamil. Rat mesencephalic neuron-glia cultures were pretreated with verapamil (0.001-1.0 μmol·L-1) for 30 min and then stimulated with LPS (5 ng·mL-1) for 7 days (Figure 1). Degeneration of dopaminergic neurons was determined by [3H]Dopamine uptake assay and counting of TH-IR neurons. [3H]Dopamine uptake assay showed that LPS (5 ng·mL-1) treatment reduced the capacity of cultures to take up dopamine to 39% of the vehicle control, and this LPS-induced reduction in [3H]dopamine was abated by pretreatment with verapamil in a concentration-dependent manner (Figure 1A). Similar to the loss of [3H]dopamine uptake, TH-IR neurons were reduced approximately 60% and verapamil (0.1 and 1 μmol·L-1) significantly attenuated the LPS-induced loss of TH-IR neurons in a concentration dependent manner (Figure 1B). Verapamil alone did not affect dopamine uptake levels or the number of TH-IR neurons at 1 μmol·L-1 (Figure 1A), however, higher concentrations of verapamil (5-10 μmol·L-1) were toxic to the cultures (Data not shown). Based on these results, 1 μmol·L-1 of verapamil was used for the remainder of our experiments. Morphologically, the dendrites of the surviving TH-IR neurons in the LPS-treated cultures were less elaborate than that of the control cultures (Figure 1C). In cultures pretreated with verapamil (1 μmol·L-1) before LPS stimulation, TH-IR neurons were more numerous and appeared less affected compared with the LPS-treated cultures (Figure 1C). Moreover, post-treatment with verapamil was still effective in protecting dopaminergic neurons against LPS-induced damage (Figure 2). Neuron-glia cultures were either treated with verapamil (1 μmol·L-1) and LPS (5 ng·mL-1) at the same time or verapamil was added at 30, 60, 120, or 180 min after the addition of LPS. Seven days later, dopamine uptake of the cultures was determined. Significant neuroprotection was observed in cultures with verapamil added up to 60 min after the addition of LPS (Figure 2).
Figure 1.
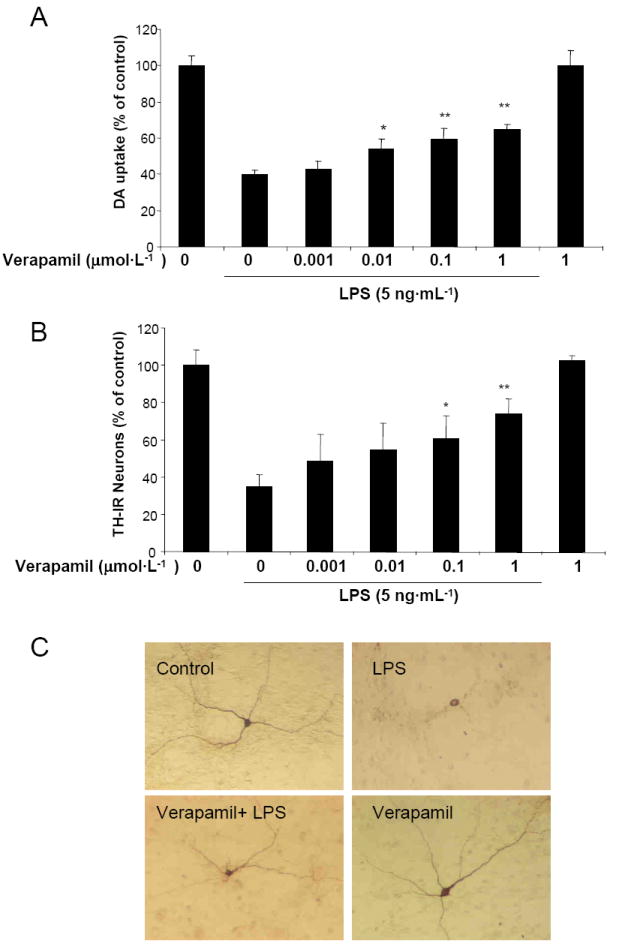
Verapamil protects dopaminergic neurons against LPS-induced neurotoxicity. Rat primary mesencephalic neuron-glia cultures were pretreated with different concentrations of verapamil for 30 min prior to treatment with 5 ng·mL-1 of LPS. Dopamine (DA) neurotoxicity was measured at 7 days following treatment using the [3H]DA uptake (A) and by counting TH-IR neurons number (B). Immunostaining images show differences in morphology (C). In control cultures, TH-IR neurons show an intricate network of TH-IR dendrites. However, TH-IR neurons in LPS-treated cultures were less numerous, with the remainder exhibiting shortened and damaged dendrites. Significant improvement in the morphology of TH-IR neurons, as judged by the intactness of cell bodies and extensiveness of dendrites, was observed in cultures pretreated with verapamil (1 μmol·L-1) prior to LPS treatment (5 ng·mL-1, 7 days). Data is expressed as the percent of the control cultures and are the mean ± SEM from three independent experiments performed with triplicate samples. * p < 0.05, ** p < 0.01, compared to LPS-treated cultures.
Figure 2.
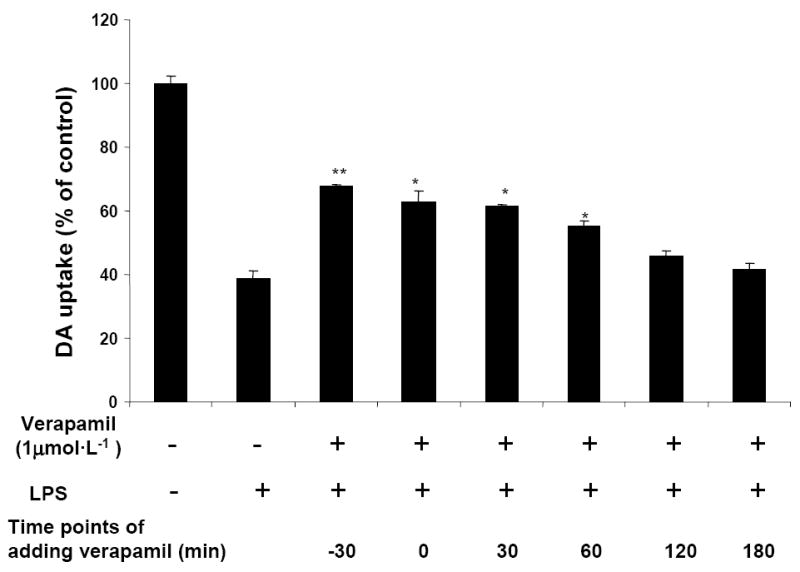
Post-treatment with verapamil protects dopamine neurons from LPS-induced neurotoxicity. Verapamil (1 μmol·L-1) was added 30 min before or at different time points after the addition of LPS (5 ng·mL-1). As controls, cultures were treated with vehicle alone. Seven days later, [3H]DA uptake was measured. Results are expressed as percentage of control cultures and are mean ± SEM of three experiments performed with triplicate. * p < 0.05, ** p < 0.01, compared to LPS-treated cultures.
3.2 Protection of LPS-induced dopaminergic neurotoxicity by verapamil is microglia-dependent
To investigate the roles of glial cells, reconstituted cultures were used by adding enriched microglia or astrocytes into neuron-enriched cultures and the effect of verapamil on the LPS-induced toxicity was evaluated. The results indicated that LPS reduced dopamine uptake capacity only in the presence of microglia, but not astroglia, and this reduction was significantly attenuated by verapamil (Figure 3). These results suggest that verapamil-elicited neuroproection against LPS-induced neurotoxicity is microglia-dependent.
Figure 3.
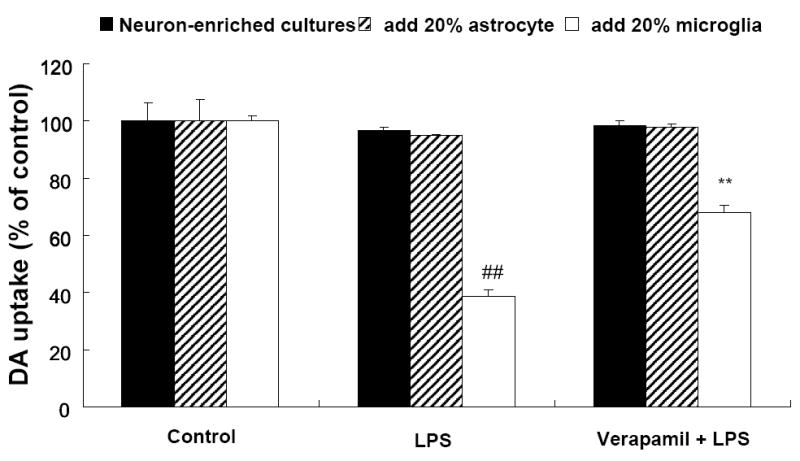
Microglia are essential for the neuroprotective effect of verapamil. Reconstituted cultures were prepared by adding 20% (1 × 105/well) astrocyte or microglia to neuron-enriched cultures (5 × 105/well). They were pretreated with verapamil (1 μmol·L-1) prior to treatment with LPS (5 ng·mL-1). [3H]DA uptake measurements were performed 7days after the indicated treatments. Results were expressed as percentage of vehicle-treated control cultures and were mean ± SEM from three independent experiments in triplicate. ##P<0.01 compared with the vehicle-treated control cultures and **P<0.001 compared with LPS-treated control cultures.
3.3 Verapamil suppresses LPS-induced release of pro-inflammatory mediators
Since verapamil attenuated microglia-mediated neurotoxicity, we speculated that the production of pro-inflammatory factors induced by LPS might be decreased by verapamil. To test this hypothesis, TNF-α and nitric oxide (NO) in neuron-glia cultures, and superoxide in microglia-enriched cultures were measured, respectively. Because the release of these pro-inflammatory factors peaked at different time points after LPS stimulation, we measured TNF-α at 3 h and nitrite (as an indicator of nitric oxide) at 24 h after LPS stimulation. As shown in Figure 4A, the production of TNF-α in cultures pretreated with verapamil (1 μmol·L-1) decreased by 29% compared with cultures treated with LPS (5 ng·mL-1) alone. As shown in Figure 4B, in neuron-glia cultures pretreated with verapamil (1 μmol·L-1) before stimulation with LPS (5 ng·mL-1), the level of nitrite was reduced by 30%. Among the pro-inflammatory factors measured, the production of superoxide from microglia (measured 30 min after LPS treatment) was the most severely affected one. Verapamil completely abolished LPS-stimulated superoxide production (Figure 4C). This finding is consistent with verapamil-mediated inhibition of NADPH oxidase, the key superoxide-generating enzyme in microglia. (see below)
Figure 4.
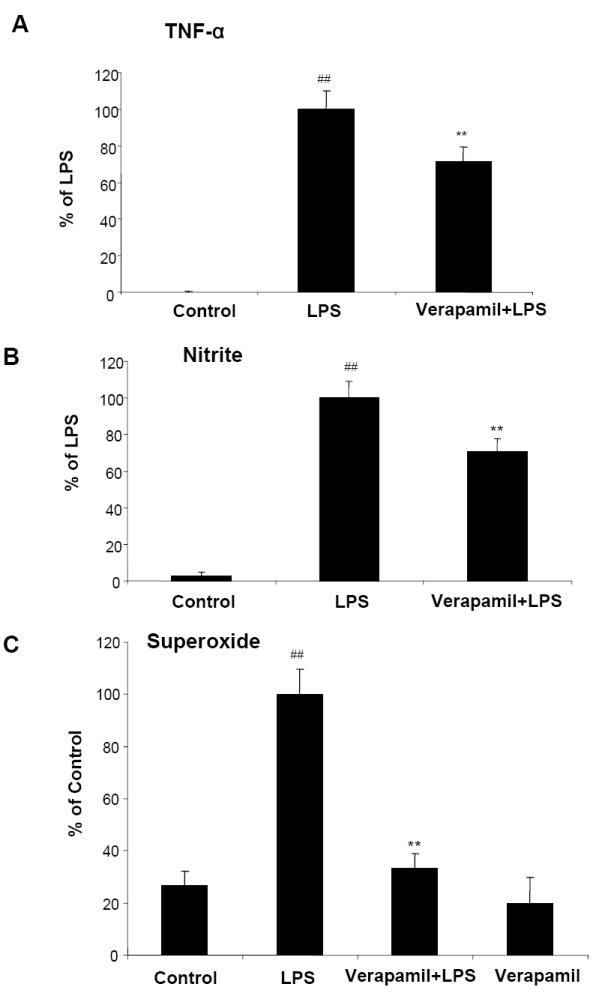
Verapamil inhibits LPS-induced production of pro-inflammatory factors in rat primary mesencephalic neuron-glia or microglia-enriched cultures. Cultures were pretreated with vehicle and verapamil (1 μmol·L-1) for 30 min before LPS (5 ng·mL-1) stimulation. Supernatant from neuron-glia cultures were collected at 3 h for TNF-α assay (A) and at 24 h for nitrite (an indicator of NO) assay (B). Levels of superoxide from microglia-enriched cultures were measured as SOD-inhibitable reduction of WST-1(C). Results were expressed as mean ± SEM from three independent experiments in triplicate. **P<0.01 compared with the LPS-treated cultures and ## compared with control.
3.4 NADPH oxidase plays a critical role in verapamil–elicited protection in LPS-induced neurodegeneration
NADPH oxidase, also called phagocyte oxidase (PHOX), is a major source of extracellular superoxide production in all the phagocytes, including microglia. In order to examine the role of PHOX in verapamil-mediated protection against LPS-induced neurodegeneration, neuron-glia cultures were prepared from PHOX-deficient (PHOX-/-) and wild-type (PHOX+/+) mice. A higher concentration of LPS (10 ng·mL-1) was used for mouse cultures. LPS treatment induced a reduction of [3H]dopamine uptake of neuron-glia cultures (60% decrease) prepared from PHOX+/+ mice (C57), which was significantly attenuated by verapamil (Figure 5). In contrast, LPS caused less decrease in [3H]dopamine uptake (30% decrease). Verapamil failed to affect LPS-induced reduction of dopamine uptake in PHOX-/- mice. One explanation for this differential effect of verapamil in wild type vs. PHOX-/- is that without the presence of PHOX, which is a target of LPS-induced toxicity, verapamil became ineffective. These findings are consistent with the idea that PHOX is critical in mediating verapamil-elicited neuroprotection.
Figure 5.
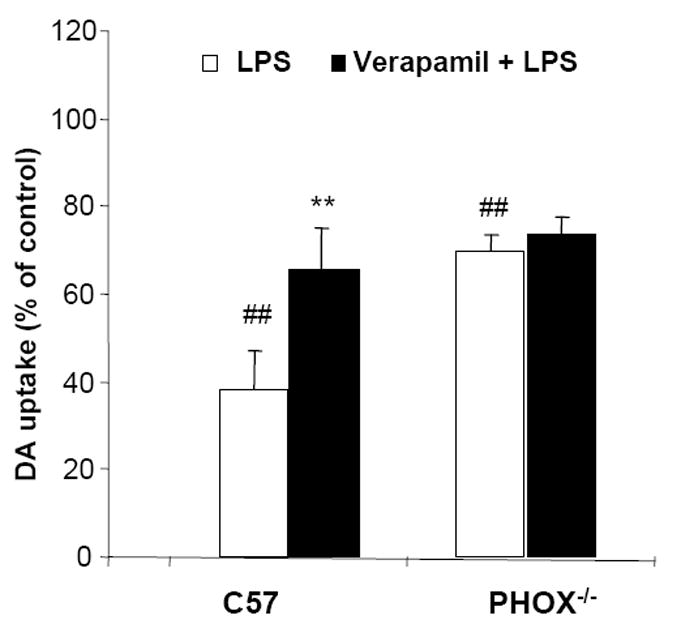
PHOX is essential in verapamil-elicited neuroprotection.
Wild-type (C57) or NADPH oxidase deficient mice (PHOX-/-) mesencephalic neuron-glia cultures were pretreated with verapamil (1 μmol·L-1) for 30 minutes prior to the addition of 10 ng·mL-1 of LPS. Seven days later, [3H]DA uptake was measured. Results are expressed as percentage of control cultures and are mean ± SEM of three experiments performed with triplicate. ## p < 0.01 compared to control cultures, ** p < 0.01, compared to LPS-treated cultures.
3.5 Verapamil binds to microglial gp91, a membrane catalytic subunit of PHOX
To further elucidate the mechanism underlying the inhibition of LPS-induced activation of PHOX by verapamil, ligand binding studies using COS7 cells, which are devoid of gp91 protein, with and without stable transfection of gp91 were performed. As shown in Figure 6, the binding capacity of [3H]Verapamil in the membrane fraction was higher in gp91-transfected cells than that of empty vector transfected control. The increased binding of verapamil to gp91 is likely associated with its inhibition of LPS-induced superoxide production, which in turn can explain the anti-inflammatory and neuroprotective effects of verapamil.
Figure 6.
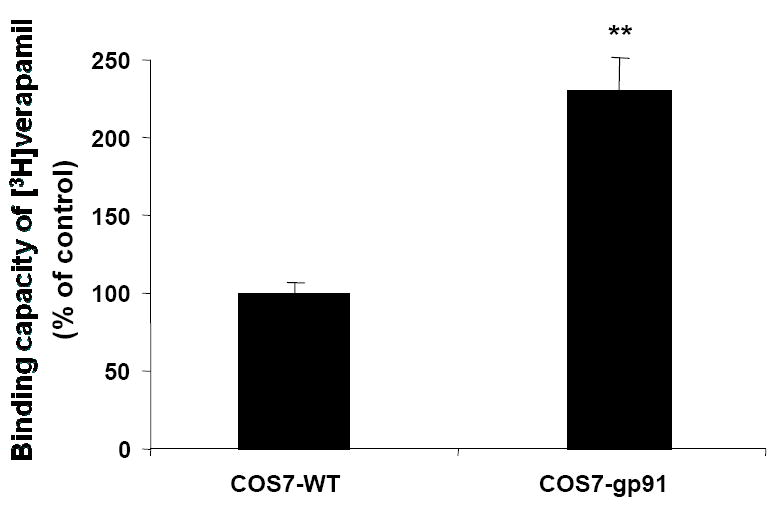
[3H]Verapamil binds to gp91, the catalytic membrane subunit of PHOX. Cell membrane fraction (500 μg protein) of either gp91-transfected COS7 (COS7-gp91) or non-transfected control (COS7-WT) membrane was incubated with 1nM [3H]Verapamil for 2 h at 4°C. The incubation was terminated by adding ice-cold assay buffer following immediate filtration through glass fiber filters. The filters were immediately washed three times with 5 mL of ice-cold assay buffer and dried in an oven at 60°C for 2 h. Radioactivity was counted using a liquid scintillation counter. Results were expressed as mean ±SEM from three independent experiments. **P<0.01 compared with the COS7 wild-type.
3.6 Verapamil stereoisomers are equi-potent in neuroprotection, inhibition of pro-inflammatory factors release and binding capacity to gp91 protein
In order to further delineate the molecular mechanism underlying verapamil-elicited anti-inflammatory and neuroprotective effects, we used two optical isomers of verapamil. It is known that the potency of S-(-)-verapamil in blocking calcium channels-associated pharmacological activities is about 10-20 fold more potent than that of R-(+)-verapamil (Busse et al., 2006; Echizen et al., 1985; Nawrath and Raschack, 1987). However, as shown in Figure 7A, racemic mixtures RS-(±)-, R-(+)- and S-(-)-verapamil isomers significantly inhibited LPS-induced DA neurotoxicity in a similar degree. In addition, results from binding studies indicated that R-(+)- and S-(-)-verapamil showed the same potency in displacing the binding of [3H]Verapamil in membrane preparation of COS7 cells transfected with gp91 (Figure 7B). Furthermore, R-(+)-verapamil and RS-(±)-verapamil are equi-potent in reducing LPS-induced production of TNF-α, NO and superoxide (Figure 7C). Taken together, these results indicate that it is unlikely that verapamil-elicited neuroprotection is mediated through blockade of calcium channels. Instead, our data strongly favor the possibility that inhibition of PHOX activation plays a critical role in the anti-inflammatory and neuroprotective effects of verapamil isomers.
Figure 7.
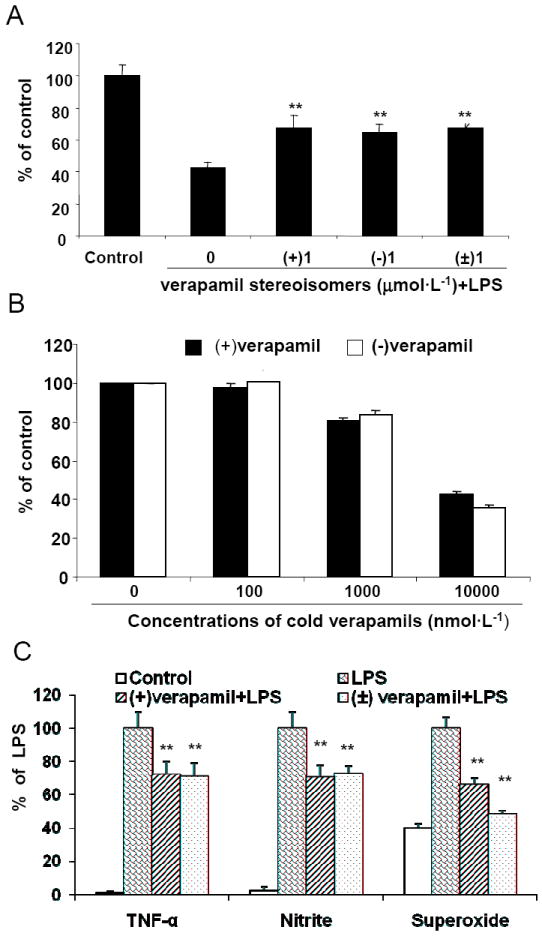
Both stereoisomers of verapamil show the same potency in neuroprotection, binding capacity to gp91 protein and inhibition of LPS-induced pro-inflammatory factors release. [3H]DA uptake assay (A); Competitive binding assay (B); and Pro-inflammatory factors (C). Results were expressed as mean ± SEM from nine (A) and three (B and C) independent experiments. **P<0.01 compared with LPS (5 ng·mL-1) treatment.
4. Discussion
In this study, we report a novel mechanism underlying the neuroprotective effect of verapamil in rodent midbrain neuron/glia cultures. We demonstrated that instead of directly acting on neurons, verapamil protected DA neurons from LPS-induced neurotoxicity by inhibiting the over-production of superoxide from microglial PHOX. Mechanistic studies revealed that verapamil possesses neuroprotective effects not only through its well-known blocking activity on L-type calcium channels of neurons, but also can be mediated through its inhibition on microglial PHOX activity. This new finding suggests a new avenue for further new applications of this series of drugs in other inflammation-related diseases.
Neuroprotective effects of verapamil have been documented in both in vitro and in vivo studies. Neuronal culture studies showed that verapamil protects against β-amyloid protein-induced cortical neuron death (Lee et al., 2005), significantly prevents amphetamine-induced apoptosis of rat cerebellar neurons (Zhou et al., 2004) and reduces hydrogen peroxide-induced oxidant injury in SH-SY5Y cells (Sarang et al., 2002). An in vivo study by Popovic et al. showed that verapamil prevents the loss of choline acetyltransferase-immunoreactive neurons in the cerebral cortex following lesions of the rat nucleus basalis magnocellularis (Popovic et al., 2006). These reports focus direct actions of verapamil on neurons and attribute its neuroprotective effect to blocking the entry of extracellular calcium into neurons which results in neuronal cell death. A recent clinical report suggests a potential neuroprotective role for centrally acting L-type calcium channel blockers of class in PD patients (Ritz et al., 2010). However, the neuroprotective mechanism of the dihydropyridine is difficult to be determined in this study. Our study strongly suggests that in addition to inhibiting calcium entry into neurons, verapamil can target microglia in the brain to attenuate the inflammatory response and provides a novel mechanism for the neuroprotective effect. This conclusion is based on the findings that S-(-)-verapamil and R-(+)-verapamil, two isomers with different affinity in binding to the calcium channel, are equi-potent in neuroprotection.
LPS has been widely used both in vitro and in vivo studies to investigate the impact of inflammation on neuron death (Qin et al., 2004), and delineate the mechanism underlying the pathogenesis of neurodegenerative diseases. Using neuron/glia and reconstituted cultures, we have clearly demonstrated the critical role of microglia in mediating the neuroprotective effect of verapamil. Results from reconstituted culture experiments by adding enriched astroglia and microglia back to enriched neuron cultures showed that verapamil-elicited neuroprotection was microglia-dependent (Figure 3). These data clearly demonstrate a novel site of action of verapamil in reducing LPS-induced neurotoxicity. Furthermore, it is worth of noting that among the neuroprotective drugs that we have studied in the midbrain neuron-glial cultures with similar mode of action, verapamil displays higher potency (0.01-1.0 μM) than others, such as memantine (1.0-10 μM), Baicalein (1-5 μM) and Curcumin (1.0-10 μM).
Results from this study also suggest that the anti-inflammatory property of verapamil isomers mediates their neuroprotective effects. Verapamil suppressed over-activation of microglia by reducing the production of superoxide, NO and cytokines such as TNF-α (Figure 4). There are several reports investigating the effects of verapamil on the production of superoxide in different cell types (Ishibashi et al., 2006; Shima et al., 2008; Sirmagul et al., 2006). Irita et al. (Irita et al., 1986) reported that verapamil inhibited superoxide production in human neutrophils, while Li et al. showed that verapamil modulates LPS-induced cytokine production in the liver (Li et al., 2006). Several reports indicated that LPS treatment can cause an increase in intracellular [Ca2+] concentrations, which is related to TNF-α production (Seabra et al., 1998; Wheeler et al., 2000); but other reports indicated that LPS induced calcium-independent inflammatory responses (Casado et al., 1997a; Casado et al., 1997b; Rodeberg and Babcock, 1996). Results from our study indicate that verapamil-mediated inhibition of superoxide production is not due to its ability to blockade of L-type calcium channels and inhibit calcium flux, but rather due to its inhibition of the superoxide-producing enzyme in microglia. This conclusion is based on the studies using two optical isomers: R-(+)- and S-(-)-verapamil. It is known that R-(+)Verapamil has much less potency than S-(-) isomer in L-type calcium channel-associated pharmacological activity (Busse et al., 2006; Echizen et al., 1985; Nawrath and Raschack, 1987). However, our results indicated that these two isomers are eqiu-potent in 1) the capacity of binding to gp91; 2) inhibition of LPS-induced PHOX activation; 3) reduction in production of LPS-induced pro-inflammatory factors and 4) reduction in LPS-induced neurotoxicity (Figure 7). Furthermore, our results showed that verapamil significantly protected LPS-induced dopamine neuron damage in cultures prepared from wild-type mice but had no significant protective effect on cells from PHOX-/- mice (Figure 5), strongly support the idea that the protective effect of verapamil is most likely mediated through the inhibition of microglial PHOX activity.
The fact that both the R-(+)- and S-(-)- isomer of verapamil possess the same potency in reducing LPS-induced neurotoxicity has important clinical implications for the use of verapamil in different diseases. The advantage of using dextrorotatory form of verapamil for new indications includes less cardiovascular side effects, because of its lower affinity in binding to L-type calcium channel. It is likely that clinical use of the R(+)-verapamil as an anti-inflammatory agent has potential therapeutic advantages. Given the effectiveness of these compounds at low concentrations and the broad spectrum of activity in inhibiting inflammatory responses, R(+)-verapamil appears to have significant potential in the treatment of chronic inflammatory neurological disorders.
Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences, U.S.A.; the Extramural grants from NIAAA (FTC) as well as The UNC Bowles Center for Alcohol Studies (YL and FTC), U.S.A.; and the National Science Council (grant NSC-96-2320-B-037-039-MY3), Taiwan.
Abbreviations
- LPS
lipopolysaccharide
- PD
Parkinson’s disease
- ROS
reactive oxygen species
- TNF-α
tumor necrosis factor-alpha
- FBS
fetal bovine serum
- TH
tyrosine hydroxylase
- DA
Dopamine
- Ver
Verapamil
- CCB
calcium channel blocker
- PHOX
phagocytic oxidase
- NADPH
nicotinamide adenine dinucleotide phosphate
- ICC
immunocytochemistry
Footnotes
Disclosures The authors have no financial conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bosson S, Kuenzig M, Schwartz SI. Verapamil improves cardiac function and increases survival in canine E. coli endotoxin shock. Circ Shock. 1985;16:307–316. [PubMed] [Google Scholar]
- Busse D, Templin S, Mikus G, Schwab M, Hofmann U, Eichelbaum M, Kivisto KT. Cardiovascular effects of (R)- and (S)-verapamil and racemic verapamil in humans: a placebo-controlled study. Eur J Clin Pharmacol. 2006;62:613–619. doi: 10.1007/s00228-006-0154-7. [DOI] [PubMed] [Google Scholar]
- Casado M, Diaz-Guerra MJ, Bosca L, Martin-Sanz P. Differential regulation of nitric oxide synthase mRNA expression by lipopolysaccharide and pro-inflammatory cytokines in fetal hepatocytes treated with cycloheximide. Biochem J. 1997a;327(Pt 3):819–823. doi: 10.1042/bj3270819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casado M, Di-G MJ, Rodrigo J, Fernandez AP, Bosca L, Martin-Sanz P. Expression of the calcium-independent cytokine-inducible (iNOS) isoform of nitric oxide synthase in rat placenta. Biochem J. 1997b;324(Pt 1):201–207. doi: 10.1042/bj3240201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deakin CD, Fagan EA, Williams R. Cytoprotective effects of calcium channel blockers. Mechanisms and potential applications in hepatocellular injury. J Hepatol. 1991;12:251–255. doi: 10.1016/0168-8278(91)90947-a. [DOI] [PubMed] [Google Scholar]
- Echizen H, Brecht T, Niedergesass S, Vogelgesang B, Eichelbaum M. The effect of dextro-, levo-, and racemic verapamil on atrioventricular conduction in humans. Am Heart J. 1985;109:210–217. doi: 10.1016/0002-8703(85)90585-x. [DOI] [PubMed] [Google Scholar]
- Fleckenstein A. History of calcium antagonists. Circ Res. 1983;52:I3–16. [PubMed] [Google Scholar]
- Gao HM, Jiang J, Wilson B, Zhang W, Hong JS, Liu B. Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: relevance to Parkinson’s disease. J Neurochem. 2002;81:1285–1297. doi: 10.1046/j.1471-4159.2002.00928.x. [DOI] [PubMed] [Google Scholar]
- Ginsberg MD. Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology. 2008;55:363–389. doi: 10.1016/j.neuropharm.2007.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchkiss RS, Bowling WM, Karl IE, Osborne DF, Flye MW. Calcium antagonists inhibit oxidative burst and nitrite formation in lipopolysaccharide-stimulated rat peritoneal macrophages. Shock. 1997;8:170–178. doi: 10.1097/00024382-199709000-00004. [DOI] [PubMed] [Google Scholar]
- Irita K, Fujita I, Takeshige K, Minakami S, Yoshitake J. Calcium channel antagonist induced inhibition of superoxide production in human neutrophils. Mechanisms independent of antagonizing calcium influx. Biochem Pharmacol. 1986;35:3465–3471. doi: 10.1016/0006-2952(86)90613-1. [DOI] [PubMed] [Google Scholar]
- Ishibashi K, Okazaki S, Hiramatsu M. Simultaneous measurement of superoxide generation and intracellular Ca2+ concentration reveals the effect of extracellular Ca2+ on rapid and transient contents of superoxide generation in differentiated THP-1 cells. Biochem Biophys Res Commun. 2006;344:571–580. doi: 10.1016/j.bbrc.2006.02.173. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Mori Y. Ca2+ channel antagonists and neuroprotection from cerebral ischemia. Eur J Pharmacol. 1998;363:1–15. doi: 10.1016/s0014-2999(98)00774-2. [DOI] [PubMed] [Google Scholar]
- Lee BY, Ban JY, Seong YH. Chronic stimulation of GABAA receptor with muscimol reduces amyloid beta protein (25-35)-induced neurotoxicity in cultured rat cortical cells. Neurosci Res. 2005;52:347–356. doi: 10.1016/j.neures.2005.04.008. [DOI] [PubMed] [Google Scholar]
- Li G, Qi XP, Wu XY, Liu FK, Xu Z, Chen C, Yang XD, Sun Z, Li JS. Verapamil modulates LPS-induced cytokine production via inhibition of NF-kappa B activation in the liver. Inflamm Res. 2006;55:108–113. doi: 10.1007/s00011-005-0060-y. [DOI] [PubMed] [Google Scholar]
- Li XM, Yang JM, Hu DH, Hou FQ, Zhao M, Zhu XH, Wang Y, Li JG, Hu P, Chen L, Qin LN, Gao TM. Contribution of downregulation of L-type calcium currents to delayed neuronal death in rat hippocampus after global cerebral ischemia and reperfusion. J Neurosci. 2007;27:5249–5259. doi: 10.1523/JNEUROSCI.0802-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Du L, Hong JS. Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J Pharmacol Exp Ther. 2000a;293:607–617. [PubMed] [Google Scholar]
- Liu B, Du L, Kong LY, Hudson PM, Wilson BC, Chang RC, Abel HH, Hong JS. Reduction by naloxone of lipopolysaccharide-induced neurotoxicity in mouse cortical neuron-glia co-cultures. Neuroscience. 2000b;97:749–756. doi: 10.1016/s0306-4522(00)00057-9. [DOI] [PubMed] [Google Scholar]
- Mustafa SB, Olson MS. Effects of calcium channel antagonists on LPS-induced hepatic iNOS expression. Am J Physiol. 1999;277:G351–360. doi: 10.1152/ajpgi.1999.277.2.G351. [DOI] [PubMed] [Google Scholar]
- Nawrath H, Raschack M. Effects of (-)-desmethoxyverapamil on heart and vascular smooth muscle. J Pharmacol Exp Ther. 1987;242:1090–1097. [PubMed] [Google Scholar]
- Popovic M, Caballero-Bleda M, Popovic N, Puelles L, van Groen T, Witter MP. Verapamil prevents, in a dose-dependent way, the loss of ChAT-immunoreactive neurons in the cerebral cortex following lesions of the rat nucleus basalis magnocellularis. Exp Brain Res. 2006;170:368–375. doi: 10.1007/s00221-005-0219-3. [DOI] [PubMed] [Google Scholar]
- Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, Liu B, Hong JS. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem. 2004;279:1415–1421. doi: 10.1074/jbc.M307657200. [DOI] [PubMed] [Google Scholar]
- Ritz B, Rhodes SL, Qian L, Schernhammer E, Olsen JH, Friis S. L-type calcium channel blockers and Parkinson disease in Denmark. Ann Neurol. 2010;67:600–606. doi: 10.1002/ana.21937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodeberg DA, Babcock GF. Role of calcium during lipopolysaccharide stimulation of neutrophils. Infect Immun. 1996;64:2812–2816. doi: 10.1128/iai.64.7.2812-2816.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarang SS, Yoshida T, Cadet R, Valeras AS, Jensen RV, Gullans SR. Discovery of molecular mechanisms of neuroprotection using cell-based bioassays and oligonucleotide arrays. Physiol Genomics. 2002;11:45–52. doi: 10.1152/physiolgenomics.00064.2002. [DOI] [PubMed] [Google Scholar]
- Schurr A. Neuroprotection against ischemic/hypoxic brain damage: blockers of ionotropic glutamate receptor and voltage sensitive calcium channels. Curr Drug Targets. 2004;5:603–618. doi: 10.2174/1389450043345209. [DOI] [PubMed] [Google Scholar]
- Seabra V, Stachlewitz RF, Thurman RG. Taurine blunts LPS-induced increases in intracellular calcium and TNF-alpha production by Kupffer cells. J Leukoc Biol. 1998;64:615–621. doi: 10.1002/jlb.64.5.615. [DOI] [PubMed] [Google Scholar]
- Shima E, Katsube M, Kato T, Kitagawa M, Hato F, Hino M, Takahashi T, Fujita H, Kitagawa S. Calcium channel blockers suppress cytokine-induced activation of human neutrophils. Am J Hypertens. 2008;21:78–84. doi: 10.1038/ajh.2007.13. [DOI] [PubMed] [Google Scholar]
- Sirmagul B, Kilic FS, Tunc O, Yildirim E, Erol K. Effects of verapamil and nifedipine on different parameters in lipopolysaccharide-induced septic shock. Heart Vessels. 2006;21:162–168. doi: 10.1007/s00380-005-0874-0. [DOI] [PubMed] [Google Scholar]
- Triggle DJ. Calcium-channel drugs: structure-function relationships and selectivity of action. J Cardiovasc Pharmacol. 1991;18(Suppl 10):S1–6. [PubMed] [Google Scholar]
- Wang T, Zhang W, Pei Z, Block M, Wilson B, Reece JM, Miller DS, Hong JS. Reactive microgliosis participates in MPP+-induced dopaminergic neurodegeneration: role of 67 kDa laminin receptor. FASEB J. 2006;20:906–915. doi: 10.1096/fj.05-5053com. [DOI] [PubMed] [Google Scholar]
- Wheeler M, Stachlewitz RF, Yamashina S, Ikejima K, Morrow AL, Thurman RG. Glycine-gated chloride channels in neutrophils attenuate calcium influx and superoxide production. FASEB J. 2000;14:476–484. doi: 10.1096/fasebj.14.3.476. [DOI] [PubMed] [Google Scholar]
- Zhou JL, Liang JH, Li CL. Inhibition of methamphetamine-induced apoptosis by the calcium channel blocker verapamil in rat cerebellar neurons. Beijing Da Xue Xue Bao. 2004;36:361–365. [PubMed] [Google Scholar]