

Abstract

The first enantioselective total syntheses of indolealk aloids of the condylocarpine type are reported. (+)-Condylocarpine, (+)-isocondylocarpine, and (+)-tubotaiwine were prepared in high enantiomeric purity (er >99:1) from (1 S,5 R)-hexahydro-1,5-methano-1 H-azocino[4,3 -b]indole-12-one 7b by way of five or six isolated intermediates.
The hexahydro-1,5-methano-1H-azocino[4,3-b]indole ring system 1 is a major fragment of Strychnos alkaloids of the strychnan and aspidopermatan types (e.g., 2–4, Figure 1) and the ring system of indole alkaloids of the uleine group (e.g., 5, Figure 1).1 During our recent total synthesis of the structurally unique indole alkaloid (−)-actinophyllic acid, we developed a concise synthesis of the hexahydro-1,5-methano-1H-azocino[4,3-b]indole ring system.2,3 A central step in this synthesis is an iron(III)-promoted intramolecular oxidative coupling of malonate and ketone enolates generated from indole piperidones 6 to deliver hexahydro-1,5-methano-1H-azocino[4,3-b]indole-12-ones 7 (eq 1). Ketone (1S,5R)-7b was prepared in enantioenriched form (er = 95:5) in 26% overall yield from 4-(tert-butoxycarbonylamino)butyric acid by way of six isolated intermediates.2,4 Herein we report the use of tetracyclic intermediate (1S,5R)-7b for the enantioselective synthesis of three representative members of the condylocarpine subtype of the aspidospermatan group of alkaloids.5,6
Figure 1.

The 1,5-methanoazocino[4,3-b]indole ring system and some representative alkaloids that possess this fragment.
 |
(1) |
Transformation of hexahydro-1,5-methano-1H-azocino[4,3-b]indole-12-ones 7 to the condylocarpine skeleton requires introduction of a two-carbon unit at C12, installation of a two-carbon bridge between C11b and N2, and dealkoxycarboxylation of the malonate unit. Our early exploratory investigations were carried out in the racemic series and focused on elaboration of C12 and N2. Several observations made during these studies provided essential insight into the chemical reactivity of the hexahydro-1,5-methano-1H-azocino[4,3-b]indole ring system (Scheme 1). Wittig reaction of ketone (±)-7a with ethylidenetriphenylphosphorane delivered ethylidene derivative (±)-8 as a single stereoisomer in 73% yield, with the Z configuration of the double bond being readily secured by 1H NMR NOE analysis. To our surprise, this product was extremely acid sensitive. For example, (±)-8 rearranged to hexahydro-6H-pyrido[4,3-b]carbazole isomer 9 when stored in CDCl3 for several days, or within 2 h at room temperature in the presence of 10 mol % of DCl (0.004 M). Related instability was observed with hexahydro-1,5-methano-1H-azocino[4,3-b]indole-12-one (±)-7b, which when exposed in CDCl3 to 0.15 M DCl for 2 h gave dihydropyrrolo[3,2-b]carbazole 10 in 55% yield.7 Under stronger acidic conditions (neat TFA), ketone (±)-7b yielded 3-hydroxycarbazole-1-carboxylic acid 11 as the major product within 4 h at room temperature. In marked contrast, structurally related hexahydro-1,5-methano-1H-azocino[4,3-b]indole diester (±)-12 does not undergo skeletal rearrangement when exposed to TFA at 0 °C or 23 °C (Scheme 1).2,8
Scheme 1.

Facile gramine-type fragmentation of 12-ethylidene and 12-oxo hexahydro-1,5-methano-1H-azocino[4,3-b]indoles and contrasting acid stability of structurally related indole derivative 12.
Gramine-type fragmentations of hexahydro-1,5-methano-1H-azocino[4,3-b]indoles to carbazoles and pyridinocarbazoles are well known.9 However, not previously recognized is the notable accelerating effect of a keto or alkylidene substituent at C12 on this fragmentation. The formation of products (±)-9 and 10 from respectively ethylidene and keto precursors (±)-8 and (±)-7b suggests that the initial step is cleavage of the C1–N bond to generate delocalized iminium ion intermediate 16 (Scheme 2). In the ethylidene series, cyclization of the pendant side chain of 16 delivers (±)-9 as a single stereoisomer.10 In the oxo series, dehydrative cyclization of the β-(tert-butoxycarbonylamino)ethyl side chain of 16 and loss of isobutylene and CO2 leads to dihydropyrrolocarbazole ester 10.11 The facile fragmentations in the keto and ethylidene series to generate intermediate 16 undoubtedly reflect activation of the C1—NBoc bond as a result of its excellent overlap with the exocyclic π-bond.12
Scheme 2.

Fragmentation of ethylidene- and keto-bridged hexahydro-1,5-methano-1H-azocino[4,3-b]indole intermediates
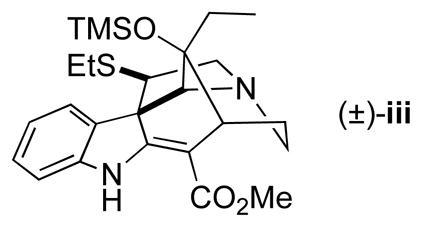
With a better understanding of the potential gramine-type fragmentation of hexahydro-1,5-methano-1H-azocino[4,3-b]indole-12-ones and their alkylidene derivatives, a successful synthetic sequence was developed in which the pyrrolidine ring of the condylocarpine skeleton was constructed using intermediates in which C12 is an sp3 carbon (Scheme 3). The syntheses began with stereoselective reduction of (1S,5R)-ketone 7b (er = 95:5)2 with sodium borohydride.13 After quenching the reduction with TFA, the reaction mixture was concentrated and the crude residue was dissolved in a 4:1 mixture of TFA and water to promote cleavage of the Boc and tert-butyl esters, and decarboxylation to yield the corresponding amino acid. Removal of TFA and water in vacuo, followed by dissolution of the residue in a 0.5 M solution of hydrochloric acid in methanol and heating at 50 °C delivered amino ester 17, a 2:1 mixture of ester epimers, in 86% overall yield from 7b. Reductive alkylation of this crude amine with 2,2-bis(ethylthio)acetaldehyde14 furnished tertiary amine 18 in 70% yield. Exposure of 18 to a suspension of dimethyl(methylthio)sulfonium tetrafluoroborate (DMTSF)15 and 4Å molecular sieves in dichloromethane at room temperature delivered pentacyclic alcohol 19 as a single stereoisomer in 75% yield.16–18 It was essential that this ring construction be carried out after dealkoxycarboxylation, as earlier extensive attempts to cyclize malonate precursors under various conditions were unsuccessful. Enantiomerically pure 19 (er >99:1) was obtained in 91% yield after separation of the highly crystalline racemate.
Scheme 3.

Total syntheses of (+)-condylocarpine, (+)-isocondylocarpine, and (+)-tubotaiwine
In three additional steps, pentacyclic intermediate 19 was converted to (+)-condylocarpine (2a) and (+)-isocondylocarpine (2b). The thioether group of 19 was cleaved with Raney nickel in THF at 50 °C to deliver the desulfurized product in 75% yield.19 After extensively screening conditions for oxidation of the secondary alcohol of this product, Albright–Goldman oxidation20 was identified as optimal for accomplishing this transformation, giving 20 in 72% yield for the 2 steps.21 Reaction of pentacyclic ketone 20 with ethyltriphenylphosphonium ylide in a mixture of THF and toluene at room temperature delivered a 1:1 mixture of enantiopure (+)-condylocarpine (2a) and (+)-isocondylocarpine (2b) in 77% combined yield. Pure samples of these interconvertible stereoisomers22 were obtained by preparative HPLC: (+)-condylocarpine (2a), [α]D +854 (c 0.39, CHCl3), and (+)-isocondylocarpine (2b), [α]D +798 (c 0.15, CHCl3). Catalytic hydrogenation23 of the initially produced mixture of 2a and 2b delivered (+)-tubotaiwine (21), [α]D +584 (c 0.91, CHCl3), in 91% yield.24–26
In summary, the first enantioselective total syntheses of indole alkaloids of the condylocarpine type have been accomplished. One important outcome of this study was the discovery that hexahydro-1,5-methano-1H-azocino[4,3-b]indoles having a carbonyl or alkylidene group as the one carbon bridge undergo facile gramine-type fragmentation in the presence of even dilute acids. This lability necessitated that the C12 carbonyl group of precursor 7b be masked by reduction during formation of the pyrrolidine ring. As a result, a synthetic sequence proceeding via five isolated intermediates was required for the elaboration of (1S,5R)-hexahydro-1,5-methano-1H-azocino[4,3-b]indole 7b to enantiopure (+)-condylocarpine (2a) and (+)-isocondylocarpine (2b).
Supplementary Material
Acknowledgments
This research was supported by the NIH Neurological Disorders & Stroke Institute (NS-12389 and the National Institute of General Medical Sciences GM-98601). Fellowship assistance for CLM (UC Irvine Chancellor’s Fellowship, Bristol–Myers Squibb Graduate Fellowship, and ACS Division of Organic Chemistry Fellowship sponsored by Amgen) and RO (Feodor Lynen Fellowship of the Alexander von Humboldt Foundation) is gratefully acknowledged. We thank Dr. Jason M. Rohde, Ironwood Pharmaceuticals, Inc., 301 Binney Street, Cambridge, MA 02142, for preliminary studies, and Dr. Joe Ziller, School of Physical Sciences, UC Irvine, for X-ray analyses. NMR, mass spectra, and X-ray analyses were obtained at UC Irvine using instrumentation acquired with the assistance of NSF and NIH Shared Instrumentation programs.
Footnotes
Supporting Information Available: Experimental details and copies of 1H and 13C NMR spectra of new compounds (PDF); CIF files for compounds (±)-20 and (±)-iii. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Cordell GA, editor. The Alkaloids. 48 Chapter 4 Academic Press; New York: 1996. [Google Scholar]; (b) Alvarez M, Joule JA. In: The Alkaloids. Cordell GA, editor. 57 Chapter 4 Academic Press; New York: 2001. [Google Scholar]
- 2.(a) Martin CL, Overman LE, Rohde JM. J Am Chem Soc. 2008;130:7568–7569. doi: 10.1021/ja803158y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Martin CL, Overman LE, Rohde JM. J Am Chem Soc. 2010;132:4894–4906. doi: 10.1021/ja100178u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For representative examples of syntheses of the hexahydro-1,5-methano-1H-azocino[4,3-b]indole ring system, see: Jackson A, Joule JA. Chem Commun. 1967:459–460.full paper: Jackson JA, Wilson NDV, Gaskell AJ, Joule JA. J Chem Soc C. 1969;19:2738–2747.Büchi G, Gould SJ, Näf F. J Am Chem Soc. 1971;93:2492–2501.Grierson DS, Harris M, Husson HP. Tetrahedron. 1983;39:3683–3694.Magnus P, Sear NL, Kim CS, Vicker N. J Org Chem. 1992;57:70–78.Gràcia J, Casamitjana N, Bonjoch J, Bosch J. J Org Chem. 1994;59:3939–3951.Micouin L, Diez A, Castells J, López D, Rubiralta M, Quirion JC, Husson HP. Tetrahedron Lett. 1995;36:1693–1696.Blechert S, Knier R, Schroers H, Wirth T. Synthesis. 1995:592–604.Saito M, Kawamura M, Hiroya K, Ogasawara K. Chem Commun. 1997:765–766.Amat M, Hadida S, Pshenichnyi G, Bosch J. J Org Chem. 1997;62:3158–3175. doi: 10.1021/jo962169u.Ergün Y, Patir S, Okay G. J Heterocycl Chem. 2002;39:315–317.Jiricek J, Blechert S. J Am Chem Soc. 2004;126:3534–3538. doi: 10.1021/ja0399021.Ishikura M, Takahashi N, Takahashi H, Yanada K. Heterocycles. 2005;66:45–50.Uludag N, Hökelek T, Patir S. J Heterocycl Chem. 2006;43:585–591.Bennasar ML, Roca T, García-Díaz D. J Org Chem. 2008;73:9033–9039. doi: 10.1021/jo801998h.
- 4.Ketones 7a and 7b are available in racemic form in four steps from dimethyl or di-tert-butyl malonate in 24% and 33% overall yield, respectively.2
- 5.For review of this group of alkaloids, see: Lounasmaa M, Somersalo P. The Condylocarpine Group of Indole Alkaloids. In: Herz W, Grisebach H, Kirby GW, Tamm Ch, editors. Progress in the Chemistry of Natural Products. Vol. 50. Springer-Verlag/Wien; New York: 1986. pp. 27–56.For the recent isolation of isocondylocarpine, see: Wu Y, Kitajima M, Kogure N, Wang Y, Zhang R, Takayama H. J Nat Med. 2009;63:283–289. doi: 10.1007/s11418-009-0334-8.
- 6.For total syntheses of (±)-condylocarpine, see: Harley-Mason J. Pure Appl Chem. 1975;41:167–174.Kuehne ME, Brook CS, Frasier DA, Xu F. J Org Chem. 1995;60:1864–1867.For studies on the synthesis of condylocarpine, see: Lounasmaa M, Somersalo P. Tetrahedron. 1986;42:1501–1509.For total syntheses of (±)-tubotaiwine, see: Dadson BA, Harley-Mason J. J Chem Soc D. 1969:665b.Kuehne ME, Frasier DA, Spitzer TD. J Org Chem. 1991;56:2696–2700.Gracia J, Bonjoch J, Casamitjana N, Amat M, Bosch J. J Chem Soc, Chem Commun. 1991:614–615.Gracia J, Casamitjana N, Bonjoch J, Bosch J. J Org Chem. 1994;59:3939–3951.Amat M, Hadida S, Pshenichnyi G, Bosch J. J Org Chem. 1997;62:3158–3175. doi: 10.1021/jo962169u.For studies on the synthesis of tubotaiwine, see: Legseir B, Henin J, Massiot G, Vercauteren J. Tetrahedron Lett. 1987;28:3573–3576.Lavilla R, Gotsens T, Rodriguez S, Bosch J. Tetrahedron. 1992;48:6445–6454.
- 7.Precursor (±)-7b was recovered in 25% yield.
- 8.In our earlier total synthesis of actinophyllic acid, products (±)-13 and (±)-14 were not purified, but directly employed in aza-Cope–Mannich transformations.2 1H NMR analysis of these crude intermediates showed no significant impurities, which we indicate in Scheme 1 as a crude yield of >90%.
- 9.To our knowledge, the first unambiguous example of such fragmentation promoted by acid was described by Husson: Besselièvre R, Husson H–P. Tetrahedron. 1983;37(Supplement 1):241–246.
- 10.The stereoisomer having a cis relationship of the angular hydrogen and the methyl substituent would be both the expected kinetic product of cyclization of (E)-ethylidene intermediate 16 and the thermodynamic product of reversible gramine-type fragmentation of product (±)-9; an axial orientation of Me substituent minimizes destabilizing A1,3 interactions with the Boc substituent.
-
11.(a) Gramine-type cleavage is likely the first step in the keto series also, because exposure of i to 0.15 M DCl in CDCl3 at room temperature for 2 h yielded a mixture of i (48%) and ii (36%). (b) In neat TFA, decarboxylative aromatization of 16 (X = O, R = t-Bu) apparently occurs rapidly leading to the eventual formation of carbazole acid 11.

- 12.For a concise discussion of the effect of adjacent C–C and C–O π-bonds on nucleophilic substitution reactions, see: Anslyn EV, Dougherty DA. Modern Physical Organic Chemistry. University Science; Sausalito, CA: 2006. pp. 653–655.
- 13.The β-tert-butoxycarbonyl group sterically shields the Si face of the ketone carbonyl group,2 resulting in hydride addition from the Re face.
- 14.Bates GS, Ramaswamy S. Can J Chem. 1983;61:2466–2475. [Google Scholar]
- 15.Trost BM, Murayama E. J Am Chem Soc. 1981;103:6529–6530. [Google Scholar]
-
16.The relative configuration of 19, and that of the secondary alcohol in precursors 17 and 18, was secured on the basis of X-ray crystallographic analysis of silyl ether derivative (±)-iii, which was prepared in an analogous fashion to 19. Crystallographic data for (±)-iii were deposited at the Cambridge Crystallographic Data Centre: CCDC 799190.
- 17.The use of 5-exo thionium ion cyclizations to form this ring of indole alkaloids is a well-established tactic in indole alkaloid synthesis, see: Gallagher T, Magnus P, Huffman JC. J Am Chem Soc. 1983;105:4750–4757.Amat M, Linares A, Bosch J. J Org Chem. 1990;55:6299–1312.Amat M, Bosch J. J Org Chem. 1992;57:5792–5796.
-
18.Methyl thioether (±)-iv was isolated with 19 as an inseparable contaminant (diagnostic 1H NMR singlet corresponding to the methyl thioether group at 1.81 ppm and LRMS analysis). This inconsequential side product likely arose by hydrolysis of DMTSF by adventitious water to give methanethiol, which underwent thiol exchange by way of the reactive thionium ion prior to cyclization.

- 19.It was advantageous to use THF as the solvent instead of methanol or ethanol, as byproducts arising from reduction of the vinylogous carbamate were suppressed in THF.
- 20.Albright JD, Goldman L. J Am Chem Soc. 1965;87:4214–4216. [Google Scholar]
- 21.Crystallographic data for a racemic sample of intermediate 20 were deposited at the Cambridge Crystallographic Data Centre: CCDC 799189.
- 22.Condylocarpine (2a) and isocondylocarpine (2b) equilibrate to a 2:1 mixture of 2a:2b when heated for 4 h at reflux in toluene or when exposed for 24 h at room temperature to acetic acid in chloroform.6b
- 23.Schumann D, Schmid H. Helv Chim Acta. 1963;46:1996–2003. [Google Scholar]
- 24.A range of optical rotations at the sodium D line has been reported for natural samples of condylocarpine (+501 to +90125 in CHCl3) and tubotaiwine (+417 to +62826 in CHCl3); citation to the highest reported rotation is provided here, and a complete summary is provided in the Supporting Information. Natural isocondylocarpine is reported to show [α]D +537 (c 0.1, CHCl3).5b
- 25.Stauffacher D. Helv Chim Acta. 1961;44:2006–2015. [Google Scholar]
- 26.Pinar M, Renner U, Hesse M, Schmid H. Helv Chim Acta. 1972;55:2972–2974. doi: 10.1002/hlca.19720550304. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.