

Abstract
Alteration in mitochondrial dynamics has been implicated in many neurodegenerative diseases. Mitochondrial apoptosis inducing factor (AIF) plays a key role in multiple cellular and disease processes. Using immunoblotting and flow cytometry analysis with Harlequin mutant mice that have a proviral insertion in the AIF gene, we first revealed that mitofusion 1 (Mfn1), a key mitochondrial fusion protein, is significantly diminished in Purkinje cells of the Harlequin cerebellum. Next, we investigated the cerebellar pathology of Harlequin mice in an age-dependent fashion, and identified a striking process of progressive and patterned Purkinje cell degeneration. Using immunohistochemistry with zebrin II, the most studied compartmentalization marker in the cerebellum, we found that zebrin II-negative Purkinje cells first started to degenerate at 7 months of age. By 11 months of age, almost half of the Purkinje cells were degenerated. Subsequently, most of the Purkinje cells disappeared in the Harlequin cerebellum. The surviving Purkinje cells were concentrated in cerebellar lobules IX and X, where these cells were positive for heat shock protein 25 and resistant to degeneration. We further showed that the patterned Purkinje cell degeneration was dependent on caspase but not poly(ADP-ribose) polymerase-1 (PARP-1) activation, and confirmed the marked decrease of Mfn1 in the Harlequin cerebellum. Our results identified a previously unrecognized role of AIF in Purkinje cell degeneration, and revealed that AIF deficiency leads to altered mitochondrial fusion and caspase-dependent cerebellar Purkinje cell loss in Harlequin mice. This study is the first to link AIF and mitochondrial fusion, both of which might play important roles in neurodegeneration.
Keywords: Purkinje cell, Mfn1, Apoptosis inducing factor, Harlequin, zebrin II, Heat shock protein 25
Introduction
Mitochondria are the power-house in most eukaryotic cells. They exist as a dynamic network that constantly remodels itself through the opposing processes of mitochondrial fusion and fission, which critically control mitochondrial morphology and function (Detmer and Chan, 2007). In addition, they play important roles in regulating cell homeostasis and cell death (McBride et al., 2006). Mitochondrial dysfunction is a critical factor in many neurodegenerative disorders, such as Huntington’s disease, Parkinson’s disease, Alzheimer’s disease, amyotrophic lateral sclerosis, and Friedreich’s ataxia (Kwong et al., 2006, Reddy, 2008), and has been implicated as an early event in most late-onset neurodegenerative diseases (Reddy, 2008).
Klein et al. (2002) reported a naturally-occurring genetic mouse model of neurodegeneration, the Harlequin mouse, which carries an X-linked recessive mutation in the apoptosis-inducing factor (AIF) gene due to a proviral insertion, resulting in an approximately 80% decrease in AIF expression. AIF is a 67 kDa flavoprotein, and is located in the mitochondrial intermembranous space in healthy cells (Susin et al., 1999a, Susin et al., 1999b). AIF shows homology with several bacterial nicotinamide adenine dinucleotide (NADH)-dependent ferrodoxin oxidoreductases (Susin et al., 1999b, Miramar et al., 2001). Under apoptotic conditions, AIF is released from the mitochondria and translocates to the nucleus where it participates in apoptosis. In the nucleus, AIF binds to DNA and induces DNA fragmentation and nuclear condensation (Susin et al., 1999a, Susin et al., 1999b, Cande et al., 2002). Because AIF mediates poly(ADP-ribose) polymerase-1 (PARP-1)-dependent cell death that can not be rescued by pan-caspase inhibitors, AIF is generally known to induce cell death via a caspase-independent mechanism (Liu et al., 1996, Yu et al., 2002). The phenotype of Harlequin mice is characterized by progressive retinal neuron defects, including the degeneration of amacrine and ganglion cells, starting at 3 months of age. In addition, cerebellar granule neuron degeneration begins at 4 months of age with progressive ataxia symptoms, and the cerebellar neuron death in the Harlequin cerebellum appears to be mediated by oxidative stress (Klein et al., 2002). However, loss of AIF leads to an increase in reactive oxygen species in various cell types in vitro (Apostolova et al., 2006) and also in cardiomyocytes of Harlequin mice (van Empel et al., 2005), suggesting that AIF deficiency might be a general mechanism of oxidative damage in different cell types. In the present study, we sought to determine whether cerebellar neurodegeneration in Harlequin mice might extend beyond the granule cell layer and involve a more general mitochondrial mechanism.
The cerebellum is an appealing model system for studying pattern formation. Despite its uniform histology, it is highly compartmentalized into transverse zones, and within each zone the cortex is further subdivided into a reproducible array of parasagittal stripes (Hawkes, 1997, Ozol et al., 1999, Armstrong and Hawkes, 2000). The most extensively studied compartment marker is zebrin II/aldolase c, which is expressed by a subset of Purkinje cells forming parasagittal stripes (Brochu et al., 1990, Ahn et al., 1994). Differential sensitivity of zebrin II-positive versus zebrin II-negative Purkinje cells to injury has been reported in several mouse mutants (Sarna and Hawkes, 2003).
We investigated whether AIF deficiency disrupts the mitochondrial fission and fusion gene expression in Harlequin mice, and demonstrated that the mitochondrial fusion gene mitofusion 1 (Mfn1) is markedly and selectively decreased in the cerebellum of Harlequin brain. Furthermore, AIF deficiency led to massive and patterned Purkinje cell degeneration respecting zebrin II boundaries in an age-dependent manner. The last surviving Purkinje neurons were heat shock protein 25 (HSP25)-expressing cells, and Purkinje cell degeneration was mediated by caspase but not PARP-1 activation. In addition, the Purkinje cell degeneration was associated with marked Mfn1 reduction. Thus, our findings identified a novel role of AIF in Purkinje cell degeneration and in mitochondrial fusion. The Harlequin mutant could be a useful late-onset neurodegenerative model to further explore new functions of AIF in mitochondrial fusion and fission.
Materials & Methods
Mice
All animal procedures conformed to institutional regulations of the University of California at Davis and guidelines of the NIH. Harlequin mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Homozygous (Hq/Hq) females and hemizygous (Hq/Y) males were obtained by mating Hq/X females with either Hq/Y or control males. Homozygous (Hq/Hq) females (N= 34) and age-matched wildtypes (N=16) were used in this study. The mice were housed in animal facilities of the University of California at Davis with a 12-h light/dark cycle and free access to food and water.
Genotype determination
Mice were genotyped using PCR with five primers: two for sex determination (SRY: 5’-TGGGACTGGTGACAATTGTC-3’ and 5’-GAGTACAGGTGTGCAGCTCT-3’), two for the wildtype AIF allele (AIF 1F: 5’AGTGTCCAGTCAAAGTACCGG-3’; AIF 1R: 5’-CTATGCCCTTCTCCATGTAGTT-3’), and one for the AIF allele harboring the proviral insertion (Harlequin allele) (AIF RV: 5’-CCCGTGTATCCAATAAAGCCTT-3’).
Antibodies
Two different mouse monoclonal anti-calbindins were used: anti-calbindin-D-28K (clone CB-955, ascites fluid, IgG1 isotype, raised against bovine kidney calbindin, Sigma, St. Louis, MO, used at 1:1,000dilution), and calbindin D-28K raised against chicken which specifically stains the 45 Ca-binding spot (Swant, Bellinzona, Switzerland,McAb 300, lot #18(F), used at 1:1,000 dilution). Both antibodies yielded Purkinje cell specific staining identical to that reported previously (Baimbridge and Miller, 1982, Brochu et al., 1990, Ozol et al., 1999). Anti-zebrin II, a mouse monoclonal antibody produced by immunization with a crude cerebellar homogenate from the weakly electric fish Apteronotus, was used directly from spent hybridoma culture medium at a concentration of 1:1,000. In the cerebellum, zebrin II immunoreactivity was restricted to a Purkinje cell subset (Brochu et al., 1990), together with very weak uniform expression in some glial cells (Walther et al., 1998). A rabbit polyclonal anti-heat shock protein 25 (anti-HSP25, 1:5,000) was purchased from StressGen (Victoria, BC, Canada: SPA-801, lot #B111411). It gave a staining pattern identical to that reported previously, where antibody absorption controls using HSP25 also abolished all immunostaining. On Western blots of cerebellar homogenate, it recognizes a single band, apparent molecular weight 25 kDa (Armstrong et al., 2000). Anti-Rabbit cleaved caspase-3 (1:50, Cell Signaling, CA) and caspase-9 (1:200, Cell Signaling, CA) antibodies were produced by immunizing animals with a synthetic peptide (KLH-coupled) corresponding to amino-terminal residues adjacent to (Asp 175) in human caspase-2 and to residues surrounding Asp353 of mouse caspase-3 respectively. Mouse monoclonal anti-calretinin (CR, 1:1,000) was raised against full-length recombinant human CR (Swant Inc. Bellinzona, Switzerland: #7699/4), and the antibody specificity was described in detail in (Schwaller et al., 1993). Affinity purified rabbit anti-Tbr2 was raised against the mouse Tbr2 synthetic peptide ((Englund et al., 2006); Chemicon, 1:1,000). Rabbit anti-phospholipase Cß4 (anti-PLCß4, 1:1,000, gift of Dr. M. Watanabe: Hokkaido University, Japan) was raised against amino acids 15-74 of the mouse PLCß4 protein fused to glutathione-S-transferase and expressed in bacteria. Control immunohistochemistry using either antibody pre-absorbed with antigen polypeptides or cerebellar sections from a PLCß4 knockout mouse yielded no significant immunostaining (Nakamura et al., 2004, Sarna et al., 2006). Anti-rabbit neurogranin (IgG) was raised against full-length recombinant rat neurogranin protein (Chemicon Inc., Temecula, CA; catalogue No. AB5620; 1:5,000). Previous studies using the same antibody have shown that neurogranin-like immunoreactivity was expressed in Golgi cells of the murine cerebellum (Singec et al., 2003). Anti-parvalbumin (ascites fluid, 1:1000, Sigma, Inc., St. Louis, MO, USA), a monoclonal antibody secreted by the PARV-19 hybridoma cell line, was produced by immunization with purified frog muscle parvalbumin. It has been shown to cross-react with parvalbumin from numerous species, including rats (Berchtold et al., 1984) and mice (Chung et al., 2007). Mouse anti-Mfn1 (Novus Biologicals, 1:200) was generated as a fusion protein against MFN1 (AAH40557, 1 a.a. ~ 742 a.a) full length recombinant protein with GST tag, between residues 348-597. Rabbit anti GFAP (Chemicon, 1:500) was raised against purified bovine GFAP protein.
Immunoblotting
Western blot analysis was carried out on total protein extracts from cerebella, cortex and olfactory bulb of adult mice. Electrophoresis was performed in SDS-polyacrylamide gel, using 30 μg of proteins per lane. The following primary antibodies were used: mouse anti-Mfn1 antibody (1:1000, Abnova), rabbit anti-Mfn2 antibody (1:1000, Sigma-Aldrich), mouse anti-Dlp1 (0.5 μg/ml, BD Transduction Laboratories), mouse anti-Opa1 (1 μg/ml, BD Transduction Laboratories), and mouse anti-β-Actin monoclonal antibody (1:1000, Santa Cruz Biotechnology). Specific immunolabeling was obtained by using horseradish peroxidase-conjugated secondary antibodies, followed by the SuperSignalWest Pico chemiluminescence detection system (Pierce, Rockford).
Flow cytometry
Cerebellar single-cell suspension was prepared as described previously (Tomomura et al., 2001). Briefly, cerebella were incubated for 20 min in Hanks’ balanced salt solution (HBSS) containing 0.05% trypsin, 3 mg/mL of bovine serum albumin, 15 mM HEPES, 3 mg/mL glucose and 1.5 mM MgSO4 (all from Sigma, St. Louis, MO, USA). Following centrifugation (200×g for 3 min), tissue was resuspended in 1 ml of DMEM (GIBCO) containing 0.7 mg of ovomucoid and 1 mg of DNase. The cells were dissociated and collected by centrifugation at 300×g for 5 min. The cellular pellets were resuspended and fixed for 20 min in 2% paraformaldehyde, permeabilized with 0.2% triton-X100 and then stained overnight at 4 °C with the following antibodies: mouse anti-Mfn1 antibody (1:200, Abnova) and rabbit anti-CaBP (1:1000, Swant)
Immunohistochemistry
Mice were deeply anaesthetized with sodium pentobarbital (100 mg/kg, i.p.) and transcardially perfused with 0.9% NaCl in 0.1 M phosphate buffered saline (PBS, pH 7.4) followed by 4% paraformaldehyde in 0.1 M PBS (pH 7.4). The brains were then removed from the skull and post-fixed in 4% paraformaldehyde at 4°C for 48 hours. The cerebella were cryoprotected through a series of buffered sucrose solutions: 10% (2 hrs), 20% (2 hrs) and 30% (overnight) and then embedded in OCT and frozen for cryosectioning. Transverse sections were cut on a cryostat at 40 μm thickness through the extent of the cerebellum and collected as free-floating sections for immunohistochemistry.
Peroxidase immunohistochemistry was carried out as described previously (Sillitoe et al., 2003). Briefly, tissue sections were washed thoroughly blocked with 10% normal goat serum (Jackson ImmunoResearch Laboratories, West Grove, PA) and then incubated in 0.1 M PBS containing 0.1% Triton-X and the primary antibody for 16-18 hours at 4°C. Sections were then incubated in horseradish peroxidase (HRP)-conjugated goat anti-rabbit or HRP-conjugated goat anti-mouse secondary antibodies (1:200 dilution in PBS; Jackson ImmunoResearch Laboratories, West Grove, PA) for 2 hours at room temperature. Diaminobenzidine (DAB, 0.5 mg/ml) was used to visualize the reaction. Finally, sections were dehydrated through an alcohol series, cleared in xylene and cover-slipped with Entellan mounting medium (BDH Chemicals, Toronto, ON, Canada).
Cerebellar sections for fluorescent immunohistochemistry were processed as described previously (Sillitoe et al., 2003). Briefly, tissue sections were washed, blocked in PBS containing 10% normal goat serum (Jackson ImmunoResearch Laboratories, West Grove, PA), and incubated in both primary antibodies overnight at room temperature. Sections were then rinsed and incubated for 2 hours at room temperature in a mixture of Alexa 546-conjugated goat anti-rabbit IgG, Alexa 488-conjugated goat anti-mouse IgG, and Alexa 643-conjugated goat anti-guinea pig IgG (Molecular Probes Inc., Eugene, OR), at 1:2,000 dilution. After several rinses in 0.1 M PBS, sections were coverslipped in non-fluorescing mounting medium (Fluorsave Reagent, Calbiochem, La Jolla, CA).
Photomicrographs were captured with a SPOT Cooled Color digital camera (Diagnostic Instruments Inc.), mounted on a Zeiss microscope and assembled in Adobe Photoshop (version 9). The images were cropped and corrected for brightness and contrast, but not otherwise manipulated. To count the relative number of Purkinje cells in both Harlequin and wild-type littermates, images were captured from 40 μm thick serial transverse sections through the cerebellar vermis. The vermis of cerebellar lobules VIII was divided from the midline into 2 compartments, and the number of immunoreactive Purkinje cell profiles was counted in each bin [8 months: Hq/Hq (N=6), +/+ (N=3); 11 months: Hq/Hq (N=7), +/+ (N=4); 14 months: Hq/Hq (N=4), +/+ (N=3)].
Results
AIF deficiency caused a marked and selective reduction of Mfn1 expression in the cerebellum
We first examined whether AIF deficiency alters the expression of mitochondrial fusion (Mfn1, Mfn2, Opa1) and fission (Dlp1) proteins using immunoblotting with brain tissues of Harlequin mice in which AIF is reduced by 80% (Klein et al., 2002). Consistent with the previous study, AIF expression level was significantly decreased in the olfactory bulb, cortex and cerebellum of Harlequin brain (Fig. 1A). Western blot analysis revealed that Mfn1 expression was markedly and selectively decreased in the cerebellum of 9-month-old Harlequin mice as compared to the age-matched wildtype, while no significant changes were identified in the expression of Mfn1 in olfactory bulb and cerebral cortex, as well as Mfn2, Opa1 and Dlp1 in olfactory bulb, cerebral cortex and cerebellum (Fig. 1A). To further confirm the Mfn1 reduction in the cerebellum, expression of Mfn1 was measured by flow cytometry analysis (Fig. 1B,C). A significant decrease of the intensity of Mfn1 immunostaining was seen in the Harlequin cerebellum (Fig. 1B,C). To reveal the cell-type specifity of the Mfn1 reduction in the cerebellum, we performed flow cytometry analysis with double-immunostaining of calcium-binding protein (CaBP), a specific marker for cerebellar Purkinje cells (Baimbridge and Miller, 1982, Ozol et al., 1999), and Mfn1. The results showed that the number of Mfn1-immuopositive Purkinje cells (blue lined rectangular regions) were significantly decreased in the Harlequin cerebellum (Fig. 1D,F) as compared to the wildtype (Fig. 1E,F).
FIGURE 1. Mfn1 expression is markedly and selectively decreased in the Harlequin cerebellum.
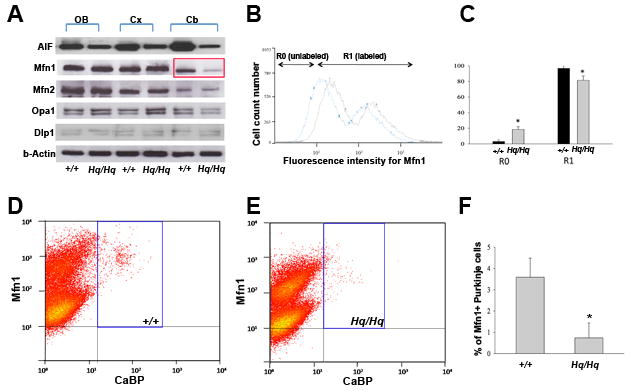
A: Immunoblot analysis of mitochondrial fusion (Mfn1, Mfn2, Opa1) and fission (Dlp1) proteins in various regions of the 9-month Harlequin brain and age-matched controls. Mfn1 protein level was markedly decreased in the cerebellum of Harlequin mice. Abbreviations: Cb = cerebellum; Cx = cerebral cortex; OB = olfactory bulb. B-C: Intensity of Mfn1 expression was quantified by flow cytometry analysis. A reduction of the Mfn1 immunostaining was seen in Harlequin cerebellum, consistent with the results by western blots (A). D-F: Measurement of Mfn1- positive, CaBP-expressing Purkinje cells by flow cytometry analysis. The X axis represents the relative fluorescence intensity of CaBP, and the Y axis represents the relative fluorescence intensity of Mfn1. Representative dot-plot illustrations of the cell suspension in the 9-month-old wildtype (D) and Harlequin (E) cerebellum. The blue-lined rectangular regions represent Mfn1 and CaBP double-immunopositive Purkinje cells. The number of Mfn1-immunopositive Purkinje cells was markedly decreased in the Harlequin cerebellum (D). Summary of the percentage of Mfn1-positive Purkinje cells out of total cells after sorting. The percentage of positive cells for Mfn1 and CaBP were quantified, and values were expressed as a percentage of total population (E). All data shown are mean ± SEM from at least three animals for each group. *p<0.05 (one-way ANOVA followed by Tukey post hoc test).
AIF deficiency resulted in remarkably small cerebellum, morphogenetic defects, and Purkinje cell pathology
Given the selective mitochondrial deficiency in Purkinje cells, we next investigated the cerebellar pathology of Harlequin mice and examined whether Purkinje cell degeneration might be a previously unrecognized cerebellar phenotype resulted from AIF deficiency. The Harlequin cerebellum exhibited a marked reduction in size when compared to the wildtype at 9 months of age as seen by cresyl violet staining (Fig. 2A,B). The basic lobular formation (e.g., the number of lobules) appeared relatively normal (Fig. 2A,B) despite the reduction in overall size. Differential defects in cerebellar morphogenesis were observed among cerebellar lobules. The posterior cerebellum (lobules VI~X) was clearly hypotropic, most significantly at the level of lobules IX and X (arrows in Fig. 2B), whereas the anterior cerebellum (I~V) exhibited a structure similar to the control (Fig. 2A,B).
FIGURE 2. General Purkinje cell pathology in the Harlequin cerebellum.

A, B: Cresyl violet staining of a sagittal section in the wildtype (A) and Harlequin cerebellum (B) at 8 months of age. Roman numerals indicate lobule numbers. The Harlequin cerebellum is greatly reduced in size (B). Foliation defects are obvious in lobules IX and X. C, D: Sagittal sections stained for anti-CaBP in the wildtype (C) and Harlequin cerebellum (D) at 8 months of age. CaBP expression is restricted to Purkinje cells, and no other structures in the cerebellum express CaBP. E, F: Higher magnification views of the regions indicated in C and D (cerebellar lobules VIII and IX). In the wildtype, CaBP uniformly stains all Purkinje cells and no gaps in Purkinje cell layer are seen (E). Extensive Purkinje cell loss is observed in lobule VIII and dorsal IX in the Harlequin cerebellum (F). G-J: Morphological Purkinje cell abnormalities in the Harlequin cerebellum. Vesicular storage material accumulation is evident in Purkinje cell soma and dendrites (G). Frequently, thick megadendrites are seen extending into the molecular layer (H). Some Purkinje cells are located ectopically, either in the molecular layer (I) or in the granule cell layer/white matter tract. Abbreviations: CN = cerebellar nuclei; gcl = granule cell layer; ml = molecular layer; pcl = Purkinje cell layer; wm = white matter. Scale bars: A = 500 μm (A-D); G = 25 μm (also applies to I); J = 50 μm.
Purkinje cells are major cerebellar neurons. Due to the dramatically reduced cerebellar size in Harlequin mice, we investigated whether the Purkinje cell population was affected. We used immunohistochemistry CaBP to reveal their spatial distribution in the Harlequin cerebellum. Peroxidase immunohistochemistry with anti-CaBP revealed strong immunoreactivity in Purkinje cell somas and their dendrites throughout all lobules in both the wildtype and Harlequin cerebellum (Fig. 2C-F). Interestingly, the Harlequin cerebellum exhibited extensive Purkinje cell loss in lobules VIII and IX compared to Purkinje cell patterning in the wildtype (Fig. 2E,F). Careful examination of CaBP-positive Purkinje cell morphology revealed several reproducible abnormalities. First, primary dendrites and Purkinje cell soma were grossly swollen (Fig. 2G). Prominent vesicular storage materials were accumulated within the cytoplasm of the somata (Fig. 2H). Secondly, the degenerating Purkinje cells often exhibited “megadendrites,” featuring massive swelling in the primary dendrite (Fig. 2H). Thirdly, Purkinje cell dendrites failed to span the width of the molecular layer (Fig. 2I). The location of Purkinje cells was also ectopic, and the cell soma was displaced into either molecular layer (Fig. 2I) or granule cell layer/white matter (Fig. 2J).
Progressive Purkinje cell degeneration in the Harlequin cerebellum
Next, we sought to determine whether altered Purkinje cell morphology affected cell survival. We found that Purkinje cell degeneration accompanied their morphological abnormalities. In the anterior region of the wildtype cerebellum, Purkinje cells were densely packed in all lobules, and no gaps were observed in the Purkinje cell and molecular layers throughout the age of 3 -14 months. CaBP-positive Purkinje cells were evenly distributed throughout the anterior vermis in the 11-month-old wildtype (Fig. 3A). At 3 months of age, anti-CaBP staining revealed no evidence of Purkinje cell degeneration in the Harlequin cerebellum. By 8 months of age, Purkinje cell degeneration was obvious in the hemisphere region of the anterior lobe (lobules I~V) in the Harlequin cerebellum (Fig. 3B, Crus1, arrows). At 11 months of age, more widespread Purkinje cell degeneration occurred and extended to the vermis (Fig. 3C). Most Purkinje cells degenerated by 14 months of age in the Harlequin cerebellum (Fig. 3D). Very few scattered Purkinje cells survived in the vermis of cerebellar lobules I-III (Fig. 3D). A similar pattern of Purkinje cell degeneration occurred in the posterior Harlequin cerebellum Purkinje cell loss was apparent at 7 months of age. By 8 months, Purkinje cell loss was obvious throughout lobules VI-VIII (Fig. 3F, J) compared to the wildtype (Fig. 3E, I). At 11 months of age, Harlequin mice exhibited widespread degeneration (Fig. 3G,K). By 14 months of age, lobules VI-VIII were nearly devoid of Purkinje cells, with only a few scattered Purkinje cells remaining (Fig. 3H,L). Quantification of Purkinje cells in the vermis of lobule VIII and IV revealed a significant decrease in the number of these cells present in Harlequin mice compared with wildtypes at 8 months, 11 months and 14 months of age (Fig. 3M, p<0.05).
FIGURE 3. Progressive Purkinje cell degeneration in the Harlequin cerebellum.

A-D: Anti-CaBP immunocytochemistry of transverse sections through the anterior lobe in the wildtype (A: 11 months) and Harlequin cerebellum (B: 8 months; C: 11 months; D: 14 months). In the wildtype cerebellum, CaBP uniformly stains all Purkinje cells. Purkinje cells are tightly packed in all lobules and no gaps between Purkinje cell soma are observed (A). In the Harlequin cerebellum, Purkinje cells are degenerated in an age-dependent manner. At 8 moths of age, Purkinje cell degeneration is apparent in the hemisphere regions (B, arrows). By 11 months of age, almost half of the Purkinje cells disappear from the anterior region of the Harlequin cerebellum (C. By 14 months, Purkinje cell degeneration is widepread. (D). E-L: Transverse cerebellar sections immunostained with anti-CaBP through the posterior vermis in the wildtype (E, I: 11 months) and Harlequin cerebellum (F, J: 8 months; G, K: 11 months; H, L: 14 months). Purkinje cell degeneration is especially apparent in lobules VII and VIII in the Harlequin mice from 8 months to 14 months of age (F, G, H). The region outlined in the rectangle in (E), (F), (G), (H) is displayed at higher magnification in (I), (J), (K), (L), respectively. M: The number of Purkinje cells is significantly decreased in an age-dependent fashion in the lobule VIII and VI of Harlequin mice (8 month: Hq/Hq (N=6), +/+ (N=3); 11 months: Hq/Hq (N=7), +/+ (N=4); 14 months: Hq/Hq (N=4), +/+ (N=3)). Abbreviations: gcl = granule cell layer; ml = molecular layer; pcl = Purkinje cell layer. Scale bars: A = 500 μm (A-D); E = 250 μm (E-H); I = 125 μm (I-L)
Zebrin II-negative Purkinje cell populations were preferentially lost at 8 months of age in the Harlequin cerebellum
Zebrin II immunocytochemistry is often used to aid in cerebellar topography demarcation in the normal rodent cerebellum. Zebrin II recognizes a single 36 kD polypeptide in a subset of Purkinje cells which reveals parasagittal stripes in the cerebellum (Brochu et al., 1990, Hawkes, 1997, Ozol et al., 1999, Armstrong and Hawkes, 2000, Sillitoe and Hawkes, 2002). There are up to seven zebrin II+ stripes in each hemicerebellum named P1+ from the midline to P7+ laterally, separated by zebrin II- stripes named P1- to P6-. Due to the patterned Purkinje cell degeneration that we observed in the Harlequin cerebellum, we decided to explore whether zebrin II-positive or -negative cells were affected. At 8 months of age, the surviving Purkinje cells formed clusters in the posterior lobules (Fig. 4A), displaying a similar expression pattern with that of zebrin II-expressing cells (Fig. 4B). Double immunostaining with anti-CaBP and anti-zebrin II revealed that most surviving Purkinje cells were zebrin II-positive (Fig. 4E arrows), suggesting that zebrin II-negative Purkinje cells preferentially degenerate at 8 months of age. Eventually, however, both the zebrin II-positive and zebrin II-negative cell populations degenerated (e.g. at 14 months, see Fig. 3D,H,L).
FIGURE 4. Purkinje cell degeneration at 8 months of age occurs preferentially in zebrin II-negative Purkinje cells and HSP25-positive Purkinje cells are most resistant to degeneration.

A: Transverse section stained for CaBP in the posterior vermis of the Harlequin cerebellum. The Purkinje cell degeneration does not appear to be random. Surviving Purkinje cells are grouped into clusters (indicated with “*”). B: Zebrin II expression in the posterior vermis of the normal mouse cerebellum. The Purkinje cell stripes are labeled as P1+ and P1- (for stripe terminology, see (Sillitoe and Hawkes, 2002)). C-E: A transverse section double-immunostained with anti-CaBP (red) and anti-zebrin II (green) in the 8 monthold Harlequin cerebellum (lobule VIII) shows that zebrin II-negative Purkinje cells preferentially die. All zebrin II-positive Purkinje cells are intact (arrows in E). F: Transverse section through the midline (dashed line in F) of ventral lobule X immunoperoxidase stained for CaBP reveals degenerating Purkinje cell clusters (indicated with “*”) in the Harlequin cerebellum. Surviving Purkinje cell clusters are labeled with “***”. G: Transverse section through the midline of ventral lobule X immunoperoxidase stained for heat shock protein 25 (HSP25). The distribution of HSP25-positive Purkinje cells appears intact, revealing 3 broad bands. HSP25 is expressed in an array of broad stripes, P1+ at the midline, P2+ and P3+ laterally on either side of the cerebellar lobule X (Armstrong et al., 2000). H-K: Double immunofluorescence labeling with anti-CaBP and HSP25 confirms that surviving Purkinje cells in ventral lobule X are actually HSP25-positive Purkinje cells. Higher magnification views of the regions in (H) are shown in (I), (J) and (K), respectively. Scale bars: B = 250 μm (A-B); E = 250 μm (C-E); F = 250 μm (F-I).
HSP25-positive Purkinje cells were most resistant and remained intact until the final stage of degeneration
We noticed that the surviving Purkinje cells primarily accumulated in the cerebellar lobule X (Fig. 4F). In this region, the small heat shock protein, HSP25, is constitutively expressed, forming parasagittal stripes (Armstrong et al., 2000). In the normal cerebellum, HSP25 is expressed only in a small subset of Purkinje cells (~2% of the total) (Armstrong et al., 2000). We investigated whether the distribution of HSP25-positive Purkinje cells was altered in the Harlequin cerebellum and whether the Purkinje cells constitutively expressing HSP25 would preferentially survive to the terminal stage of degeneration. In the normal cerebellum, HSP25-immunoreactive Purkinje cells form three clusters. Despite the reduced size of the Harlequin cerebellum, the overall expression pattern of HSP25 in cerebellar lobule X appeared normal in the mutant (Fig. 4G). At 11 months of age, anti-CaBP staining revealed three surviving Purkinje cell clusters (Fig. 4F, indicated with “***”) that appeared to correspond to HSP25-positive Purkinje cells (Fig. 4G) in the Harlequin cerebellum. Double immunofluorescence staining with anti-CaBP and anti-HSP25 confirmed that surviving Purkinje cells were HSP25-positive Purkinje cells (Fig. 4H-K).
Purkinje cell degeneration in the Harlequin cerebellum was dependent on caspase activation
AIF is known to mediate caspase-independent cell death induced by PARP-1 activation (Yu et al., 2002). Thus, we investigated whether PARP-1 might be involved in Purkinje cell degeneration in the Harlequin cerebellum. Immunocytochemistry with an anti-PARP-1 antibody revealed no PARP-1 expression in the cerebellum of wildtype or Harlequin mice aged 3 - 11 months (Fig. 5A,B), indicating that Purkinje cell degeneration in the Harlequin cerebellum might not be mediated by PARP-1 activation. Lack of PARP-1 immunoreactivity led us to believe that AIF-mediated degeneration was occurring via a caspase-dependant mechanism rather than a caspase independent mechanism. Thus, we investigated caspase activation in the Harlequin cerebellum. We performed immunostaining with activated caspase-3 and caspase-9 antibodies on transverse sections of the anterior hemisphere of the cerebellum on 8 month old Harlequin mice. We found numerous activated caspase-3 and caspase-9 positive cells present in the Purkinje cell and granular layers (Fig. 5F-H), whereas no caspase expression was observed in age-matched wildtype mice (Fig. 5C-E). The Harlequin mice lacked expression of activated caspase-3 and caspase-9 in the molecular layer and white matter tract (Fig. 5F-H). Double-label immunofluorescence staining confirmed that many of the activated caspase-3 and caspase-9-positive cells were CaBP-positive Purkinje cells (Fig. 5I-K). These data indicate that the extensive Purkinje cell degeneration in the Harlequin cerebellum is mediated by a caspase-dependent signaling pathway.
FIGURE 5. Purkinje cell degeneration in the Harlequin cerebellum is dependant on caspase activation.

A, B: Transverse section peroxidase stained for a PARP-1 antibody at 8 months of age in the wildtype (A) and Harlequin cerebellum (B). In both cases, no PARP-1 immunoreactivity is observed in the cerebellar cortex. To rule out a malfunction of the antibody, PARP-1 immunoreactivity in cerebellar nuclei (CN) neurons are shown in the inserts. C-H: Anterior hemisphere sections are immunostained with caspase-3 (C, D, F, G) and caspase-9 (E, H) in the wildtype (C-E) and Harlequin cerebellum (F-H). Caspase-3 and caspase-9 are not expressed at 8 months of age in the wild type cerebellum (C-E). Conversely, a dramatic elevated expression of caspase-3 and -9 is observed both in Purkinje cell and granule cell layers of the age-matched Harlequin cerebellum (F-H). I, J: Transverse section double immunofluorescence stained with anti-CaBP (green) and anti-caspase-3 (red) at 8 months of age in the Harlequin cerebellum shows that most Purkinje cells co-express caspase-3 (arrowheads in J). Consistent with the single immunostaining results shown in (C, D), there is no caspase-3 expression in wildtype Purkinje cells (I). K: Double immunofluorescence labeling with CaBP (red) and caspase-9 (green) reveals that degenerating Purkinje cells also express caspase-9 (arrowheads). Abbreviations: CN=cerebellar nuclei neurons; gcl = granule cell layer; ml = molecular layer; pcl = Purkinje cell layer; wm = white matter. Scale bars: A = 125 μm (A,B, D,E,G,H);.C = 250 μm (C,F); K = 25 μm (I-K).
Mfn1 expression in Purkinje cell was significantly reduced in Purkinje cells of Harlequin mice
Mfn1 was uniformly expressed in the soma and dendrites of Purkinje cells in wildtype mice at 7 and 11 months of age (Fig. 6A,D,F,G). In contrast, it was only expressed in the soma of Purkinje cells in the Harlequin cerebellum and diminished in the neurites at 9 months of age (Fig. 6C). By 11 months of age, Mfn1 expression was completely depleted from Purkinje cells in the Harlequin cerebellum (Fig. 6E,H,I). Double immunostaining with anti-Mfn1 and caspase-3 showed that Mfn1-immunoreactive surviving Purkinje cells did not express caspase-3, indicating that diminished Mfn1 and altered mitochondrial fusion might contribute to the demise of the degenerating cerebellar Purkinje cells in Harlequin mice (Fig. 6J-L).
FIGURE 6. Mfn1 expression is reduced in the Harlequin cerebellum.

A-I: Transverse sections stained with anti-Mfn1 in the wildtype (A, D, F, G) and Harlequin cerebellum (B, C, E, H, I). Mfn1 is uniformly expressed in the soma and dendrites of the Purkinje cells in the anterior vermis (A) and anterior hemisphere region (D) at 7 and 11 months, respectively. In the Harlequin cerebellum, Mfn1 expression in Purkinje cells is almost diminished at 11 months (E, H, I) compared to wildtype (D,F,G). Also note that Mfn1 is exclusively expressed in ectopically located degenerating Purkinje cell soma at 9 months of age in the Harlequin cerebellum (C). J-L: Double immunostaining with anti-Mfn1 (green) and anti-caspase-3 (red) shows that Mfn1-immunoreactive Purkinje cells do not express caspase-3 at 9 months of age. Abbreviations: gcl = granule cell layer; ml = molecular layer; pcl = Purkinje cell layer; wm = white matter. Scale bars: A = 100 μm (A-C); D = 250 μm (also applies to E); F = 100 μm (also applies to H); J = 125 μm (J-L).
Massive granule neuron degeneration occurred in the posterior cerebellum
Cerebellar defects in Harlequin mice were not restricted to Purkinje cells. A previous study indicated that granule cell degeneration was prominent in the Harlequin cerebellum (Klein et al., 2002). Similarly, we observed massive granule cell degeneration in the posterior lobules of the Harlequin cerebellum at 8 months of age (Fig. 7C,D) compared with their wildtype counterparts (Fig. 7A,B). Interestingly, only granule cells in the posterior lobules degenerated, whereas granule neurons in the anterior lobules appeared to be spared (Fig. 7C).
FIGURE 7. Granule cell degeneration is confined to the posterior lobules.

Cresyl violet stained sagittal sections from the wildtype (A, B) and Harlequin cerebellum (C, D) at 8 months of age. The region outlined by the rectangle in (A) and (C) is shown at higher magnification in (B) and (D), respectively. The granule cell degeneration is mainly restricted to the posterior lobules in the Harlequin mice (C, D), while numerous compact granule cells are seen in the wildtype cerebellum (A, B). Roman numerals indicate lobule numbers. Scale bar: A = 250 μm (also applies to C).
Other cerebellar neurons were relatively unaffected in Harlequin mice
The cerebellum contains multiple neuron populations. We performed immunohistochemistry for multiple neuron-specific markers to determine if other neuronal populations were similarly affected by AIF reduction. We found that massive neuronal degeneration was limited to the Purkinje and granule cells and did not extend to other cerebellar neurons in the Harlequin mice. The anatomical aspects of cerebellar deep nuclei neurons, which are the sole output in the cerebellum, were entirely normal; however, the overall area of the cerebellar nuclei area was reduced compared to the wildtype (Fig. 8A,B). The distribution of unipolar brush cells, as identified by CR (Sekerkova et al., 2004), PLCb4 (Chung et al., 2009a, Chung et al., 2009b) or Tbr2 (Englund et al., 2006) was not altered in the Harlequin cerebellum. These cells appeared densely packed in the granular layer as identified by CR (Fig. 8C,D), PLCb4 (Fig. 8E,F) or Tbr2 (Fig. 8G,H). In the normal adult mouse cerebellar cortex, neurogranin immunoreactivity is restricted to the somata and dendritic arbors of Golgi cells (Singec et al., 2003). The presence of neurogranin-positive Golgi cells in lobule VI remained largely unchanged in Harlequin mice (Fig. 8I,J). Parvalbumin, a marker of basket and stellate cells in the molecular layer (Bastianelli, 2003) also appeared similar between the wildtype and Harlequin cerebellum (Fig. 8K,L). Thus, despite a marked size reduction of the mutant cerebellum and the massive degeneration of Purkinje and granule neurons, other cerebellar neurons were relatively unaffected in Harlequin mice.
FIGURE 8. Many aspects of cerebellar cell type distribution are unaffected in Harlequin mice.

A, B: Transverse section of the cerebellar deep nuclei stained with cresyl violet in the wildtype (A) and Harlequin cerebellum (B). Despite the reduced size in the Harlequin mutant, the basic anatomical structure of cerebellar nuclei is normal. C, D: Immunoperoxidase staining for calretinin in a sagittal section through lobules VIII-X in the wildtype (C) and Harlequin cerebellum (D). Calretinin-immunopositive unipolar brush cells are preferentially located in lobules IX and X. Roughly similar distribution patterns are observed in the wildtype and Harlequin mice. E, F: Transverse section through lobule VII-VIII immunostained for PLCb4 in the wildtype (E) and Harlequin cerebellum (F). In both cases, PLCb4-positive unipolar brush cells are tightly packed in granule cell layer. Note that PLCb4 is also expressed in a subset of Purkinje cells (= zebrin II-negative subset: (Sarna et al., 2006)). G, H: Transverse section through lobule IX immunoperoxidase stained for Tbr2. No significant difference is observed between the wildtype and Harlequin mutant. I, J: Immunoperoxidase staining of a transverse section through lobules VI-VIII in the wildtype (I) and Harlequin cerebellum (J) reveals that anti-neurogranin immunoreactivity is detected in Golgi cells. The number of neurogranin-positive Golgi cells in lobule VI is roughly unchanged. K, L: Peroxidase stained transverse section with anti-parvalbumin immunostaining in the wildtype (K) and Harlequin cerebellar cortex (L). No systematic differences are seen between the wildtype and Harlequin cerebellum. Abbreviations: Aint = anterior interposed nucleus; CR = calretinin; CV = cresyl violet; Lat = Lateral nucleus; NG = neurogranin; Parv = parvalbumin; PLCb4 = phospholipase C beta 4; Tbr1 = T brain factor 1. Scale bar: A = 500 μm (A-L).
Cerebellar astrogliosis was prominent in Harlequin mice
Astrogliosis is a hallmark of many neurodegenerative diseases (Maragakis and Rothstein, 2006). Due to the massive Purkinje cell degeneration present in the Harlequin cerebellum, we assessed the distribution of GFAP-positive astrocytes in these mice. Weak and uniform GFAP expression was observed near the Purkinje cell layer in the wildtype cerebellum (Fig. 9A-C). In contrast, the Harlequin cerebellum displayed prominent GFAP-immunoreactivity in the Purkinje cells layer, with astrocytic processes extending into the molecular layer (Fig. 9D-F). Activated astrocytes formed several clusters in anterior lobules of the Harlequin cerebellum (indicated with “*” in Fig. 9E), similar to the patterning of CaBP-positive Purkinje cells. Thus, we then examined the topographical relationship between the activated astrocytes and degenerating Purkinje cell clusters. Double immunofluorescence staining with anti-CaBP and anti-GFAP revealed that GFAP-positive astrocytes were located in close proximity to degenerating Purkinje cell clusters (Fig. 9G-L).
FIGURE 9. Astrogliosis is prominent in the Harlequin cerebellum.

A-F: Transverse sections through the anterior part of the cerebellum immunoperoxidase-stained with anti-GFAP in the wildtype (A-C) and Harlequin cerebellum (D-F). Few GFAP-positive cells are present in the wildtype cerebellum (A-C). There is a marked increase in GFAP-immunoreactivity in the Harlequin cerebellum (D-F). The region outlined in the rectangle in (A) and (D) is demonstrated at higher magnification in (B) and (E), respectively. Roman numerals indicate lobule numbers. G-L: Double immunostaining with anti-CaBP (green) and anti-GFAP (red) illustrates that GFAP-positive astrocytes are mainly associated with degenerated Purkinje cell clusters. The distributions of surviving Purkinje cell clusters and GFAP-positive astrocyte clusters are strictly non-overlapping and mutually exclusive. Abbreviations: gcl = granule cell layer; ml = molecular layer; pcl = Purkinje cell layer. Scale bars: A = 250 μm (also applies to D); B = 125 μm (E,G-I); L = 50 μm (J-L).
Discussion
In this study, we revealed a novel function of AIF in regulating mitochondrial fusion and Purkinje cell degeneration. Using Harlequin mutant mice, we showed that AIF deficiency leads to down-regulation of cerebellar Mfn1 and Purkinje cell neurodegeneration. We further demonstrated that the striking process of progressive and patterned Purkinje cell degeneration was dependent on caspase but not PARP-1 activation. Our results indicate that the altered mitochondrial fusion in Harlequin mice make them a good candidate for mechanistic studies on mitochondrial fusion and fission as well as AIF-related oxidative stress and cell death studies. The present study is the first to link the regulation of AIF and Mfn1, and to reveal the striking process of progressive and patterned Purkinje cell degeneration in Harlequin mice. These results have important implications in linking AIF and mitochondrial fusion in neurodegeneration.
Purkinje cell death is often observed as a common feature in many neurological disorders including autism, Huntington’s disease, Alzheimer’s disease, multiple system atrophy, epilepsy, and alcoholism (Sarna and Hawkes, 2003). Many of these diseases are accompanied by some aspects of motor dysfunction. We characterized cerebellar defects in Harlequin mutant mice, which display progressive cerebellar ataxia. We observed a striking age-dependent Purkinje cell degeneration, and nearly all Purkinje cells were lost by 14 months of age in the Harlequin cerebellum. The Purkinje cell loss in the Harlequin cerebellum appeared to be highly patterned. The time course of Purkinje cell degeneration could be separated into three phases: First, zebrin II-positive Purkinje cells consistently survived longer than zebrin II-negative cells; second, within the zebrin II-positive population, the Purkinje cells in lobule X (NZ) were more resistant than other lobules; and third, within lobule X, Purkinje cells expressing HSP25 survived the longest.
The patterned Purkinje cell loss has been described in several naturally-occurring mutants, such as acid sphingomyelinase null (Sarna et al., 2001), and BALB/c npcnih (Sarna and Hawkes, 2003, Sarna et al., 2003). In murine models of the neurodegenerative diseases Niemann-Pick type C – BALB/c npcnih and C57BLKs/J spm, zebrin II-negative Purkinje cells die first, leaving a pattern of surviving stripes that subsequently also degenerate, except for those that constitutively express HSP25 (Sarna et al., 2001, Sarna et al., 2003). Likewise, in the Purkinje cell degeneration (pcd) mouse cerebellar cortex, Purkinje cells start to degenerate from postnatal day (P) 20 and continue so that approximately 90% of them are lost by P45 and nearly all are gone by P60 (Landis and Mullen, 1978). The few surviving Purkinje cells also express HSP25, reminiscent of those seen in the BALB/c npcnih cerebellum (Sarna et al., 2003). Taken together, our data combined with these data strongly suggest that Purkinje cells show a subtype-dependent susceptibility to injury or degeneration (Sarna and Hawkes, 2003).
In the Harlequin cerebellum, the Purkinje cell loss was progressive. At the terminal stage of degeneration, the surviving Purkinje cells were concentrated in lobule IX and X of the posterior vermis. Many Purkinje cells in these lobules expressed HSP25. Previous studies have shown that HSP25 and its rat homolog HSP27 protected against various forms of stress or injury in many different cell types. For example, overexpression of HSP25 or HSP27 protected L929 and CCL39 fibroblasts against hydrogen peroxide-induced cytoskeletal disruption and cell death (Mehlen et al., 1995, Huot et al., 1996), and overexpression of HSP25 in transfected myoblasts produced dose-dependent protection against hydrogen peroxide-induced damage (Escobedo et al., 2004). In addition, rat sensory neurons transfected with human HSP27 mRNA significantly reduced apoptotic cell death (Lewis et al., 1999, Wagstaff et al., 1999). These results suggest that HSP25 expression affords protection and increases cell survival in vitro and in vivo. HSP25 is expressed in two different zones: CZ (cerebellar lobules VI and VII) and NZ (lobules IX and X). Many HSP25-expressing Purkinje cells in NZ are clearly protective and survive the longest in the Harlequin cerebellum. Conversely, the HSP25-positive Purkinje cells in the CZ are not preferentially protected from the degeneration. The mechanism underlying this phenomenon is unknown and may be due in part to location. Sparing of cells within the NZ but not in CZ has also been described in other neurological mouse mutants. Wassef et al. (1987) first reported the spatiotemporal pattern of Purkinje cell degeneration and the preferential survival of Purkinje neurons located in cerebellar lobules IX and X in DW/J-Pas mice. In addition, mouse models of Niemann Pick type A/B and C exhibited similar data with Harlequin mice, and surviving Purkinje cells in NZ were associated with HSP25 expression (Sarna et al., 2001, Sarna et al., 2003). In addition, Purkinje cells in cerebellar lobule X were more resistant to injury in spinocerebellar ataxia type 6 (Yang et al., 2000). Taken together, the constitutive HSP25 expression may afford protection against cell injury in the cerebellum, suggesting that HSP25 induction in Purkinje cells may be a potential therapeutic strategy.
Our data showed that Purkinje cell degeneration in the Harlequin cerebellum was dependent on caspase activation. AIF is well known to mediate cell death induced by PARP-1 activation (Liu et al., 1996, Susin et al., 1999b, Cande et al., 2002, Yu et al., 2002). Neither PARP-1 nor AIF was detected in the Harlequin cerebellum (Fig. 5). Thus, extensive Purkinje cell loss in the Harlequin cerebellum is likely to be mediated by the normal physiological function of AIF within mitochondria. Oxidative stress has been implicated as a key factor in the pathogenesis of Harlequin neurodegeneration (van Empel et al., 2006). Although AIF does not seem to be physically involved with components of the respiratory complex, it has been suggested that the vulnerability of Harlequin mice to oxidative stress is due to the participation of AIF in mitochondrial respiration by handling the reactive oxygen species released from the respiratory chain (Vahsen et al., 2004). Several in vitro and in vivo studies support this hypothesis. In AIF knockout embryonic stem cells and various cell lines with RNAi-mediated AIF knockdown, the function of the mitochondrial respiratory chain, particularly complex I, is disrupted, e.g. decreased protein expression levels of the complex I subunits p17, p20, and p39 (Vahsen et al., 2004, Urbano et al., 2005, Apostolova et al., 2006). In addition, Bénit et al. (Benit et al., 2008) demonstrated that complex I activity was reduced to approximately 50% of the control value in the Harlequin cerebellum. Complex I deficiency was first observed around 1 month of age and persisted until 6 months (Benit et al., 2008). The extensive Purkinje cell degeneration occurred at 7 months of age, and the severe ataxia phenotype was observed at 6 months. Thus, cerebellar function maintained for several weeks despite mitochondrial complex I deficiency, suggesting that unknown factors are likely to be involved in the Harlequin phenotype. In the present study, we observed the sequential events of neurodegeneration in the Harlequin cerebellum, i.e. granule cell degeneration followed by progressive and patterned Purkinje cell degeneration. Our results suggest that complex I deficiency might be involved in early granule cell degeneration and that later Purkinje cell degeneration might result from a deficiency of mitochondria fusion. Mitochondria are highly dynamic cellular powerhouses that play critical roles in cell death. In recent years, a mitochondrial network concept has been articulated; mitochondria do not exist as distinct, individual, static, autonomous organelles. Rather, they form a highly dynamic network in the cell, via the balance of the opposing mitochondrial fusion and fission processes (Chan, 2006, Chen and Chan, 2009). We observed diminished expression of Mfn1 in Harlequin Purkinje cells, which might alter the balance of mitochondrial fusion and fission. This balance is crucial for mitochondrial morphology and function, and previous studies demonstrated that the mitochondrial fusion and fission machinery could be “hijacked” to propagate apoptotic signals or oxidative damage (Karbowski et al., 2006). Further studies are warranted to elucidate the functional role of AIF in mitochondrial fusion and fission, and to explore whether altered mitochondrial machinery due to AIF deficiency propagates oxidative stress and subsequent neuron degeneration or whether the altered mitochondrial processes are a “side effect” of neuron degeneration. Thus, the Harlequin mouse could serve as not only a model for late-onset cerebellar neurodegeneration studies but also as a tool for studying the effects of mitochondrial fusion and fission on neurodegeneration and cell death.
Acknowledgments
These studies were supported by grants to W.D. from the NIH (RO1 NS059043 and RO1 ES015988), National Multiple Sclerosis Society, Feldstein Medical Foundation and Shriners Hospitals for Children. We thank Dr. Richard Hawkes (University of Calgary, Alberta, Canada) for providing zebrin II antibody.
Footnotes
We declare no conflicts of interest related to this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn AH, Dziennis S, Hawkes R, Herrup K. The cloning of zebrin II reveals its identity with aldolase C. Development. 1994;120:2081–2090. doi: 10.1242/dev.120.8.2081. [DOI] [PubMed] [Google Scholar]
- Apostolova N, Cervera AM, Victor VM, Cadenas S, Sanjuan-Pla A, Alvarez-Barrientos A, Esplugues JV, McCreath KJ. Loss of apoptosis-inducing factor leads to an increase in reactive oxygen species, and an impairment of respiration that can be reversed by antioxidants. Cell Death Differ. 2006;13:354–357. doi: 10.1038/sj.cdd.4401776. [DOI] [PubMed] [Google Scholar]
- Armstrong CL, Hawkes R. Pattern formation in the cerebellar cortex. Biochem Cell Biol. 2000;78:551–562. [PubMed] [Google Scholar]
- Armstrong CL, Krueger-Naug AM, Currie RW, Hawkes R. Constitutive expression of the 25-kDa heat shock protein Hsp25 reveals novel parasagittal bands of purkinje cells in the adult mouse cerebellar cortex. J Comp Neurol. 2000;416:383–397. doi: 10.1002/(sici)1096-9861(20000117)416:3<383::aid-cne9>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Baimbridge KG, Miller JJ. Immunohistochemical localization of calcium-binding protein in the cerebellum, hippocampal formation and olfactory bulb of the rat. Brain Res. 1982;245:223–229. doi: 10.1016/0006-8993(82)90804-6. [DOI] [PubMed] [Google Scholar]
- Bastianelli E. Distribution of calcium-binding proteins in the cerebellum. Cerebellum. 2003;2:242–262. doi: 10.1080/14734220310022289. [DOI] [PubMed] [Google Scholar]
- Benit P, Goncalves S, Dassa EP, Briere JJ, Rustin P. The variability of the harlequin mouse phenotype resembles that of human mitochondrial-complex I-deficiency syndromes. PLoS One. 2008;3:e3208. doi: 10.1371/journal.pone.0003208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berchtold MW, Celio MR, Heizmann CW. Parvalbumin in non-muscle tissues of the rat. Quantitation and immunohistochemical localization. J Biol Chem. 1984;259:5189–5196. [PubMed] [Google Scholar]
- Brochu G, Maler L, Hawkes R. Zebrin II: a polypeptide antigen expressed selectively by Purkinje cells reveals compartments in rat and fish cerebellum. J Comp Neurol. 1990;291:538–552. doi: 10.1002/cne.902910405. [DOI] [PubMed] [Google Scholar]
- Cande C, Cohen I, Daugas E, Ravagnan L, Larochette N, Zamzami N, Kroemer G. Apoptosis-inducing factor (AIF): a novel caspase-independent death effector released from mitochondria. Biochimie. 2002;84:215–222. doi: 10.1016/s0300-9084(02)01374-3. [DOI] [PubMed] [Google Scholar]
- Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Chen H, Chan DC. Mitochondrial dynamics--fusion, fission, movement, and mitophagy--in neurodegenerative diseases. Hum Mol Genet. 2009;18:R169–176. doi: 10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S, Zhang Y, Van Der Hoorn F, Hawkes R. The anatomy of the cerebellar nuclei in the normal and scrambler mouse as revealed by the expression of the microtubule-associated protein kinesin light chain 3. Brain Res. 2007;1140:120–131. doi: 10.1016/j.brainres.2006.01.100. [DOI] [PubMed] [Google Scholar]
- Chung SH, Marzban H, Watanabe M, Hawkes R. Phospholipase Cbeta4 expression identifies a novel subset of unipolar brush cells in the adult mouse cerebellum. Cerebellum. 2009a;8:267–276. doi: 10.1007/s12311-009-0092-x. [DOI] [PubMed] [Google Scholar]
- Chung SH, Sillitoe RV, Croci L, Badaloni A, Consalez G, Hawkes R. Purkinje cell phenotype restricts the distribution of unipolar brush cells. Neuroscience. 2009b;164:1496–1508. doi: 10.1016/j.neuroscience.2009.09.080. [DOI] [PubMed] [Google Scholar]
- Detmer SA, Chan DC. Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol. 2007;8:870–879. doi: 10.1038/nrm2275. [DOI] [PubMed] [Google Scholar]
- Englund C, Kowalczyk T, Daza RA, Dagan A, Lau C, Rose MF, Hevner RF. Unipolar brush cells of the cerebellum are produced in the rhombic lip and migrate through developing white matter. J Neurosci. 2006;26:9184–9195. doi: 10.1523/JNEUROSCI.1610-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobedo J, Pucci AM, Koh TJ. HSP25 protects skeletal muscle cells against oxidative stress. Free Radic Biol Med. 2004;37:1455–1462. doi: 10.1016/j.freeradbiomed.2004.07.024. [DOI] [PubMed] [Google Scholar]
- Hawkes R. An anatomical model of cerebellar modules. Prog Brain Res. 1997;114:39–52. doi: 10.1016/s0079-6123(08)63357-9. [DOI] [PubMed] [Google Scholar]
- Huot J, Houle F, Spitz DR, Landry J. HSP27 phosphorylation-mediated resistance against actin fragmentation and cell death induced by oxidative stress. Cancer Res. 1996;56:273–279. [PubMed] [Google Scholar]
- Karbowski M, Norris KL, Cleland MM, Jeong SY, Youle RJ. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 2006;443:658–662. doi: 10.1038/nature05111. [DOI] [PubMed] [Google Scholar]
- Klein JA, Longo-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Frankel WN, Bronson RT, Ackerman SL. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419:367–374. doi: 10.1038/nature01034. [DOI] [PubMed] [Google Scholar]
- Kwong JQ, Beal MF, Manfredi G. The role of mitochondria in inherited neurodegenerative diseases. J Neurochem. 2006;97:1659–1675. doi: 10.1111/j.1471-4159.2006.03990.x. [DOI] [PubMed] [Google Scholar]
- Landis SC, Mullen RJ. The development and degeneration of Purkinje cells in pcd mutant mice. J Comp Neurol. 1978;177:125–143. doi: 10.1002/cne.901770109. [DOI] [PubMed] [Google Scholar]
- Lewis SE, Mannion RJ, White FA, Coggeshall RE, Beggs S, Costigan M, Martin JL, Dillmann WH, Woolf CJ. A role for HSP27 in sensory neuron survival. J Neurosci. 1999;19:8945–8953. doi: 10.1523/JNEUROSCI.19-20-08945.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Maragakis NJ, Rothstein JD. Mechanisms of Disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol. 2006;2:679–689. doi: 10.1038/ncpneuro0355. [DOI] [PubMed] [Google Scholar]
- McBride HM, Neuspiel M, Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol. 2006;16:R551–560. doi: 10.1016/j.cub.2006.06.054. [DOI] [PubMed] [Google Scholar]
- Mehlen P, Kretz-Remy C, Briolay J, Fostan P, Mirault ME, Arrigo AP. Intracellular reactive oxygen species as apparent modulators of heat-shock protein 27 (hsp27) structural organization and phosphorylation in basal and tumour necrosis factor alpha-treated T47D human carcinoma cells. Biochem J. 1995;312:367–375. doi: 10.1042/bj3120367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miramar MD, Costantini P, Ravagnan L, Saraiva LM, Haouzi D, Brothers G, Penninger JM, Peleato ML, Kroemer G, Susin SA. NADH oxidase activity of mitochondrial apoptosis-inducing factor. J Biol Chem. 2001;276:16391–16398. doi: 10.1074/jbc.M010498200. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Sato K, Fukaya M, Araishi K, Aiba A, Kano M, Watanabe M. Signaling complex formation of phospholipase Cbeta4 with metabotropic glutamate receptor type 1alpha and 1,4,5-trisphosphate receptor at the perisynapse and endoplasmic reticulum in the mouse brain. Eur J Neurosci. 2004;20:2929–2944. doi: 10.1111/j.1460-9568.2004.03768.x. [DOI] [PubMed] [Google Scholar]
- Ozol K, Hayden JM, Oberdick J, Hawkes R. Transverse zones in the vermis of the mouse cerebellum. J Comp Neurol. 1999;412:95–111. [PubMed] [Google Scholar]
- Reddy PH. Mitochondrial medicine for aging and neurodegenerative diseases. Neuromolecular Med. 2008;10:291–315. doi: 10.1007/s12017-008-8044-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarna J, Miranda SR, Schuchman EH, Hawkes R. Patterned cerebellar Purkinje cell death in a transgenic mouse model of Niemann Pick type A/B disease. Eur J Neurosci. 2001;13:1873–1880. doi: 10.1046/j.0953-816x.2001.01564.x. [DOI] [PubMed] [Google Scholar]
- Sarna JR, Hawkes R. Patterned Purkinje cell death in the cerebellum. Prog Neurobiol. 2003;70:473–507. doi: 10.1016/s0301-0082(03)00114-x. [DOI] [PubMed] [Google Scholar]
- Sarna JR, Larouche M, Marzban H, Sillitoe RV, Rancourt DE, Hawkes R. Patterned Purkinje cell degeneration in mouse models of Niemann-Pick type C disease. J Comp Neurol. 2003;456:279–291. doi: 10.1002/cne.10522. [DOI] [PubMed] [Google Scholar]
- Sarna JR, Marzban H, Watanabe M, Hawkes R. Complementary stripes of phospholipase Cbeta3 and Cbeta4 expression by Purkinje cell subsets in the mouse cerebellum. J Comp Neurol. 2006;496:303–313. doi: 10.1002/cne.20912. [DOI] [PubMed] [Google Scholar]
- Schwaller B, Buchwald P, Blumcke I, Celio MR, Hunziker W. Characterization of a polyclonal antiserum against the purified human recombinant calcium binding protein calretinin. Cell Calcium. 1993;14:639–648. doi: 10.1016/0143-4160(93)90089-o. [DOI] [PubMed] [Google Scholar]
- Sekerkova G, Ilijic E, Mugnaini E. Time of origin of unipolar brush cells in the rat cerebellum as observed by prenatal bromodeoxyuridine labeling. Neuroscience. 2004;127:845–858. doi: 10.1016/j.neuroscience.2004.05.050. [DOI] [PubMed] [Google Scholar]
- Sillitoe RV, Benson MA, Blake DJ, Hawkes R. Abnormal dysbindin expression in cerebellar mossy fiber synapses in the mdx mouse model of Duchenne muscular dystrophy. J Neurosci. 2003;23:6576–6585. doi: 10.1523/JNEUROSCI.23-16-06576.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillitoe RV, Hawkes R. Whole-mount immunohistochemistry: a high-throughput screen for patterning defects in the mouse cerebellum. J Histochem Cytochem. 2002;50:235–244. doi: 10.1177/002215540205000211. [DOI] [PubMed] [Google Scholar]
- Singec I, Knoth R, Ditter M, Frotscher M, Volk B. Neurogranin expression by cerebellar neurons in rodents and non-human primates. J Comp Neurol. 2003;459:278–289. doi: 10.1002/cne.10600. [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Brenner C, Larochette N, Prevost MC, Alzari PM, Kroemer G. Mitochondrial release of caspase-2 and -9 during the apoptotic process. J Exp Med. 1999a;189:381–394. doi: 10.1084/jem.189.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999b;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Tomomura M, Rice DS, Morgan JI, Yuzaki M. Purification of Purkinje cells by fluorescence-activated cell sorting from transgenic mice that express green fluorescent protein. Eur J Neurosci. 2001;14:57–63. doi: 10.1046/j.0953-816x.2001.01624.x. [DOI] [PubMed] [Google Scholar]
- Urbano A, Lakshmanan U, Choo PH, Kwan JC, Ng PY, Guo K, Dhakshinamoorthy S, Porter A. AIF suppresses chemical stress-induced apoptosis and maintains the transformed state of tumor cells. EMBO J. 2005;24:2815–2826. doi: 10.1038/sj.emboj.7600746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahsen N, Cande C, Briere JJ, Benit P, Joza N, Larochette N, Mastroberardino PG, Pequignot MO, Casares N, Lazar V, Feraud O, Debili N, Wissing S, Engelhardt S, Madeo F, Piacentini M, Penninger JM, Schagger H, Rustin P, Kroemer G. AIF deficiency compromises oxidative phosphorylation. EMBO J. 2004;23:4679–4689. doi: 10.1038/sj.emboj.7600461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Empel VP, Bertrand AT, van der Nagel R, Kostin S, Doevendans PA, Crijns HJ, de Wit E, Sluiter W, Ackerman SL, De Windt LJ. Downregulation of apoptosis-inducing factor in harlequin mutant mice sensitizes the myocardium to oxidative stress-related cell death and pressure overload-induced decompensation. Circ Res. 2005;96:e92–e101. doi: 10.1161/01.RES.0000172081.30327.28. [DOI] [PubMed] [Google Scholar]
- van Empel VP, Bertrand AT, van Oort RJ, van der Nagel R, Engelen M, van Rijen HV, Doevendans PA, Crijns HJ, Ackerman SL, Sluiter W, De Windt LJ. EUK-8, a superoxide dismutase and catalase mimetic, reduces cardiac oxidative stress and ameliorates pressure overload-induced heart failure in the harlequin mouse mutant. J Am Coll Cardiol. 2006;48:824–832. doi: 10.1016/j.jacc.2006.02.075. [DOI] [PubMed] [Google Scholar]
- Wagstaff MJ, Collaco-Moraes Y, Smith J, de Belleroche JS, Coffin RS, Latchman DS. Protection of neuronal cells from apoptosis by Hsp27 delivered with a herpes simplex virus-based vector. J Biol Chem. 1999;274:5061–5069. doi: 10.1074/jbc.274.8.5061. [DOI] [PubMed] [Google Scholar]
- Walther EU, Dichgans M, Maricich SM, Romito RR, Yang F, Dziennis S, Zackson S, Hawkes R, Herrup K. Genomic sequences of aldolase C (Zebrin II) direct lacZ expression exclusively in non-neuronal cells of transgenic mice. Proc Natl Acad Sci U S A. 1998;95:2615–2620. doi: 10.1073/pnas.95.5.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassef M, Sotelo C, Cholley B, Brehier A, Thomasset M. Cerebellar mutations affecting the postnatal survival of Purkinje cells in the mouse disclose a longitudinal pattern of differentially sensitive cells. Dev Biol. 1987;124:379–389. doi: 10.1016/0012-1606(87)90490-8. [DOI] [PubMed] [Google Scholar]
- Yang Q, Hashizume Y, Yoshida M, Wang Y, Goto Y, Mitsuma N, Ishikawa K, Mizusawa H. Morphological Purkinje cell changes in spinocerebellar ataxia type 6. Acta Neuropathol. 2000;100:371–376. doi: 10.1007/s004010000201. [DOI] [PubMed] [Google Scholar]
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]