

Abstract
The grail of protein science is the connection between structure and function. For myoglobin (Mb) this goal is close. Described as only a passive dioxygen storage protein in texts, we argue here that Mb is actually an allosteric enzyme that can catalyze reactions among small molecules. Studies of the structural, spectroscopic, and kinetic properties of Mb lead to a model that relates structure, energy landscape, dynamics, and function. Mb functions as a miniature chemical reactor, concentrating and orienting diatomic molecules such as NO, CO, O2, and H2O2 in highly conserved internal cavities. Reactions can be controlled because Mb exists in distinct taxonomic substates with different catalytic properties and connectivities of internal cavities.
Proteins perform most of the
work in living systems. An understanding of their function with
predictive power will advance fields from biology to medicine and
bioengineering. The road to such an understanding requires detailed
studies of one or a few selected proteins. Myoglobin (Mb) is a good
candidate for this endeavor. Although the textbook role of Mb is only
the storage of dioxygen, it has long been known to react with a wide
variety of small molecules (1). The facts that NO and CO are intimately
involved in the regulation of cellular function (2–5), that
H2O2, O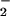 ,
and ONOO− are central to oxidative damage (6)
and perhaps regulation of tissue (7), and that Mb exists in muscles in
high concentration (8) suggest that the physiological role of Mb may be
multifaceted, involving more than simple storage and transport of
dioxygen. Indeed, other roles have been proposed. Mb may facilitate
diffusion of O2 (8), mediate oxidative
phosphorylation (8), protect against oxidative damage (9, 10), and
inactivate enzymes (11). Mb also may increase the effectiveness of NO
as a signaling molecule by enhancing the NO concentration gradients
(12, 13). Particularly exciting is the recent discovery of a highly
conserved neuroglobin in the brain, with an amino acid sequence similar
to Mb (14). The low concentration of neuroglobin suggests a catalytic
function.
,
and ONOO− are central to oxidative damage (6)
and perhaps regulation of tissue (7), and that Mb exists in muscles in
high concentration (8) suggest that the physiological role of Mb may be
multifaceted, involving more than simple storage and transport of
dioxygen. Indeed, other roles have been proposed. Mb may facilitate
diffusion of O2 (8), mediate oxidative
phosphorylation (8), protect against oxidative damage (9, 10), and
inactivate enzymes (11). Mb also may increase the effectiveness of NO
as a signaling molecule by enhancing the NO concentration gradients
(12, 13). Particularly exciting is the recent discovery of a highly
conserved neuroglobin in the brain, with an amino acid sequence similar
to Mb (14). The low concentration of neuroglobin suggests a catalytic
function.
The structure of Mb is shown in Fig. 1a. The backbone (blue) forms eight α-helixes, arranged in the Mb fold, wrapped around a heme group (green), along with His-93 (below) and His-64 (above). The redox-active amino acids also are displayed, methionine in yellow, tyrosine in red, and tryptophan in purple. Not highlighted, but important in determining the reactivity, are water molecules and the protonatable groups, which appear throughout the protein. Four cavities within the protein, determined by xenon binding studies using x-ray diffraction (15), are shown as black spheres. These cavities are not defects, they are involved in the binding process,
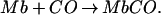 |
1 |
They may, however, also be essential for other reactions, as illustrated in Fig. 1b, where a NO molecule trapped in the xenon cavities of Mb is depicted as interacting with a dioxygen molecule bound at the heme iron atom (16–18),
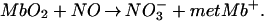 |
2 |
Fig. 1b suggests that Mb is built like a miniature chemical reactor, with the iron as active center, the heme and the Xe1 cavities as chambers, and the heme group as a controlling diaphragm. Moreover, the passage between Xe4 and the heme cavity may be constructed so that the NO orientation is optimal for a reaction. The information describing this reactor comes from studies of ligand binding.
Figure 1.
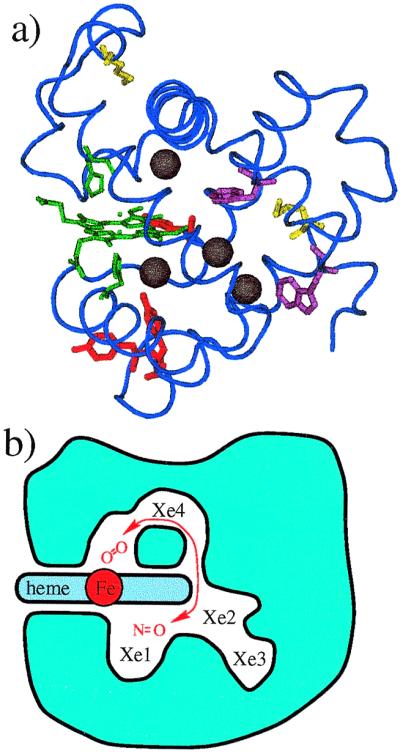
(a) The sperm whale Mb skeleton, with side chains Tyr-103, -146, and -151, Trp-7 and -14, Met-55 and -131, His-64 and -93, and Xe1, Xe2, Xe3, and Xe4 from ref. 15. (b) Cartoon of Mb as NO is reacting with a bound O2. The heme cavity is on the distal (Upper), the Xe1 cavity on the proximal (Lower) side of the heme. A dioxygen molecule is shown bound covalently to the iron atom in the heme cavity. A NO molecule can move from the Xe1 cavity via the path shown to react with the O2.
Ligand Binding, Structure, and Energy Landscape
Studies of the binding of CO and O2 have dominated Mb research. If Mb is an allosteric enzyme, then these studies did not target the important goal. They led, however, to the formulation of relevant concepts. The investigations were performed over wide ranges of temperature (4 to 330 K) and time (fs to ks) and with a wide variety of tools.
Kinetic experiments show that the path of a CO molecule coming from the solvent (S) and moving to the binding site at the heme iron (A) can be approximated with a sequential reaction model (19–21),
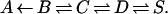 |
3 |
The kinetic data prove that at physiological temperatures the states B, C, D, and S are in pre-equilibrium. Before binding at the heme iron, the CO returns to the solvent about 40 times. The formation of the covalent bond at the iron, B→A, consequently is rate limiting. Quantum computation has led to an understanding of this step (22). The kinetic data also show that CO in D is enthalpically bound by about 10 kJ/mol. Thus, CO is concentrated by about a factor 50 in D as compared to the same volume in the solvent.
For many years, the location of the states B, C, and D in Eq. 3 could only be guessed. Recent advances in cryo-crystallography (23–26) provide this information. The results are used in Fig. 1b and are given in a more realistic form in Fig. 2. In B, the CO remains in the heme cavity, approximately parallel to the heme plane, about 4 Å from the iron. In Fig. 1b, B is located where the arrow from Xe4 points to the dioxygen. In C, the CO most likely occupies Xe4. In D the CO sits in the largest cavity, Xe1, as has also been inferred from kinetic experiments under a few bar of Xe (27). The pathway for CO, shown in Fig. 1 for NO, also is supported by binding studies under intense light both with the wild-type Mb (28) and the proximal mutant L89I (29). The cavities are not defects, but are of functional importance as can be deduced from the facts that they are conserved in Mb over a wide range of species (30) and that Xe1, Xe2, and the heme cavity contain potentially reactive residues, as listed in Table 1.
Figure 2.

X-ray-determined structure of left Mb at pH 5 (Protein DataBank file 1spe, from ref. 38), and right Mb at pH 7 (Protein Data Bank file 1a6g, from Ref. 23). His-64 (Upper Left) has moved outside the heme pocket in A0, and smaller changes are visible throughout the protein. The B-site is labeled B, and the xenon cavities 1, 2, 3, and 4 are labeled with numbers.
Table 1.
Amino acids lining the cavities in sperm whale Mb
| Heme cavity | Xe1 | Xe2 | Xe3 | Xe4 |
|---|---|---|---|---|
| Leu-29 | Leu-89 | Leu-72 | Trp-7 | Gly-25 |
| Phe-46 | His-93 | Leu-104 | Ile-75 | Ile-28 |
| His-64 | Leu-104 | Ile-107 | Leu-76 | Leu-29 |
| Val-68 | Phe-138 | Ser-108 | Gly-80 | Gly-65 |
| Ile-107 | Ile-142 | Leu-135 | His-82 | Val-68 |
| Heme | Tyr-146 | Phe-138 | Ala-134 | Leu-69 |
| Heme | Arg-139 | Leu-137 | Leu-72 | |
| Heme | Phe-138 | Ile-107 | ||
| Ile-111 |
Amino acids adjacent to cavities. Residues in boldface are conserved among most mammals as well as the human hemoglobin chains. Only two residues, Phe-43 and His-93, are conserved in all Mbs (30), while all listed residues except Ile-142 and Ile-28 are conserved among a list of 15 mammals that are identical over only 59% of the sequence. The tertiary structure is conserved.
Armed with these results we now justify the scenario introduced earlier for the rapid oxidation of NO by MbO2 in Fig. 1b. NO, like CO, will be weakly bound in the xenon pockets, mostly Xe1, and can visit the B site dozens of times before exiting the protein. On each visit to B, NO has a chance to react. The entrance to B is determined by the three amino acids that form the bottleneck between C and B. The bottleneck may be designed to orient and polarize the NO and the O2 for optimal reaction. These observations are equally valid if reaction (2) occurs through intermediate NOx compounds.
The binding of CO tells us even more about protein structure and dynamics. Binding of CO at low temperatures is nonexponential in time and implies that Mb cannot be in a unique structure (19). Many additional experiments indeed prove that proteins exist in an ensemble of structures, described by an energy landscape (31–33). A given primary sequence can fold into a very large number of somewhat different structures, each called a conformational substate. The complete characterization of a substate requires about 3,000 coordinates to describe the positions of all atoms of the protein and the hydration shell. The energy landscape is organized hierarchically, with valleys within valleys within valleys (34). The highest two tiers are sketched in Fig. 3a as a tree diagram and in Fig. 3b as a one-dimensional cross section through the energy landscape. The highest tier is evident from the vibrational spectrum of CO bound to the iron, shown in Fig. 3c. It displays three distinct peaks at stretch frequencies of 1,967 cm −1, 1,947 cm−1, and 1,929 cm−1, denoted as A0, A1, and A3. They are called taxonomic substates because they can be studied and described individually. Within each taxonomic substate, Mb assumes a very large number of slightly different conformations or statistical substates, broadening each of the peaks and leading to the nonexponential time dependence within each taxonomic substate (35). Temperature-derivative spectroscopy shows that the barriers between states in Eq. 3 depend on the taxonomic substate (28, 29).
Figure 3.

(a) Tree diagram of the taxonomic substate of Mb and its numerous statistical substates. (b) Cross section through the hierarchical energy landscape. (c) Infrared spectrum of CO bound to the iron atom of Mb, showing the stretch frequencies of the three A substates. Allostery is a consequence of the existence of these taxonomic substates.
Taxonomic and statistical substates are both important for protein dynamics. Protein motions are transitions between substates; these are relevant for protein relaxation and for motion of the CO into and out of the Mb (20, 35, 36). Both types of substates must be considered when searching for the transition state of an enzymatic reaction; the average value of, say, the redox potential may not reliably predict yields of redox reactions. It may be necessary to perform experiments in nonphysiological conditions to obtain significant populations of the relevant taxonomic substates.
Allostery and Taxonomic Substates
An allosteric protein can exist in two or more different conformations, with different reactive properties. The relative population of the different conformations is determined by activators (37). Multimeric proteins such as hemoglobin are considered to be the prototypes of allosteric systems, whereas Mb usually is assumed to be nonallosteric. The observation that the association rate coefficient for CO binding at physiological temperatures is about four times larger at pH 5 than at pH 9 (21) casts doubts on this assumption. The pH dependence occurs because the barrier between the states B and A, which is rate limiting at physiological temperatures, is different in the three taxonomic substates; it is smallest in A0 and largest in A3 (35). The differences are due to different structures, mainly the position of the distal histidine, His-64 (see Fig. 2). In A0, the charged histidine is not in the heme pocket, but extends into the solvent (38). The association rate coefficient is proportional to the rate coefficient for the final step, B→A (19, 21), which speeds up in the A0 state (35) because His-64 no longer blocks access to the CO binding site at the iron (22). If the shift in the populations of the taxonomic substates were caused only by pH, the different binding rates for CO could be ascribed to a “direct interaction” caused by the change in charge on the distal histidine. Transitions among the substates can, however, be induced not only by changes in pH (39), but also in temperature (40), pressure (41), or by addition of a variety of salts such as iodide, citrate, or thiocyanate (42). Furthermore, the pathways leading from D to A differ for the three substates, strengthening the argument that the taxonomic states involve correlated changes of the entire protein (28). We expect that the energy landscape of the protein remains largely unchanged when considering other reactions. The same cavities, taxonomic substates, and effects on the populations of taxonomic substates will be present. Different catalysis rates of the different taxonomic substates then produce allostery, as predicted by ref. 31. We consequently consider Mb to be an allosteric protein and remark that allostery thus does not require multimeric proteins.
Mb as an Allosteric Enzyme
Enzymes catalyze specific reactions. In allosteric enzymes, these reactions are controlled by activators. Two concepts that have emerged from the study of CO binding are important for the role of Mb as an allosteric enzyme, the conserved and connected cavities and the taxonomic substates. The cavities can speed up a bimolecular reaction by increasing the concentration of both reactants, one by covalently binding to the iron, the other by concentrating it in the cavities. The reaction rate then can be controlled and further increased by the shaping of the connections between the cavities and by orienting and polarizing the reactants. Moreover, competing reactions can be reduced or eliminated. In chemistry, confinement of reactive species currently is being explored as a way of achieving precise control of reactions (43). As shown here, nature discovered this trick long ago and shaped Mb like a chemical reactor for single molecules, as sketched in Fig. 1b. The second property, the existence of taxonomic substates with different reactive properties, permits the protein to control reactions by changing the population of these substates in response to activators.
Mb is able to catalyze a variety of redox reactions. To ascertain if Mb, through its taxonomic substates, can control such reactions, we have measured the time course and the pH dependence of the reaction of nitrite and MbO2:
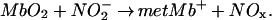 |
4 |
This reaction is a one-electron oxidation of Mb by the nitrite ion, resulting in another oxide of nitrogen and consuming two protons.
We have performed a preliminary study of the kinetics of reaction 4 as a function of pH by measuring the optical absorption spectrum after mixing MbO2 with NaNO2. Because MbO2 is unstable at low pH, a pH jump from pH 8 was done during the mixing with nitrite. Conversion of MbO2 to metMb+ was observed, and the kinetics of this process are shown in Fig. 4 as the absorbance at 421 nm. The data show a lag phase and three phases of reaction kinetics, all with a strong pH dependence, suggesting that several different intermediates are involved. The overall rate, λtotal, can be expressed as:
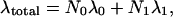 |
5 |
where λ0 and λ1 are the rate coefficients for reaction 4 in the substates A0 and A1, respectively, and where N0 and N1 are the corresponding normalized populations. The data can be fit approximately with λ0 = 10 s−1 and λ1 = 0, and with a population ratio given by the Henderson–Hasselbalch relation with pK = 4.2. This pK characterizes the A0⇌A1 transition (39). It is not clear whether the rate is faster in the A0 state because His-64 delivers a proton to the active site or because the more open pocket structure of the A0 conformation (see Fig. 2) facilitates access by water molecules that are able to donate the two protons necessary for reaction 4. In either case, effects that shift the equilibrium between A0 and A1 (temperature, salts, pressure) will influence the reaction rate.
Figure 4.

Kinetics of MbO2 oxidation by nitrite. (a)
Kinetics of oxidation of 20 μM MbO2, observed when
MbO2 is mixed with 0.1% NO (wt/vol)
in a 50 mM sodium acetate/HCl buffer ≈5 min after exposure of
deoxyMb to air to produce MbO2. Complete UV-Vis optical
absorbance spectra were obtained on a dual-beam OLIS (Jefferson, GA)
stopped-flow apparatus. Filling of the chamber required 20 ms.
(b) pH dependence of these kinetics.
(wt/vol)
in a 50 mM sodium acetate/HCl buffer ≈5 min after exposure of
deoxyMb to air to produce MbO2. Complete UV-Vis optical
absorbance spectra were obtained on a dual-beam OLIS (Jefferson, GA)
stopped-flow apparatus. Filling of the chamber required 20 ms.
(b) pH dependence of these kinetics.
The relative slowness of reaction 4 compared to reaction 2 is because delivery of protons, and possibly electrons, is required; when measured in vivo, rather than in dilute solution, the kinetics are likely to be quite different from those presented here. It has been shown for peroxidase and nitrogen chemistry of Mb that physiological concentrations of reductants such as NADH, ascorbate, and glutathione, as well as NO, will influence the reaction rates and product yields (9, 10, 16, 44). The observation that lactate, at the tens of mM level, affects both the optical absorption spectrum and the oxygen affinity of Mb (45) suggests that an allosteric mechanism is at work, as lactate can change its concentration from 2 to 20 mM between rested and exhausted muscle (46). Although normally requiring a dehydrogenase enzyme to be redox-active, we expect the negatively charged lactate molecule binding near the heme to increase the heme reduction potential and perhaps, by directly reducing Fe IV and Fe V, influence reactions involving this species, as observed with ascorbate (9).
Whether the nitrogen chemistry of muscle is affected because Mb regulates the accessibility of diatomic molecules, electrons, and protons in an environment-dependent manner ultimately will require physiology experiments. In mice, it has been shown that removal of Mb results in multiple compensatory mechanisms involving the NO-related chemistry of the cell (47, 48). In bacteria, flavo-hemoglobins likewise have been implicated in the NO-related chemistry (49, 50). Clearly there are numerous possibilities for Mb to influence the course of the redox pathways of the muscle cell.
The detailed chemistry of NO as it interacts with components of the cellular environment is complex and not understood (13, 51, 52). The ability of Mb to concentrate small ligands and exist in several taxonomic substates and oxidation states suggests that it is involved in the NO-related chemistry of skeletal and cardiac muscle. The nonuniform spacial and temporal nature of NO chemistry, and high protein concentrations in the muscle cell greatly complicate comparisons between in vivo and in vitro experiments. Despite the extensive work that already has been done, two major challenges lie ahead. The first challenge is to explain quantitatively, just as for the binding of CO, the many redox reactions of Mb in terms of structure, energy landscape, and dynamics. Because electron and proton motion is necessary for most enzymatic reactions, a fundamental understanding of Mb may aid the understanding of a wide variety of enzyme reactions. The second challenge is to connect the microscopic properties of Mb to the physiological needs of life. Mb, the model protein for the study of ligand binding, may thus become the model protein for understanding enzymatic reactions and elucidating the networks by which NO chemistry affects the biochemistry of life.
Acknowledgments
We thank Cliff Unkefer for use of his stopped-flow apparatus, Brian Dyer for biochemistry facilities, Greg Petsko for the coordinate file used to make Fig. 1a, and Peter Wolynes and Ilme Schlichting for helpful discussions. This work was performed under the auspices of the U.S. Department of Energy through the Center for Nonlinear Studies at the Los Alamos National Laboratory and National Institutes of Health Grant GM 36298 (to J.T.G.).
Abbreviation
- Mb
myoglobin
References
- 1.Antonini E, Brunori M. Hemoglobin and Myoglobin in their Reactions with Ligands. Amsterdam: North–Holland; 1971. [Google Scholar]
- 2.Murad F. Angew Chem Int Ed. 1999;38:1856–1868. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<1856::AID-ANIE1856>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 3.Furchgott R F. Angew Chem Int Ed. 1999;38:1870–1880. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<1870::AID-ANIE1870>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 4.Ignarro L J. Angew Chem Int Ed. 1999;38:1882–1892. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<1882::AID-ANIE1882>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 5.Snyder S H, Jaffrey S R, Zakhary R. Brain Res Rev. 1998;26:167–175. doi: 10.1016/s0165-0173(97)00032-5. [DOI] [PubMed] [Google Scholar]
- 6.Berlett B S, Stadtman E R. J Biol Chem. 1997;272:20313–20316. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki Y J, Ford G D. J Mol Cell Cardiol. 1999;31:345–353. doi: 10.1006/jmcc.1998.0872. [DOI] [PubMed] [Google Scholar]
- 8.Wittenberg J B, Wittenberg B A. Annu Rev Biophys Biophys Chem. 1990;19:217–241. doi: 10.1146/annurev.bb.19.060190.001245. [DOI] [PubMed] [Google Scholar]
- 9.Galaris D, Cadenas E, Hochstein P. Arch Biochem Biophys. 1989;273:497–504. doi: 10.1016/0003-9861(89)90509-2. [DOI] [PubMed] [Google Scholar]
- 10.Nakamura M, Nakamura S. Biophys Biochim Acta. 1996;1289:329–335. doi: 10.1016/0304-4165(95)00161-1. [DOI] [PubMed] [Google Scholar]
- 11.Miura T, Muraoka S, Fujimoto Y. Chem Biol Interact. 1999;123:51–61. doi: 10.1016/s0009-2797(99)00124-6. [DOI] [PubMed] [Google Scholar]
- 12.Lancaster J R., Jr Proc Natl Acad Sci USA. 1994;91:8137–8141. doi: 10.1073/pnas.91.17.8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu X, Miller M J S, Joshi M S, Thomas D D, Lancaster J R., Jr Proc Natl Acad Sci USA. 1998;95:2175–2179. doi: 10.1073/pnas.95.5.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burmester T, Weich B, Reinhardt S, Hankein T. Nature (London) 2000;407:520–523. doi: 10.1038/35035093. [DOI] [PubMed] [Google Scholar]
- 15.Tilton R F, Kuntz I D, Jr, Petsko G A. Biochemistry. 1984;23:2849–2857. doi: 10.1021/bi00308a002. [DOI] [PubMed] [Google Scholar]
- 16.Doyle M P, Hoekstra J W. J Inorg Biochem. 1981;14:351–357. doi: 10.1016/s0162-0134(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 17.Eich R F, Li T S, Lemon D D, Doherty D H, Curry S R, Aitken J F, Mathews A J, Johnson K A, Smith R D, Phillips G N, et al. Biochemistry. 1996;35:6976–6983. doi: 10.1021/bi960442g. [DOI] [PubMed] [Google Scholar]
- 18.Arnold E V, Bohle D S, Jordan P A. Biochemistry. 1999;38:4750–4756. doi: 10.1021/bi982729e. [DOI] [PubMed] [Google Scholar]
- 19.Austin R H, Beeson K W, Eisenstein L, Frauenfelder H, Gunsalus I C. Biochemistry. 1975;14:5355–5373. doi: 10.1021/bi00695a021. [DOI] [PubMed] [Google Scholar]
- 20.Beece D, Eisenstein L, Frauenfelder H, Good D, Marden M C, Reinisch L, Reynolds A H, Sorensen L B, Yue K T. Biochemistry. 1980;19:5147–5157. doi: 10.1021/bi00564a001. [DOI] [PubMed] [Google Scholar]
- 21.Doster W, Beece D, Bowne S F, DiIorio E E, Eisenstein L, Frauenfelder H, Reinisch L, Shyamsunder E, Winterhalter K H, Yue K T. Biochemistry. 1982;21:4831–4839. doi: 10.1021/bi00263a001. [DOI] [PubMed] [Google Scholar]
- 22.McMahon B H, Stojković B P, Hay P J, Martin R L, Garcia A E. J Chem Phys. 2000;113:6831–6850. [Google Scholar]
- 23.Schlichting I, Berendzen J, Phillips G N, Jr, Sweet R M. Nature (London) 1994;371:808–812. doi: 10.1038/371808a0. [DOI] [PubMed] [Google Scholar]
- 24.Hartmann H, Zinser S, Komninos P, Schneider R T, Nienhaus G U, Parak F. Proc Natl Acad Sci USA. 1996;93:7013–7016. doi: 10.1073/pnas.93.14.7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chu K, Vojtchovsky J, McMahon B H, Sweet R M, Berendzen J, Schlichting I. Nature (London) 2000;403:921–923. doi: 10.1038/35002641. [DOI] [PubMed] [Google Scholar]
- 26.Ostermann A, Waschipky R, Parak F G, Nienhaus G U. Nature (London) 2000;404:205–208. doi: 10.1038/35004622. [DOI] [PubMed] [Google Scholar]
- 27.Scott E E, Gibson Q H. Biochemistry. 1997;36:11909–11911. doi: 10.1021/bi970719s. [DOI] [PubMed] [Google Scholar]
- 28.Nienhaus G U, Mourant J R, Chu K, Frauenfelder H. Biochemistry. 1994;33:13413–13430. doi: 10.1021/bi00249a030. [DOI] [PubMed] [Google Scholar]
- 29.Abadan Y, Chien E Y T, Chu K, Eng C D, Nienhaus G U, Sligar S G. Biophys J. 1995;68:2497–2504. doi: 10.1016/S0006-3495(95)80432-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suzuki T, Imai K. Cell Mol Life Sci. 1998;54:979–1004. doi: 10.1007/s000180050227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frauenfelder H, Sligar S G, Wolynes P G. Science. 1991;254:1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 32.Frauenfelder H, Wolynes P G, Austin R H. Rev Mod Phys. 1999;71:S419–S430. [Google Scholar]
- 33.McMahon B H, Müller J D, Wraight C A, Nienhaus G U. Biophys J. 1998;74:2567–2587. doi: 10.1016/S0006-3495(98)77964-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ansari A, Berendzen J, Bowne S F, Frauenfelder H, Iben I E T, Sauke T B, Shyamsunder E, Young R D. Proc Natl Acad Sci USA. 1985;82:5000–5004. doi: 10.1073/pnas.82.15.5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson J B, Lamb D C, Frauenfelder H, Müller J D, McMahon B H, Nienhaus G U, Young R D. Biophys J. 1996;71:1563–1573. doi: 10.1016/S0006-3495(96)79359-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jackson T A, Lim M, Anfinrud P A. Chem Phys. 1994;180:131–140. [Google Scholar]
- 37.Wyman J, Gill S J. Binding and Linkage. Mill Valley, CA.: University Science Books; 1990. [Google Scholar]
- 38.Yang F, Phillips G N., Jr J Mol Biol. 1996;256:762–774. doi: 10.1006/jmbi.1996.0123. [DOI] [PubMed] [Google Scholar]
- 39.Müller J D, McMahon B H, Chien E Y T, Sligar S G, Nienhaus G U. Biophys J. 1999;77:1036–1051. doi: 10.1016/s0006-3495(99)76954-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Young R D, Frauenfelder H, Johnson J B, Lamb D C, Nienhaus G U, Philipp R, Scholl R. Chem Phys. 1991;158:315–327. [Google Scholar]
- 41.Frauenfelder H, Alberding N A, Ansari A, Braunstein D, Cowen B R, Hong M K, Iben I E T, Johnson J B, Luck S, Marden M C, et al. J Phys Chem. 1990;94:1024–1037. [Google Scholar]
- 42.Müller J D, McMahon B H, Nienhaus G U. Am Phys Soc Bull. 1997;42:504. [Google Scholar]
- 43.Rouhi A M. Chem Eng News. 2000;August 21:40–47. [Google Scholar]
- 44.Jourd'heuil D, Mills L, Miles A M, Grisham M B. Nitric Oxide Biol Chem. 1998;2:37–44. doi: 10.1006/niox.1998.0167. [DOI] [PubMed] [Google Scholar]
- 45.Giardina B, Ascenzi P, Clementi M E, DeSanctis G, Rizzi M, Coletta M. J Biol Chem. 1996;271:16999–17001. doi: 10.1074/jbc.271.29.16999. [DOI] [PubMed] [Google Scholar]
- 46.Milligan C L, Girard S S. J Exp Biol. 1993;180:175–193. [Google Scholar]
- 47.Godecke A, Flogel U, Zanger K, Ding Z P, Hirchenhain J, Decking U K M, Schrader J. Proc Natl Acad Sci USA. 1999;96:10495–10500. doi: 10.1073/pnas.96.18.10495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garry D J, Meeson A, Yan Z, Williams R S. Cell Mol Life Sci. 2000;57:896–898. doi: 10.1007/PL00000732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Membrillo-Hernández J, Coopamah M D, Anjum M F, Stevanin T M, Kelly A, Hughes M N, Poole R K. J Biol Chem. 1999;274:748–754. doi: 10.1074/jbc.274.2.748. [DOI] [PubMed] [Google Scholar]
- 50.Gardner P R. J Biol Chem. 2000;275:31581–31587. doi: 10.1074/jbc.M004141200. [DOI] [PubMed] [Google Scholar]
- 51.Groves J T. Curr Opin Chem Biol. 1999;3:226–235. doi: 10.1016/S1367-5931(99)80036-2. [DOI] [PubMed] [Google Scholar]
- 52.Wink D A, Mitchell J B. Free Radical Biol Med. 1998;25:434–456. doi: 10.1016/s0891-5849(98)00092-6. [DOI] [PubMed] [Google Scholar]