

Abstract
Background
Recognition of biological patterns holds promise for improved identification of patients at risk for myocardial infarction (MI) and death. We hypothesized that identifying high- and low-risk patterns from a broad spectrum of hematologic phenotypic data related to leukocyte peroxidase-, erythrocyte- and platelet-related parameters may better predict future cardiovascular risk in stable cardiac patients than traditional risk factors alone.
Methods and Results
Stable patients (n=7369) undergoing elective cardiac evaluation at a tertiary care center were enrolled. A model (PEROX) that predicts incident 1-year death and MI was derived from standard clinical data combined with information captured by a high-throughput peroxidase-based hematology analyzer during performance of a complete blood count with differential. The PEROX model was developed using a random sampling of subjects in a derivation cohort (n=5895) and then independently validated in a nonoverlapping validation cohort (n=1474). Twenty-three high-risk (observed in ≥10% of subjects with events) and 24 low-risk (observed in ≥10% of subjects without events) patterns were identified in the derivation cohort. Erythrocyte- and leukocyte (peroxidase)-derived parameters dominated the variables predicting risk of death, whereas variables in MI risk patterns included traditional cardiac risk factors and elements from all blood cell lineages. Within the validation cohort, the PEROX model demonstrated superior prognostic accuracy (78%) for 1-year risk of death or MI compared with traditional risk factors alone (67%). Furthermore, the PEROX model reclassified 23.5% (P<0.001) of patients to different risk categories for death/MI when added to traditional risk factors.
Conclusion
Comprehensive pattern recognition of high- and low-risk clusters of clinical, biochemical, and hematologic parameters provided incremental prognostic value in stable patients having elective diagnostic cardiac catheterization for 1-year risks of death and MI.
Keywords: atherosclerosis, biological markers, cardiovascular diseases, hematology, myocardial infarction
Despite recent advances in both our understanding of the pathophysiology of cardiovascular disease and our ability to image atherosclerotic plaque, accurate determination of risk in stable cardiac patients remains a challenge. The clinically unidentified high-risk patient who does not undergo aggressive risk factor modification and experiences a major adverse cardiac event is of great concern.1,2 Similarly, more accurate identification of low-risk subjects is needed to refocus finite healthcare resources on those who stand most to benefit. Most current clinical risk assessment tools involve algorithms developed from epidemiology-based studies of untreated primary prevention populations and are limited in their application to a higher-risk and medicated cardiology outpatient setting.3 An area of active investigation is the incorporation of combinations of novel biological markers, genetic polymorphisms, or noninvasive imaging approaches for additive prognostic value.4-7 Despite considerable interest, efforts to incorporate more holistic array-based phenotyping technologies (eg, genomic, proteomic, metabolomic, expression array) for improved cardiac risk stratification remain in their infancy and have yet to be translated into efficient and robust platforms amenable to the high-throughput demands of clinical practice.
Blood is a complex but integrated sensor of physiological homeostasis. Perturbations in blood composition and blood cell function are seen in both acute and chronic inflammatory conditions. Elevated leukocyte count (both neutrophils and monocytes) has long been associated with cardiovascular morbidity and mortality.8,9 Leukocyte adhesion, activation, and degranulation and release of peroxidase-containing granules are key steps in the inflammatory process and have been implicated in the development and progression of cardiovascular atheroma.10 Myeloperoxidase, an abundant leukocyte granule protein enriched within culprit lesions,11 is mechanistically linked with multiple stages of cardiovascular disease,12 including modification of lipoproteins,13-15 creation of proinflammatory lipid mediators,14,16 regulation of protease cascades,17,18 and modulation of nitric oxide bioavailability and vascular tone.19-21 Systemic myeloperoxidase levels are increased in patients presenting with chest pain22 and suspected acute coronary syndromes23 who subsequently experience near-term adverse cardiovascular events, and alterations in leukocyte intracellular peroxidase activity are seen in patients with cardiovascular disease.24,25 Similarly, erythrocytes are critical mediators of both oxygen delivery to tissues and regulation of nitric oxide delivery and bioavailability within the vascular compartment,26 and platelets are essential participants in atherothrombotic disease.27,28 Thus, numerous mechanistic and epidemiological ties exist between various components and activities of circulating leukocytes, erythrocytes, and platelets with processes critical to both vascular homeostasis and progression of cardiovascular disease.24,25,28-33 Here, we hypothesize that data derived from a common, high-throughput, peroxidase-based hematology analyzer (including leukocyte-, erythrocyte- and platelet-related parameters beyond standard complete blood count and differential) can provide a broad spectrum of novel data incremental to existing clinical risk assessment in predicting future cardiovascular risks.
Methods
Study Sample
GeneBank is an institutional review board–approved prospective cohort study at the Cleveland Clinic with enrollment from 2002 to 2006. Patients were eligible for inclusion if they were undergoing elective diagnostic cardiac catheterization, were ≥18 years of age, and were both stable and without active chest pain at time of enrollment. All subjects with a positive cardiac troponin T test (≥0.03 ng/mL) on enrollment blood draw immediately before catheterization were excluded from the study. Indications for catheterization included history of positive or equivocal stress test (46%), to rule out significant coronary artery disease in the presence of cardiac risk factors (63%), catheterization before surgery or intervention (24%), recent but historical myocardial infarction (MI; 7%), prior coronary artery bypass or percutaneous intervention with recurrence of symptoms (37%), history of cardiomyopathy (3%), or remote history of acute coronary syndrome (0.9%). All subjects gave written informed consent approved by the institutional review board.
Collection of Specimens and Clinical Data
Patients were interviewed with the use of a standardized demographics and clinical history questionnaire. Blood samples were taken from the femoral artery at the onset of the catheterization procedure before the administration of heparin, collected into an EDTA tube, and stored either on ice or at 4°C until transfer to laboratory (typically within 2 hours) for immediate hematology analyzer analysis and subsequent processing and storage of plasma at −80°C. Basic metabolic panel, fasting lipid profile, and high-sensitivity C-reactive protein levels were measured on the Abbott Architect platform (Abbott Laboratories, Abbott Park, Ill) in a core laboratory. Samples were identified by barcode only, and all laboratory personnel remained blinded to clinical data. Follow-up telephone interviews were performed by research personnel to track patient outcomes at 1 year, with all events (death and MI) adjudicated and confirmed by source documentation.
Comprehensive Hematology Analyses
Hematology analyses were performed with an Advia 120 hematology analyzer (Siemens, New York, NY). This hematology analyzer functions as a flow cytometer, using in situ peroxidase cytochemical staining to generate a complete blood count and differential based on flow cytometry analysis of whole anticoagulated blood. All hematology measurements used in the present study were generated automatically by the analyzer during routine performance of a complete blood count and differential and did not require any additional sample preparation or processing steps. However, additional steps must be taken to ensure that the data are saved and extracted appropriately (as outlined in the Materials section in the online-only Data Supplement) because not all measurements are routinely reported. All leukocyte-, erythrocyte-, and platelet-related parameters derived from both cytograms and absorbance data were extracted from instrument data (DAT) files by blinded laboratory technicians. All hematology parameters used demonstrated reproducible results (with SD from mean ≤30%) on replicate intraday and interday (>10 times) analyses. Examples of a leukocyte cytogram and a table listing all hematology analyzer elements recovered and used for analysis are included in the Materials section in the online-only Data Supplement.
Statistical Analyses and Construction of the PEROX Score
An initial 7466 subjects were consented for hematology analyses. Of these, 7369 (98.7%) were included in statistical analyses. The 97 subjects not included in statistical analyses were excluded because they were lost to follow-up, they subsequently asked to be withdrawn from the study, or their hematology laboratory data failed to meet quality control parameters (eg, platelet clumping or hemolyzed sample). The initial data set was stratified on the basis of whether a patient experienced an adjudicated event (nonfatal MI or death) by 1 year after enrollment. Randomization using a uniform distribution method was performed to randomly select 80% of patients (derivation cohort) for model building, and the remaining 20% (validation cohort) was set aside for model testing and validation before statistical analyses. Mean and median differences were assessed with the Student t test and Mann-Whitney U test, respectively. For the purpose of ranking, univariate hazard ratios (HRs) were generated for continuous variables or logarithmically transformed continuous variables (if not normally distributed).
To establish an individual subject’s risk, a score was developed (PEROX) by initially identifying binary variable pairs that form reproducible high-risk (observed in ≥10% of subjects with events) and low-risk (observed in ≥10% of subjects without events) patterns for death or MI at 1-year using the logical analysis of data method.34-36 Using this combinatorics and optimization-based mathematical method, we derived a single calculated value for an individual’s overall 1-year risk for death or MI from a weighted integer sum of high- and low-risk patterns present. Briefly, logical analysis of data was first used to identify binary variable pairs that form reproducible positive and negative predictive patterns for risk for death or MI at 1 year. Variables were included on the basis of clinical significance, perceived potential informativeness, reproducibility (for hematology parameters) as monitored in interday and intraday replicates, and nonredundancy as assessed by cluster analysis performed within leukocyte, erythrocyte, and platelet subgroups. More details on logical analysis of data variable selection and analyses are provided in the Materials section in the online-only Data Supplement. Criteria for the development of the PEROX model included 3 equal proportions for each hematology parameter, 2 variables per pattern, and a minimal prevalence of 10% of the events for high-risk and 10% of nonevents for low-risk patterns. Patterns were generated through the use of logical analysis of data software (http://pit.kamick.free.fr/lemaire/software-lad.html)34-36 and tuned for both homogeneity and prevalence to obtain best accuracy on cross-validation experiments. The weights for each positive and negative pattern were determined by the following: 1 divided by the number of high-risk patterns and −1 divided by the number of low-risk patterns, respectively. An overall risk score for a patient was calculated by the sum of positive and negative pattern weights. A maximum score of 1 would be calculated in a patient with only positive patterns; a minimum score of −1 would be present in a patient with only negative patterns. The original score range was adjusted from ±1 to a range of 0 to 100 by assuming 50 (rather than 0) as the midpoint of equal variance. The PEROX score was thus calculated as follows: 50×[(1/23 possible high-risk patterns)×(No. of actual high-risk patterns)−(1/24 possible low-risk patterns)×(No. of low-risk patterns)]+50. The reproducibility of the PEROX score was assessed by examining multiple replicate samples from multiple subjects both within and between days, revealing intraday and interday coefficients of variance of 53±0.4% and 10±2% (mean±SD), respectively. A more detailed explanation of how the PEROX score was built and a complete list of all hematology analyzer variables used within the PEROX score (including an example calculation using patient data) are provided in the Materials section in the online-only Data Supplement.
Validation of PEROX Score and Comparisons
Kaplan-Meier survival curves for PEROX model tertiles were generated within the validation cohort for the 1-year outcomes, including death, nonfatal MI, or either outcome, and compared by log-rank test. Cox proportional-hazards regression was used for time-to-event analysis to calculate HRs and 95% confidence intervals (CIs) for 1-year outcomes of death, MI, or either outcome. Cubic splines (with 95% CIs) were generated to examine the relationship between PEROX model and 1-year outcomes from the derivation cohort, superimposed with absolute 1-year event rates observed in the validation cohort.
Receiver-operating characteristic (ROC) curves were plotted and area under the curve (AUC) was estimated for 1-year outcomes for the validation cohort with the use of risk scores assigned by the PEROX model along with traditional risk factors (including age, gender, smoking, low-density lipoprotein [LDL] cholesterol, high-density lipoprotein [HDL] cholesterol, systolic blood pressure, and history of diabetes mellitus) and compared with risk models incorporating traditional risk factors alone. To obtain an unbiased estimate of AUC, resampling (250 bootstrap samples from the validation cohort) was performed. For each bootstrap sample, AUC values were calculated for traditional risk factors with and without PEROX. AUCs were compared by using a method of comparing correlated ROC curves to calculate P values for each bootstrap sample.37 The Friedman test blocked on replicate was also used to compare AUCs of 250 bootstrap samples.38 In addition, the net reclassification improvement (NRI) was determined by assessing net improvement in risk classification (higher predicted risk in subjects with events at 1 year, lower predicted risk in subjects without events at 1 year) using a ratio of 6:3:1 for low-, medium-, and high-risk categories.39 Consistency of risk stratification was also evaluated by applying ROC analyses to models composed of traditional risk factors alone or in combination with the PEROX risk score within the entire cohort, as well as within primary prevention and secondary prevention subgroups. Statistical analyses were performed with SAS 8.2 (SAS Institute, Inc, Cary, NC) and R 2.8.0 (Vienna, Austria). Values of P<0.05 were considered statistically significant.
Results
Clinical and laboratory parameters used in the development of the PEROX model are shown in Table 1 and were similar between the derivation and validation cohorts. One-year event rates for incident nonfatal MI or death, individually and as a composite, did not significantly differ between the derivation and validation cohorts (P=0.37 for MI; P=0.50 for death; P=1.00 for MI or death). Many traditional cardiac risk factors such as elevations in total cholesterol, LDL cholesterol, and triglycerides predicted 1-year death or MI as expected. Reduced diastolic blood pressure and body mass index were associated with a decrease in risk, likely reflecting confounding by indication bias whereby patients with a higher prevalence of comorbidities are more likely to be taking medication or undergoing aggressive interventions. Multiple statistically significant HRs were observed between various leukocyte, erythrocyte, and platelet parameters and incident 1-year risks for nonfatal MI and death in univariate analyses, consistent with multiple prior individual reported associations with various hematologic parameters30-33 (see the Materials section in the online-only Data Supplement).
Table 1.
Clinical and Laboratory Parameters
| Derivation Cohort (n=5895) | Validation Cohort (n=1474) | Death at 1 y, HR (95% CI) | MI at 1 y, HR (95% CI) | |
|---|---|---|---|---|
| Traditional risk factors | ||||
| Age, y* | 64.1±11.3 | 64.1±10.9 | 1.88 (1.65–2.14)† | 1.14 (0.99–1.32) |
| Male, n (%)* | 4021 (68) | 1024 (69) | 0.93 (0.73–1.18) | 1.21 (0.88–1.66) |
| History of hypertension, n (%)* | 4335 (74) | 1075 (73) | 1.67 (1.24–2.25)† | 1.53 (1.07–2.19)† |
| Current smoking, n (%)* | 770 (13) | 162 (11)† | 0.90 (0.63–1.29) | 1.28 (0.87–1.89) |
| History of smoking, n (%) | 3869 (66) | 995 (68) | 1.35 (1.04–1.74)† | 0.90 (0.67–1.20) |
| Diabetes mellitus, n (%)* | 2054 (35) | 544 (37) | 2.09 (1.66–2.62)† | 1.55 (1.17–2.06)† |
| History of CVD, n (%) | 4056 (71) | 1017 (71) | 2.95 (1.85, 4.70)† | 2.41 (1.39–4.19)† |
| Laboratory measurements | ||||
| Fasting blood glucose, mg/dL* | 111±47 | 112±43 | 1.23 (1.13–1.33)† | 1.27 (1.16–1.39)† |
| Creatinine, mg/dL* | 1.1 (0.8–1.1) | 1.1 (0.8–1.1) | 1.57 (1.48–1.67)† | 1.22 (1.09–1.37)† |
| Potassium, mmol/L* | 4.2 (4.0–4.5) | 4.2 (4.0–4.5) | 1.10 (1.04–1.17)† | 0.97 (0.84–1.12) |
| C-reactive protein, mg/dL* | 3.0 (1.7–5.9) | 3.0 (1.6–5.5) | 1.92 (1.71–2.16)† | 1.21 (1.05–1.40)† |
| Total cholesterol, mg/dL | 176±43 | 178±43 | 0.71 (0.62–0.81)† | 0.93 (0.80–1.07) |
| LDL cholesterol, mg/dL | 100±36 | 101±36 | 0.78 (0.69–0.89)† | 0.97 (0.84–1.13) |
| HDL cholesterol, mg/dL* | 46±14 | 46±14 | 0.84 (0.74–0.95)† | 0.71 (0.60–0.84)† |
| Triglycerides, mg/dL* | 160±119 | 163±120 | 0.82 (0.71–0.96)† | 1.07 (0.96–1.19) |
| Clinical characteristics | ||||
| Systolic blood pressure, mm Hg* | 135±21 | 136±22† | 0.96 (0.85–1.07) | 1.17 (1.02–1.34)† |
| Diastolic blood pressure, mm Hg | 75±12 | 75±13 | 0.81 (0.73–0.90)† | 0.97 (0.85–1.12) |
| Body mass index, kg/m2* | 30±6 | 30±6 | 0.78 (0.68–0.89)† | 0.90 (0.78–1.05) |
| Aspirin use, n (%) | 4270 (72) | 1087 (73) | 0.64 (0.51–0.81)† | 0.93 (0.68–1.27) |
| Statin use, n (%) | 3450 (59) | 869 (59) | 0.82 (0.65–1.03) | 0.70 (0.53–0.92)† |
| Events | ||||
| Death at 1 y, n (%) | 242 (4) | 54 (4) | ||
| MI at 1 y, n (%) | 148 (3) | 44 (3) |
CVD indicates cardiovascular disease. Data are shown as mean±SD for normally distributed continuous variables, median (interquartile range) for nonnormally distributed continuous variables, or number in category (percent of total in category) for categorical variables. HRs were calculated per SD (for normally distributed variables). For variables with nonnormal distribution (creatinine, potassium, C-reactive protein), values were log transformed, and HRs were calculated per log of SD.
Variables ultimately included in the PEROX model.
P<0.05.
Comprehensive Hematologic Profile Patterns Identify Patient Risk for MI or Death
In the derivation cohort, 23 high-risk patterns (Table 2) were identified in patients who were more likely to experience death (>3.6-fold risk) or MI (>1.4-fold risk) over the ensuing year. Unique discriminating patterns in those who died included variables derived from multiple erythrocyte- and leukocyte (peroxidase)-related parameters, as well as plasma levels of C-reactive protein. High-risk patterns for MI included multiple erythrocyte, leukocyte (peroxidase), and platelet parameters; traditional risk factors; and blood chemistries (Table 2). Variables common to both high-risk death and MI patterns included age, hypertension, mean red blood cell hemoglobin concentration, hemoglobin concentration distribution width, hypochromic erythrocyte cell count, and peroxidase y sigma (a peroxidase-based measure of neutrophil size distribution).
Table 2.
High-Risk Patterns in PEROX Model for 1-Year Death or MI
| Pattern | n | Rate, % | HR (95% CI) | |
|---|---|---|---|---|
| Death, high risk | ||||
| 1 | Hgb content distribution width >3.93 and RBC hgb concentration mean <35.07 | 815 | 13* | 4.94 (3.88–6.30) |
| 2 | Hypochromic RBC count >189 and Hgb content distribution width >3.93 | 658 | 13* | 4.47 (3.48–5.73) |
| 3 | Mean corpuscular Hgb concentration <34.38 and perox d/D <0.89 | 466 | 14* | 4.46 (3.42–5.81) |
| 4 | Hypochromic RBC count >189 and macrocytic RBC count >192 | 588 | 13* | 4.37 (3.39–5.64) |
| 5 | Mean corpuscular Hgb concentration <33.00 and mononuclear central x channel <14.38 | 422 | 14* | 4.37 (3.33–5.74) |
| 6 | Age >67 and hematocrit <36.45 | 515 | 13* | 4.08 (3.13–5.32) |
| 7 | Mononuclear polymorphonuclear valley <18.50 and peroxidase y sigma >9.48 | 474 | 13* | 3.85 (2.93–5.07) |
| 8 | Mononuclear central x channel <14.38 and peroxidase y mean >19.02 | 494 | 12* | 3.68 (2.80–4.85) |
| 9 | C-reactive protein >13.75 and history of hypertension | 531 | 12* | 3.63 (2.77–4.76) |
| MI, high risk | ||||
| 1 | Mean platelet concentration >27.89 and potassium <3.85 | 332 | 5† | 2.17 (1.33–3.56) |
| 2 | Triglycerides <130 and age >76 | 464 | 5† | 1.94 (1.23–3.04) |
| 3 | RBC distribution width >13.83 and lymphocyte count >1.75 | 371 | 5† | 1.93 (1.18–3.17) |
| 4 | Hypochromic RBC count >56 and diabetes mellitus | 1212 | 4† | 1.91 (1.37–2.68) |
| 5 | Body mass index <24.7 and neutrophil count <3.58 | 446 | 4† | 1.91 (1.20–3.03) |
| 6 | Systolic blood pressure >150 and history of hypertension | 1163 | 4† | 1.89 (1.35–2.66) |
| 7 | Polymorphonuclear cluster x axis mode >29.87 and RBC distribution width >13.22 | 729 | 4† | 1.80 (1.22–2.67) |
| 8 | Hgb distribution width >2.69 and peroxidase y sigma >8.59 | 842 | 4† | 1.79 (1.23–2.61) |
| 9 | Platelet concentration distribution width <5.39 and RBC hgb concentration mean <34.69 | 870 | 4† | 1.79 (1.23–2.60) |
| 10 | Mean corpuscular hemoglobin >32.60 and male | 500 | 4† | 1.78 (1.13–2.81) |
| 11 | Lymphocyte count <0.96 and potassium >4.4 | 387 | 4† | 1.73 (1.04–2.87) |
| 12 | Platelet concentration distribution width >6.04 and monocyte count >0.46 | 119 | 4† | 1.7 (0.71–4.06) |
| 13 | Neutrophil cluster mean y <71.19 and current smoker | 447 | 4† | 1.69 (1.04–2.74) |
| 14 | Mean platelet concentration >23.19 and basophil count >0.12 | 178 | 3† | 1.36 (0.61–3.03) |
Hgb indicated hemoglobin; RBC, red blood cell. Shown above are high-risk patterns present in the population, with n representing the number of patients in the derivation cohort in each pattern. The event rate within each pattern and HR (95% CI) are shown for each pattern based on univariate Cox models for ranking purposes. Units for each variable are shown in Table 1.
Death rate.
MI rate.
An additional 24 low-risk patterns (Table 3) were observed in patients less likely to experience death (<0.34-fold risk) or MI (<0.57-fold risk). Variables that were shared between low-risk patterns for both death and MI risk included C-reactive protein levels, absolute neutrophil count, mean platelet concentration (a flow cytometry determined index of platelet granule content), and monocyte/polymorphonuclear valley (a measure of separation among clusters of peroxidase-containing cell populations). In general, the low-risk patterns for incident 1-year death and MI risk are dominated by multiple diverse hematology analyzer variables of all 3 blood cell types (erythrocyte, leukocyte, and platelet) and age.
Table 3.
Low-Risk Patterns in PEROX Model for 1-Year Death or MI
| Pattern | n | Rate, % | HR (95% CI) | |
|---|---|---|---|---|
| Death, low risk | ||||
| 1 | RBC hgb concentration mean >35.07 and hematocrit >42.25 | 1443 | 1* | 0.18 (0.10–0.31) |
| 2 | Macrocytic RBC count <192 and age <67 | 2283 | 1* | 0.22 (0.15–0.32) |
| 3 | RBC Hgb concentration mean >35.07 and RBC count >4.42 | 1494 | 1* | 0.24 (0.15–0.38) |
| 4 | Mean platelet concentration >27.52 and age <67 | 1651 | 1* | 0.24 (0.16–0.38) |
| 5 | Peroxidase y sigma <8.10 and age <67 | 1982 | 1* | 0.26 (0.17–0.38) |
| 6 | C-reactive protein <4.0 and hematocrit >42.25 | 1688 | 1* | 0.26 (0.17–0.40) |
| 7 | Hematocrit >42.25 and perox d/D >0.89 | 1972 | 1* | 0.27 (0.18–0.40) |
| 8 | Mononuclear polymorphonuclear valley >18.50 and age <67 | 1750 | 1* | 0.27 (0.18–0.41) |
| 9 | RBC Hgb concentration mean >35.07 and white blood cell count <5.86 | 1436 | 1* | 0.30 (0.19–0.46) |
| 10 | Neutrophil count <3.96 and age <67 | 1697 | 2* | 0.34 (0.23–0.49) |
| MI, low risk | ||||
| 1 | No history of cardiovascular disease and RBC distribution width <13.22 | 919 | 1† | 0.31 (0.15–0.63) |
| 2 | Lymphocyte/large unstained cell threshold <44.50 and blasts <0.51% | 946 | 1† | 0.34 (0.17–0.66) |
| 3 | Systolic blood pressure <134 and basophil count <0.03 | 743 | 1† | 0.34 (0.16–0.73) |
| 4 | Platelet clumps >41 and fasting blood glucose <92.5 | 782 | 1† | 0.37 (0.18–0.76) |
| 5 | Hemoglobin distribution width <2.69 and hypochromic RBC count <14 | 891 | 1† | 0.41 (0.22–0.77) |
| 6 | Hypochromic RBC count <14 and Neutrophil count <5.83 | 1159 | 1† | 0.43 (0.25–0.74) |
| 7 | Mononuclear central x channel <12.70 and neutrophil y cluster mean >69.30 | 841 | 1† | 0.44 (0.23–0.82) |
| 8 | Mononuclear polymorphonuclear valley >14.50 and creatinine <0.75 | 910 | 1† | 0.44 (0.24–0.81) |
| 9 | No history of cardiovascular disease and systolic blood pressure <134 | 756 | 1† | 0.44 (0.23–0.86) |
| 10 | No. of peroxidase-saturated cells <0.01 and neutrophil count <4.69 | 781 | 1† | 0.47 (0.25–0.90) |
| 11 | HDL cholesterol >59 and mean platelet concentration <28.56 | 830 | 1† | 0.49 (0.27–0.90) |
| 12 | Mononuclear central x channel <12.70 and C-reactive protein <5.31 | 896 | 1† | 0.49 (0.27–0.88) |
| 13 | Mononuclear central x channel <12.70 and basophil count <0.07 | 961 | 1† | 0.54 (0.31–0.93) |
| 14 | No history of cardiovascular disease and neutrophil cluster mean x <66.07 | 1261 | 2† | 0.57 (0.36–0.92) |
RBC indicates red blood cell; Hgb, hemoglobin. Shown are low-risk patterns present in the population, with n representing the number of patients in derivation cohort in each pattern. The event rate within each pattern and HR (95% CI) are shown for each pattern based on univariate Cox models for ranking purposes. Units for each variable are shown in Table 1.
Death rate.
MI rate.
A composite PEROX model for prediction of incident 1-year death or nonfatal MI risk was generated within the derivation cohort by summing individual high- and low-risk patterns for death and MI individually. The reproducibility of the PEROX model was assessed by examining multiple replicate samples from multiple subjects both within and between days, revealing intraday and interday coefficients of variance of 5±0.4% and 10±2% (mean±SD), respectively. Stability of high- and low-risk patterns used for construction of the PEROX score and model validation analyses with the Somers D rank correlation40 and Hosmer-Lemeshow statistic41 are provided in the Materials section in the online-only Data Supplement.
The PEROX Model Predicts Incident 1-Year Risks for Nonfatal MI and Death
Within the derivation cohort, the PEROX model ROC curve analyses for the 1-year end points of death, MI, and the composite of death or MI demonstrated AUCs of 80%, 66%, and 75%, respectively. For the composite end point, an ROC curve potential cut point was identified that was virtually identical to the top tertile cut point within the derivation cohort.
Initial characterization of the performance of the PEROX score within the validation cohort included time-to-event analysis for death, MI, or the composite of either event using risk score tertiles to stratify subjects into equivalently sized groups of low, medium, and high risk (Figure 1A through 1C). For each outcome monitored, increasing cumulative event rates were noted over time within increasing tertiles (log-rank P<0.001 for each outcome). Figure 1D through 1F demonstrates the relationship between predicted (and 95% CI) absolute 1-year event rates estimated by PEROX score within the validation cohort. Also shown are actual event rates plotted in deciles of PEROX scores for both the derivation and validation cohorts. Observed event rates from the derivation cohort were similar to those observed in the validation cohort (Figure 1D through 1F), and strong tight positive associations were noted between increasing risk score and risk for experiencing nonfatal MI, death, or the composite adverse outcome.
Figure 1.
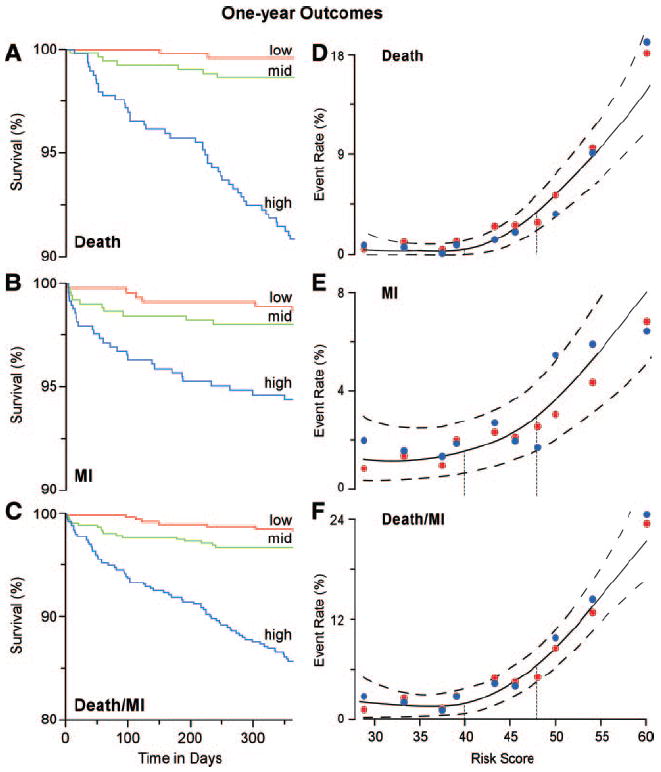
Kaplan-Meier curves and composite risk for 1-year outcomes based on tertiles of PEROX risk score in the validation cohort. Kaplan-Meier curves for cumulative probability of death (A), MI (B), or either event (C) according to low, medium, and high tertiles of PEROX score. Spline curves (solid line) with 95% CIs (dashed line) showing association between cumulative event (y axis) for death (D), MI (E), and death or MI (F) for PEROX score (x axis) are shown. Also illustrated are the absolute event rates per decile of PEROX score within the derivation (red solid circle) and validation (blue solid circle) cohorts. Vertical dotted lines indicate the tertile cut points separating low (<40), medium (≥40 to <48), and high (≥48) PEROX scores.
Relative Performance of the PEROX Model for Accurate Risk Assessment and Reclassification of Patients
In additional analyses within the validation cohort, ROC curve analyses were performed comparing the accuracy of traditional cardiac risk factors alone and with PEROX for the prediction of 1-year death or MI. Traditional risk factors alone showed modest accuracy (AUC=67%) for 1-year death or MI, whereas the addition of the PEROX risk score to traditional risk factors significantly increased prognostic accuracy (AUC=78%; P<0.001). To further evaluate the validity of the PEROX score, resampling (250 bootstrap samples from the validation cohort, n=1474) was performed and ROC analyses for each bootstrap sample was calculated for the prediction of 1-year death or MI risk. Compared with traditional risk factors alone, the PEROX score demonstrated superior prognostic accuracy among subjects within the independent validation cohort (Figure 2). When PEROX risk score categories were defined by tertiles (in which approximately equal proportions of subjects within the entire cohort are stratified into each risk bin), the 1-year event rate for death/MI among subjects stratified within high– versus low–PEROX-risk groups was 14% versus 2%, a 7-fold risk gradient. Results of Cox proportional-hazards regression for time-to-event analyses within the validation cohort (n=1434) are shown in Table 4 and reveal that the PEROX risk score significantly predicts the major adverse cardiac end points of death, MI, or the composite end point even after adjustment for traditional risk factors. Subjects with a high (top tertile) PEROX risk category relative to low (bottom tertile) PEROX risk show an HR of 6.5 (95% CI, 4.9 to 8.6) for 1-year death/MI. The potential clinical utility of the PEROX risk score was further compared with traditional risk factors in reclassifying patients into risk groups. As shown in Table 5, adding the PEROX score significantly improves risk classification at the 1-year follow-up for death (NRI=19.4%; P<0.001), MI (NRI=15.6; P=0.002), or both events (NRI=23.5; P<0.001) compared with traditional risk factors alone. These findings are consistent among either primary or secondary prevention subjects (Table 6).
Figure 2.
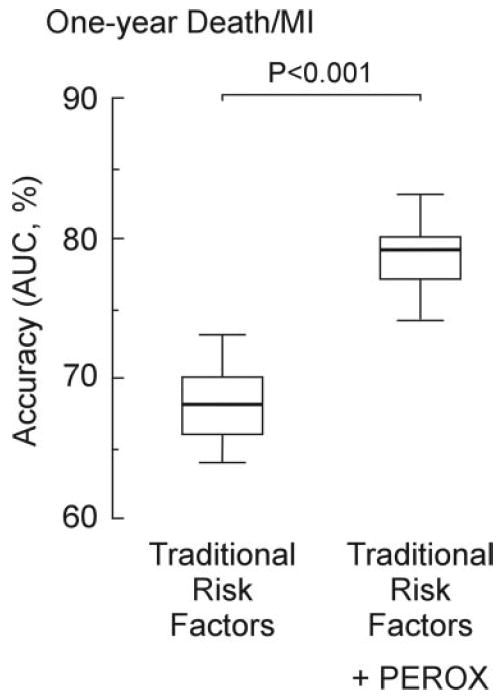
Validation analysis of PEROX risk score. Models were assessed for their association with 1-year incident risk of MI or death. Models were made up of traditional risk factors alone (including age, gender, smoking, LDL cholesterol, HDL cholesterol, systolic blood pressure, and history of diabetes mellitus) vs traditional risk factors plus PEROX score. Resampling (250 bootstrap samples from the validation cohort, n=1474) was performed. All data analyses, including ROC analyses and AUC determinations, were separately recalculated at each resampling for models with/without PEROX score. The AUCs calculated from the bootstrap samples are compared by the use of side-by-side box plots in which boxes represent interquartile ranges (defined as the difference between the first and third quartiles) and whiskers represent the 5th and 95th percentile values.
Table 4.
Unadjusted and Adjusted HR of PEROX Risk Scores for Adverse Cardiac Events at the 1-Year Follow-Up
| HR (95% CI) | P | |
|---|---|---|
| Death | ||
| Unadjusted | 3.68 (2.72–4.96) | <0.001 |
| Adjusted | 3.74 (2.61–5.36) | <0.001 |
| MI | ||
| Unadjusted | 1.77 (1.31–2.38) | <0.001 |
| Adjusted | 2.00 (1.40–2.87) | <0.001 |
| Death/MI | ||
| Unadjusted | 2.57 (2.06–3.21) | <0.001 |
| Adjusted | 2.76 (2.14–3.57) | <0.001 |
Multivariate Cox models were constructed within the validation cohort (n=1434) for the end points of death, MI, or the composite of death or MI using either the PEROX risk score alone or the PEROX risk score adjusted for traditional risk factors including age, gender, smoking, LDL cholesterol, HDL cholesterol, systolic blood pressure, and history of diabetes mellitus. The HRs shown correspond to 1-SD increment.
Table 5.
Reclassification Among Subjects Who Experienced Versus Did Not Experience Adverse Clinical Event at the 1-Year Follow-Up
| IDI |
Event-Specific Reclassification |
|||
|---|---|---|---|---|
| % | P | NRI, % | P | |
| Death | ||||
| Without PEROX | … | … | … | … |
| With PEROX | 0.316 | <0.001 | 0.194 | <0.001 |
| MI | ||||
| Without PEROX | … | … | … | … |
| With PEROX | 0.140 | <0.001 | 0.156 | 0.002 |
| Death/MI | ||||
| Without PEROX | … | … | … | … |
| With PEROX | 0.220 | <0.001 | 0.235 | <0.001 |
Both NRI and integrated discrimination improvement (IDI) were used to quantify improvement in model performance. P values compare models with and without PEROX risk scores. Both models were adjusted for traditional risk factors including age, gender, smoking, LDL cholesterol, HDL cholesterol, systolic blood pressure, and history of diabetes mellitus. Cutoff values for NRI estimation used a ratio of 6:3:1 for low-, medium-, and high-risk categories. The risk of adverse cardiac events was estimated with the Cox model.
Table 6.
AUC Values of Models With/Without PEROX Risk Scores for Adverse Cardiac Events at the 1-Year Follow-Up Stratified According to Primary Versus Secondary Prevention Status
| Primary Prevention (n=1859) | Secondary Prevention (n=5510) | |
|---|---|---|
| Death events, n | 40 | 256 |
| Without PEROX | 69 | 70 |
| With PEROX | 81 | 80 |
| P | 0.009 | <0.001 |
| MI events, n | 23 | 169 |
| Without PEROX | 58 | 62 |
| With PEROX | 71 | 68 |
| P | 0.072 | 0.007 |
| Death/MI events, n | 63 | 416 |
| Without PEROX | 64 | 65 |
| With PEROX | 78 | 75 |
| P | <0.001 | <0.001 |
ROC curves and AUCs were calculated for 1-year death, MI, and combined death or MI end points. ROC curves for the models with/without PEROX were constructed, and the corresponding AUC values were compared. One-year predicted probabilities of an adverse cardiac event were estimated from the Cox model. P values shown represent a comparison of AUC values estimated from models with and without PEROX risk score among primary prevention or secondary prevention subjects within the whole cohort (n=7369). Both models were adjusted for traditional risk factors including age, gender, smoking, LDL cholesterol, HDL cholesterol, systolic blood pressure, and history of diabetes mellitus.
Discussion
Studies by our group25 and Buffon et al24 previously implicated intracellular peroxidase content of leukocytes in cardiovascular risk stratification of patients. From these preceding observations and the numerous mechanistic links between myeloperoxidase,12,42 monocytes,43,44 and neutrophils45 for atherosclerosis and acute coronary syndromes, we hypothesized that data derived from a peroxidase-based hematology analyzer would harbor clinically useful information related to cardiovascular disease prognosis. As the analyses unfolded, it became clear that patterns generated by a combination of clinical information and alternative hematology measures could provide significant incremental value. In particular, review of the components contributing to the high- and low-risk patterns that contribute to the PEROX model reveals that a number of erythrocyte- and leukocyte-related phenotypes, as well as a smaller number of platelet-related parameters, provided prognostic value in identifying individuals in this population at both increased and decreased risk for near-term adverse cardiac events. The present studies show that alterations in multiple subtle phenotypes within leukocyte, erythrocyte, and platelet lineages can provide prognostic information relevant to cardiovascular health and atherothrombotic risk, consistent with the numerous mechanistic links to cardiovascular disease pathogenesis for each of these hematopoietic lineages. Hematology analyzers are some of the most commonly used instruments within hospital laboratories. Our studies show that information already captured by these instruments during routine use (but not typically reported) can aid in the clinical assessment of a stable cardiology patient, dramatically improving the accuracy with which subjects can be risk classified at both the high- and low-risk ends of the spectrum. Blood is a dynamic integrated sensor of the physiological state. A hematology analyzer profile serves as a holistic assessment of a broad spectrum of phenotypes related to multiple diverse and mechanistically relevant cell types from which we can recognize patterns, like fingerprints, that provide clinically useful information in the evaluation of cardiovascular risk in subjects.
Another intriguing finding in the present studies is how much hematology parameters, especially from erythrocyte and leukocyte lineages, contribute to the prognostic value of the PEROX model. This observation strongly underscores the growing appreciation that atherosclerosis is a systemic disease, with parameters in the blood combined with biochemical profiles of systemic inflammation being strongly linked to disease pathogenesis. Although many of the patterns identified as low-and high-risk traits within subjects are of unclear biological meaning, a large number are made up of elements with recognizable mechanistic connections to disease pathogenesis. As a group, all patterns reported appear to be robust, reproducible, and present in multiple independent samplings of the independent validation cohort. The identification of reproducible high- and low-risk patterns among the clinical, laboratory, and hematologic parameters monitored further indicates the presence of underlying complex relationships between multiple hematologic parameters, clinical and metabolic parameters, and cardiovascular disease pathogenesis.
Much interest focuses on the idea that array-based phenotyping will play an ever-increasing role in the future of preventive medicine, serving as a powerful method to improve risk classification of subjects and ultimately individualize tailored therapies. Rather than use research-based arrays (genomic, proteomic, metabolomic, expression array) that are no doubt powerful and extremely useful, we decided to use a robust, high-throughput workhorse of clinical laboratory medicine that is already in broad clinical use: the hematology analyzer. The hematology analyzer selected is commonly available worldwide and has the added advantage of being a flow cytometer that uses in situ peroxidase cytochemical staining for identifying and quantifying leukocytes, an added phenotypic dimension relevant to disease pathogenesis. Although the precise risk score developed here should be considered only proof of concept, the holistic approach taken illustrates that in the outpatient cardiology setting, these hematologic data may add prognostic information.
Limitations of the study are worth noting. Foremost, although the study population examined is large and replication of the findings is observed within multiple samplings of an entirely distinct, nonoverlapping validation cohort of subjects, the generalizability of these findings to other populations remains to be tested. The population examined, although stable and without evidence of myocardial necrosis (ie, cardiac troponin T negative), was nonetheless selected by referral for elective diagnostic coronary angiography. Furthermore, although the PEROX score predicted incident MI risk, it is notable that the PEROX offers lower discrimination for MI than for death. In this regard, it would be helpful to know the causes of death and the C statistic for cardiovascular mortality because only all-cause mortality data were available. It should also be noted that the present study represents a large, single-center study with ≈90% whites and a high degree of cardiovascular risk factors and comorbidities, some of which (eg, left ventricular function, measures of fitness or frailty, pulmonary diseases, performance of stress test) were not available in all subjects and therefore not included in the model. Several alternative populations (such as community-based or previously untreated populations) will be particularly interesting to examine with the PEROX model. Similarly, patients in whom complete blood count is already being measured (eg, those presenting with chest pain and suspected acute coronary syndromes) represent a particularly attractive cohort to monitor, given the high-throughput nature of the hematology analyzer and the existing availability of the data. One might hypothesize that additional platelet parameters, for example, might add to rapid risk screening in such a cohort. Whereas the process for generating the PEROX score may be applied elsewhere, in each setting, it is essential to incorporate additional key clinical variables when available, and it may be necessary to modify the PEROX model or to recalibrate the PEROX score based on local distributions of clinical and hematologic variables.
Conclusion
The results from the present studies suggest that the expanded use of more comprehensive hematology analyzer profiling of blood holds promise for improved risk stratification, more efficient targeting of preventive risk reduction efforts, and potential for monitoring of therapeutic responses in the future.
CLINICAL PERSPECTIVE.
Array-based phenotyping may improve risk classification of subjects and help tailor therapies. In this study, rather than use research-based arrays (genomic, proteomic, metabolomic, expression array), we used a robust and clinically validated high-throughput workhorse of clinical laboratory medicine, the hematology analyzer, to generate a comprehensive array of hematologic data. Using an established data-mining methodology (logical analysis of data), we generated a risk score (the PEROX score) that is based on reproducible high- and low-risk patterns of combinations of clinical and biochemical/hematology variables among subjects undergoing elective diagnostic cardiac catheterization. Within an independent nonoverlapping validation cohort, the PEROX risk score demonstrated superior prognostic accuracy for 1-year risk of death or myocardial infarction compared with traditional risk factors alone and significant net reclassification index. The results from the present studies suggest that expanded use of existing tools in profiling clinical and blood patterns holds promise for improved risk stratification.
Supplementary Material
Acknowledgments
This work is dedicated to the memory of Dr Peter L. Hammer, a true scholar and innovator. We thank Drs John Barnard, Steven Chen, and David Van Wagoner for helpful discussions, as well as Jamie Murphy for assistance in the preparation of the manuscript.
Sources of Funding This study was supported by National Institutes of Health grants P01 HL076491-055328, P01 HL077107-050004, and P01 HL087018-020001 and the Case Western Reserve University/Cleveland Clinic Clinical and Translational Science Award (grant 1KL2RR024990). Acquisition of the Advia 120 instrument was made possible in part by a gift from Herbert and Judith Harvey. Support for performance of fasting lipid profiles, basic metabolic panel, and high-sensitivity C-reactive protein was provided by Abbott Laboratories. Some of the calibrator (Cal-Chex H) for the hematology analyzer was generously provided by Streck (Omaha, Neb).
Footnotes
Disclosures Drs Hazen and Brennan report that they are named as coinventors on pending patents filed by the Cleveland Clinic that refer to the use of biomarkers in inflammatory and cardiovascular disorders. Dr Hazen also is the scientific founder of Cleveland Heart Labs Inc; has received research grant support from Abbott Laboratories, Lipo-science, and Cleveland Heart Labs; and has received honoraria and consulting fees from Abbott Laboratories, Merck, Pfizer, Cleveland Heart Labs, Esperion, Takeda, and AstraZeneca. Dr Tang has received research grant support from Abbott Laboratories. The other authors report no conflicts.
References
- 1.Naghavi M, Falk E, Hecht HS, Jamieson MJ, Kaul S, Berman D, Fayad Z, Budoff MJ, Rumberger J, Naqvi TZ, Shaw LJ, Faergeman O, Cohn J, Bahr R, Koenig W, Demirovic J, Arking D, Herrera VL, Badimon J, Goldstein JA, Rudy Y, Airaksinen J, Schwartz RS, Riley WA, Mendes RA, Douglas P, Shah PK. From vulnerable plaque to vulnerable patient, part III: executive summary of the Screening for Heart Attack Prevention and Education (SHAPE) Task Force report. Am J Cardiol. 2006;98:2H–15H. doi: 10.1016/j.amjcard.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 2.Maisel AS, Bhalla V, Braunwald E. Cardiac biomarkers: a contemporary status report. Nat Clin Pract Cardiovasc Med. 2006;3:24–34. doi: 10.1038/ncpcardio0405. [DOI] [PubMed] [Google Scholar]
- 3.See R, Lindsey JB, Patel MJ, Ayers CR, Khera A, McGuire DK, Grundy SM, de Lemos JA. Application of the screening for Heart Attack Prevention and Education Task Force recommendations to an urban population: observations from the Dallas Heart Study. Arch Intern Med. 2008;168:1055–1062. doi: 10.1001/archinte.168.10.1055. [DOI] [PubMed] [Google Scholar]
- 4.Wang TJ, Gona P, Larson MG, Tofler GH, Levy D, Newton-Cheh C, Jacques PF, Rifai N, Selhub J, Robins SJ, Benjamin EJ, D’Agostino RB, Vasan RS. Multiple biomarkers for the prediction of first major cardiovascular events and death. N Engl J Med. 2006;355:2631–2639. doi: 10.1056/NEJMoa055373. [DOI] [PubMed] [Google Scholar]
- 5.Kathiresan S, Melander O, Anevski D, Guiducci C, Burtt NP, Roos C, Hirschhorn JN, Berglund G, Hedblad B, Groop L, Altshuler DM, Newton-Cheh C, Orho-Melander M. Polymorphisms associated with cholesterol and risk of cardiovascular events. N Engl J Med. 2008;358:1240–1249. doi: 10.1056/NEJMoa0706728. [DOI] [PubMed] [Google Scholar]
- 6.Detrano R, Guerci AD, Carr JJ, Bild DE, Burke G, Folsom AR, Liu K, Shea S, Szklo M, Bluemke DA, O’Leary DH, Tracy R, Watson K, Wong ND, Kronmal RA. Coronary calcium as a predictor of coronary events in four racial or ethnic groups. N Engl J Med. 2008;358:1336–1345. doi: 10.1056/NEJMoa072100. [DOI] [PubMed] [Google Scholar]
- 7.Gaziano TA, Young CR, Fitzmaurice G, Atwood S, Gaziano JM. Laboratory-based versus non-laboratory-based method for assessment of cardiovascular disease risk: the NHANES I Follow-up Study cohort. Lancet. 2008;371:923–931. doi: 10.1016/S0140-6736(08)60418-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Danesh J, Collins R, Appleby P, Peto R. Association of fibrinogen, C-reactive protein, albumin, or leukocyte count with coronary heart disease: meta-analyses of prospective studies. JAMA. 1998;279:1477–1482. doi: 10.1001/jama.279.18.1477. [DOI] [PubMed] [Google Scholar]
- 9.Rana JS, Boekholdt SM, Ridker PM, Jukema JW, Luben R, Bingham SA, Day NE, Wareham NJ, Kastelein JJ, Khaw KT. Differential leucocyte count and the risk of future coronary artery disease in healthy men and women: the EPIC-Norfolk Prospective Population Study. J Intern Med. 2007;262:678–689. doi: 10.1111/j.1365-2796.2007.01864.x. [DOI] [PubMed] [Google Scholar]
- 10.Packard RR, Libby P. Inflammation in atherosclerosis: from vascular biology to biomarker discovery and risk prediction. Clin Chem. 2008;54:24–38. doi: 10.1373/clinchem.2007.097360. [DOI] [PubMed] [Google Scholar]
- 11.Sugiyama S, Okada Y, Sukhova GK, Virmani R, Heinecke JW, Libby P. Macrophage myeloperoxidase regulation by granulocyte macrophage colony-stimulating factor in human atherosclerosis and implications in acute coronary syndromes. Am J Pathol. 2001;158:879–891. doi: 10.1016/S0002-9440(10)64036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nicholls SJ, Hazen SL. Myeloperoxidase and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2005;25:1102–1111. doi: 10.1161/01.ATV.0000163262.83456.6d. [DOI] [PubMed] [Google Scholar]
- 13.Podrez EA, Schmitt D, Hoff HF, Hazen SL. Myeloperoxidase-generated reactive nitrogen species convert LDL into an atherogenic form in vitro. J Clin Invest. 1999;103:1547–1560. doi: 10.1172/JCI5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang R, Brennan ML, Shen Z, MacPherson JC, Schmitt D, Molenda CE, Hazen SL. Myeloperoxidase functions as a major enzymatic catalyst for initiation of lipid peroxidation at sites of inflammation. J Biol Chem. 2002;277:46116–46122. doi: 10.1074/jbc.M209124200. [DOI] [PubMed] [Google Scholar]
- 15.Zheng L, Settle M, Brubaker G, Schmitt D, Hazen SL, Smith JD, Kinter M. Localization of nitration and chlorination sites on apolipoprotein A-I catalyzed by myeloperoxidase in human atheroma and associated oxidative impairment in ABCA1-dependent cholesterol efflux from macrophages. J Biol Chem. 2005;280:38–47. doi: 10.1074/jbc.M407019200. [DOI] [PubMed] [Google Scholar]
- 16.Thukkani AK, McHowat J, Hsu FF, Brennan ML, Hazen SL, Ford DA. Identification of alpha-chloro fatty aldehydes and unsaturated lysophosphatidylcholine molecular species in human atherosclerotic lesions. Circulation. 2003;108:3128–3133. doi: 10.1161/01.CIR.0000104564.01539.6A. [DOI] [PubMed] [Google Scholar]
- 17.Weiss SJ, Peppin G, Ortiz X, Ragsdale C, Test ST. Oxidative autoactivation of latent collagenase by human neutrophils. Science. 1985;227:747–749. doi: 10.1126/science.2982211. [DOI] [PubMed] [Google Scholar]
- 18.Askari AT, Brennan ML, Zhou X, Drinko J, Morehead A, Thomas JD, Topol EJ, Hazen SL, Penn MS. Myeloperoxidase and plasminogen activator inhibitor 1 play a central role in ventricular remodeling after myocardial infarction. J Exp Med. 2003;197:615–624. doi: 10.1084/jem.20021426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abu-Soud HM, Hazen SL. Nitric oxide is a physiological substrate for mammalian peroxidases. J Biol Chem. 2000;275:37524–37532. doi: 10.1074/jbc.275.48.37524. [DOI] [PubMed] [Google Scholar]
- 20.Vita JA, Brennan ML, Gokce N, Mann SA, Goormastic M, Shishehbor MH, Penn MS, Keaney JF, Jr, Hazen SL. Serum myeloperoxidase levels independently predict endothelial dysfunction in humans. Circulation. 2004;110:1134–1139. doi: 10.1161/01.CIR.0000140262.20831.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baldus S, Heitzer T, Eiserich JP, Lau D, Mollnau H, Ortak M, Petri S, Goldmann B, Duchstein HJ, Berger J, Helmchen U, Freeman BA, Meinertz T, Munzel T. Myeloperoxidase enhances nitric oxide catabolism during myocardial ischemia and reperfusion. Free Radic Biol Med. 2004;37:902–911. doi: 10.1016/j.freeradbiomed.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 22.Brennan ML, Penn MS, Van Lente F, Nambi V, Shishehbor MH, Aviles RJ, Goormastic M, Pepoy ML, McErlean ES, Topol EJ, Nissen SE, Hazen SL. Prognostic value of myeloperoxidase in patients with chest pain. N Engl J Med. 2003;349:1595–1604. doi: 10.1056/NEJMoa035003. [DOI] [PubMed] [Google Scholar]
- 23.Baldus S, Heeschen C, Meinertz T, Zeiher AM, Eiserich JP, Munzel T, Simoons ML, Hamm CW. Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation. 2003;108:1440–1445. doi: 10.1161/01.CIR.0000090690.67322.51. [DOI] [PubMed] [Google Scholar]
- 24.Buffon A, Biasucci LM, Liuzzo G, D’Onofrio G, Crea F, Maseri A. Widespread coronary inflammation in unstable angina. N Engl J Med. 2002;347:5–12. doi: 10.1056/NEJMoa012295. [DOI] [PubMed] [Google Scholar]
- 25.Zhang R, Brennan ML, Fu X, Aviles RJ, Pearce GL, Penn MS, Topol EJ, Sprecher DL, Hazen SL. Association between myeloperoxidase levels and risk of coronary artery disease. JAMA. 2001;286:2136–2142. doi: 10.1001/jama.286.17.2136. [DOI] [PubMed] [Google Scholar]
- 26.Schechter AN, Gladwin MT. Hemoglobin and the paracrine and endocrine functions of nitric oxide. N Engl J Med. 2003;348:1483–1485. doi: 10.1056/NEJMcibr023045. [DOI] [PubMed] [Google Scholar]
- 27.Davi G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med. 2007;357:2482–2494. doi: 10.1056/NEJMra071014. [DOI] [PubMed] [Google Scholar]
- 28.Podrez EA, Byzova TV, Febbraio M, Salomon RG, Ma Y, Valiyaveettil M, Poliakov E, Sun M, Finton PJ, Curtis BR, Chen J, Zhang R, Silverstein RL, Hazen SL. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med. 2007;13:1086–1095. doi: 10.1038/nm1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Z, Nicholls SJ, Rodriguez ER, Kummu O, Horkko S, Barnard J, Reynolds WF, Topol EJ, DiDonato JA, Hazen SL. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat Med. 2007;13:1176–1184. doi: 10.1038/nm1637. [DOI] [PubMed] [Google Scholar]
- 30.Tonelli M, Sacks F, Arnold M, Moye L, Davis B, Pfeffer M. Relation between red blood cell distribution width and cardiovascular event rate in people with coronary disease. Circulation. 2008;117:163–168. doi: 10.1161/CIRCULATIONAHA.107.727545. [DOI] [PubMed] [Google Scholar]
- 31.Thompson SG, Kienast J, Pyke SD, Haverkate F, van de Loo JC. Hemostatic factors and the risk of myocardial infarction or sudden death in patients with angina pectoris: European Concerted Action on Thrombosis and Disabilities Angina Pectoris Study Group. N Engl J Med. 1995;332:635–641. doi: 10.1056/NEJM199503093321003. [DOI] [PubMed] [Google Scholar]
- 32.Morange PE, Bickel C, Nicaud V, Schnabel R, Rupprecht HJ, Peetz D, Lackner KJ, Cambien F, Blankenberg S, Tiret L. Haemostatic factors and the risk of cardiovascular death in patients with coronary artery disease: the AtheroGene study. Arterioscler Thromb Vasc Biol. 2006;26:2793–2799. doi: 10.1161/01.ATV.0000249406.92992.0d. [DOI] [PubMed] [Google Scholar]
- 33.Danesh J, Collins R, Peto R, Lowe GD. Haematocrit, viscosity, erythrocyte sedimentation rate: meta-analyses of prospective studies of coronary heart disease. Eur Heart J. 2000;21:515–520. doi: 10.1053/euhj.1999.1699. [DOI] [PubMed] [Google Scholar]
- 34.Lauer MS, Alexe S, Pothier Snader CE, Blackstone EH, Ishwaran H, Hammer PL. Use of the logical analysis of data method for assessing long-term mortality risk after exercise electrocardiography. Circulation. 2002;106:685–690. doi: 10.1161/01.cir.0000024410.15081.fd. [DOI] [PubMed] [Google Scholar]
- 35.Crama YHP, Ibaraki T. Cause-effect relationships and partially defined Boolean functions. Ann Operations Res. 1988;16:299–326. [Google Scholar]
- 36.Boros E, Hammer PL, Ibaraki T, Kogan A. Logical analysis of numerical data. Math Programming. 1997;79:163–190. [Google Scholar]
- 37.Hanley JA, McNeil BJ. A method of comparing the areas under receiver operating characteristic curves derived from the same cases. Radiology. 1983;148:839–843. doi: 10.1148/radiology.148.3.6878708. [DOI] [PubMed] [Google Scholar]
- 38.Hollander M, Wolfe D. Nonparametric Statistical Methods. New York, NY: John Wiley & Sons; 1973. [Google Scholar]
- 39.Pencina MJ, D’Agostino RB, Sr, D’Agostino RB, Jr, Vasan RS. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med. 2008;27:157–172. doi: 10.1002/sim.2929. [DOI] [PubMed] [Google Scholar]
- 40.Potters L, Purrazzella R, Brustein S, Fearn P, Leibel SA, Kattan MW. A comprehensive and novel predictive modeling technique using detailed pathology factors in men with localized prostate carcinoma. Cancer. 2002;95:1451–1456. doi: 10.1002/cncr.10869. [DOI] [PubMed] [Google Scholar]
- 41.Hosmer DW, Hosmer T, Le Cessie S, Lemeshow S. A comparison of goodness-of-fit tests for the logistic regression model. Stat Med. 1997;16:965–980. doi: 10.1002/(sici)1097-0258(19970515)16:9<965::aid-sim509>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 42.Morrow DA, Sabatine MS, Brennan ML, de Lemos JA, Murphy SA, Ruff CT, Rifai N, Cannon CP, Hazen SL. Concurrent evaluation of novel cardiac biomarkers in acute coronary syndrome: myeloperoxidase and soluble CD40 ligand and the risk of recurrent ischaemic events in TACTICS-TIMI 18. Eur Heart J. 2008;29:1096–1102. doi: 10.1093/eurheartj/ehn071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Loscalzo J. The macrophage and fibrinolysis. Semin Thromb Hemost. 1996;22:503–506. doi: 10.1055/s-2007-999051. [DOI] [PubMed] [Google Scholar]
- 44.Navab M, Ananthramaiah GM, Reddy ST, Van Lenten BJ, Ansell BJ, Fonarow GC, Vahabzadeh K, Hama S, Hough G, Kamranpour N, Berliner JA, Lusis AJ, Fogelman AM. The oxidation hypothesis of atherogenesis: the role of oxidized phospholipids and HDL. J Lipid Res. 2004;45:993–1007. doi: 10.1194/jlr.R400001-JLR200. [DOI] [PubMed] [Google Scholar]
- 45.Naruko T, Ueda M, Haze K, van der Wal AC, van der Loos CM, Itoh A, Komatsu R, Ikura Y, Ogami M, Shimada Y, Ehara S, Yoshiyama M, Takeuchi K, Yoshikawa J, Becker AE. Neutrophil infiltration of culprit lesions in acute coronary syndromes. Circulation. 2002;106:2894–2900. doi: 10.1161/01.cir.0000042674.89762.20. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.