

Abstract
X-ray diffraction and other biophysical tools reveal features of the atomic structure of an amyloid-like crystal. Sup35, a prion-like protein in yeast, forms fibrillar amyloid assemblies intrinsic to its prion function. We have identified a polar peptide from the N-terminal prion-determining domain of Sup35 that exhibits the amyloid properties of full-length Sup35, including cooperative kinetics of aggregation, fibril formation, binding of the dye Congo red, and the characteristic cross-β x-ray diffraction pattern. Microcrystals of this peptide also share the principal properties of the fibrillar amyloid, including a highly stable, β-sheet-rich structure and the binding of Congo red. The x-ray powder pattern of the microcrystals, extending to 0.9-Å resolution, yields the unit cell dimensions of the well-ordered structure. These dimensions restrict possible atomic models of this amyloid-like structure and demonstrate that it forms packed, parallel-stranded β-sheets. The unusually high density of the crystals shows that the packed β-sheets are dehydrated, despite the polar character of the side chains. These results suggest that amyloid is a highly intermolecularly bonded, dehydrated array of densely packed β-sheets. This dry β-sheet could form as Sup35 partially unfolds to expose the peptide, permitting it to hydrogen-bond to the same peptide of other Sup35 molecules. The implication is that amyloid-forming units may be short segments of proteins, exposed for interactions by partial unfolding.
Amyloid is a fibrillar deposit formed in vivo by various human proteins when they undergo a partial unfolding and misassembly (for reviews, see refs. 1 and 2). Its formation is observed in some 20 human diseases (3, 4), including Alzheimer's disease, type II diabetes mellitus, and the transmissible spongiform encephalopathies or prion diseases (5). A protein fibril is identified as amyloid by its structural and tinctorial properties (6): amyloid fibrils are unbranched, range from 60 to 120 Å in diameter, and bind the dye Congo red. The fibrils also produce a characteristic “cross-β” x-ray diffraction pattern, consistent with a model in which stacked β-sheets form parallel to the fiber axis having their individual β-strands perpendicular to the fiber axis (7, 8). Although there is no canonical amino acid sequence for the known amyloidogenic proteins, sequence segments rich in polar residues such as asparagine and glutamine are common. The native three-dimensional structures of the amyloidogenic proteins are also dissimilar, despite the common fibrillar structure (6). Amyloid fibrils are unusually stable, and there is presently no effective treatment for amyloid diseases.
The yeast protein Sup35 offers a convenient model for studying both amyloid formation and the prion-like transmission of protein conformation. Although Sup35 is not a homolog of the mammalian prion protein (PrP) implicated in the spongiform encephalopathies, it shares with PrP the ability to transmit its aberrant fold and therefore may be referred to as a prion-like protein (9, 10). Sup35's normal cellular role is to terminate translation (11). Yeast harboring the [PSI+] determinant exhibit increased translational read-through of stop codons (12), presumably due to the aggregation and consequent loss of function of Sup35. Sup35 is a 685-residue protein. The first 123 residues constitute the prion-determining domain (PrD) (13), which contains a series of nine-residue repeats with consensus sequence PQGGYQQYN, similar to the human prion protein repeats (PHGGGWGQ). The PrD is sufficient for [PSI+] propagation (14) and readily forms amyloid fibrils in vitro (15, 16). The ability of the PrD to seed amyloid formation is highly species-dependent and hence sequence-specific (17).
Because Sup35 forms fibrils in vitro and because inhomogeneous fibrils are not amenable to crystallization, we prepared several chimeras and fragments of Sup35 with the goal of crystallization for structural studies. We also studied short peptides from the N terminus of the PrD, as this region is critical for maintenance of the [PSI+] phenotype in vivo (18) and is likely to reveal the atomic basis of aggregation of Sup35.
Materials and Methods
Molecular Cloning.
All of the Sup35 constructs with N-terminal histidine tags were provided by Susan Lindquist (University of Chicago) and have been described (16). The glutathione S-transferase (GST)-Sup35 (1–251) chimera (19) was prepared by PCR with the N-terminal primer, 5′-CATGGATCCCCCCTCGATCTAGCAACAATGTCGGATTCAAACCAA-3′ and C-terminal primer, 5′-CTGGAATTCTTAAACAACTTCGTCATCCAC-3′, using the full-length Sup35 DNA in vector pJC45 (16, 20) as a template. GST-Sup35(1–251)-His was created by PCR using the same template and N-terminal primer as for GST-Sup35(1–251) and the C-terminal primer 5′-CTGGAATTCTTAATGATGATGATGATGATGAACAACTTCGTCATCCAC-3′. The PCR products were subcloned as BamHI–EcoRI fragments into the pGEX-2T vector (Amersham Pharmacia).
Protein Expression and Purification.
All proteins with N-terminal histidine tags were expressed in Escherichia coli BL21/DE3 cells (Novagen). GST-fusion proteins were expressed in strain ARI 814 (21). Electrocompetent bacterial cells were transformed with expression plasmid and grown in Luria broth supplemented with 100 μg/ml ampicillin. At an OD600 of 0.5–0.7, protein expression was induced with 1 mM isopropyl β-d-thiogalactoside (Fisher Scientific). After 2–3 h of induction, cells were harvested by centrifugation and frozen for up to 2 weeks. For all but the GST-fusion proteins, induced cells were lysed by sonication in 30 mM Tris⋅HCl and 300 mM NaCl (pH 8) with 1 mM phenylmethylsulfonyl fluoride protease inhibitor (Sigma). For the proteins with an N-terminal histidine tag, the soluble cell lysate was incubated with nickel-NTA agarose (Qiagen, Chatsworth, CA) and stirred at 4°C for 30 min. Purification follows the methods of Glover et al. (16). For the GST-fusion proteins, cells were lysed in PBS with 0.1 mM phenylmethylsulfonyl fluoride, 0.2 mM EDTA, and a protease inhibitor mixture containing 20 μg/ml benzamidine and 10 μg/ml phenanthroline. The soluble cell lysate was incubated with GST-Sepharose (Amersham Pharmacia) for 30 min at 4°C and then poured into a disposable column. The column was washed twice with 4 column volumes of PBS and eluted with 10–25 mM glutathione (Sigma, G-4251), 50 mM Tris⋅HCl, and 14 mM β-mercaptoethanol (Fisher Scientific), pH 7. Protein concentrations were determined by using the Bio-Rad microassay, using BSA as a standard.
Electron Microscopy (EM).
Samples were prepared by applying a drop of protein or peptide solution to a carbon-coated Parlodion support film on a glow-discharged copper grid. The grids then were rinsed with three drops of distilled water, stained with uranyl acetate, and dried. All specimens were examined on a Hitachi H-7000 microscope at an accelerating voltage of 75 kV.
Fibril Inhibition Assay.
Freshly purified GST-Sup35-His protein solutions at a concentration of approximately 0.5 mg/ml were divided into 12 equal aliquots. The following conditions were tested for their ability to inhibit fibril formation: (a) 1 M NaCl, (b) 10% glycerol, (c) 0.5 M sucrose, (d) pH adjusted to 6.5, (e) pH adjusted to 2.6, (f) 10% isopropanol, (g) 150 mM NaCl, (h) 12.5 mM glutamine, (i) 12.5% DMSO, (j) 1% β-d-octyl glucoside, (k) 0.05% Congo red, and (l) no additive. All samples were stored at 4°C. A 400-μl aliquot from each fraction was taken at 1, 2, and 6 days to test for fibril formation. The fibril inhibition assay is based on the assumption that the majority of fibrils in a fibrillar sample will not pass through a 0.2-μm pore filter. Each aliquot was divided in two; half was filtered, the other half not filtered. The samples then were acetone-precipitated and prepared for SDS/PAGE. Fibril inhibition was indicated by less than 10% difference between protein concentration before and after filtration. Additionally, lower-order oligomers were detected by native PAGE.
Peptide Synthesis.
The peptide GNNQQNY was synthesized at the Biopolymer Synthesis Center at the California Institute of Technology (Pasadena) by automated solid-phase t-Boc chemistry. The peptides SQNGNQQRG, GNNQQNYQQ, NNQQNYQQ, and GNNQQNYQR were synthesized at the University of Southern California (Los Angeles) peptide facility. Peptides were purified by C18 reverse-phase HPLC, and their molecular weights were confirmed by matrix-assisted laser desorption ionization–time of flight mass spectrometry.
X-Ray Diffraction from Fibrils.
Peptide fibrils grown from a 1 mg/ml solution in water were oriented by repeated pipetting against the wall of a glass capillary tube. Fibrils mounted in a sealed glass capillary tube were exposed to copper Kα X-radiation from a Rigaku RU-200 x-ray generator operating at 50 kV and 100 mA. Data were collected at room temperature for 20 min with 2° oscillations on a Rigaku R-AxisIIC imaging plate detector.
Turbidity Measurement.
Turbidity was monitored by absorbance at 400 nm by using a Hewlett–Packard HP8453 UV-visible spectrophotometer equipped with a multicell transporter. All solutions were filtered immediately before the first measurement. Measurements were recorded while stirring once every 5 min for 20 h. Turbidity curves were generated by applying a three-point sliding window average to the raw data. Turbidity of a 0.33 mg/ml GNNQQNY solution was monitored with and without added Sup35 (approximately 1 μg/ml final protein concentration).
Crystallization of GNNQQNY.
Lyophilized GNNQQNY peptide was dissolved in water to a final concentration of 10 mg/ml peptide and immediately filtered (0.2 μm pore size). Microcrystals of GNNQQNY were grown in batch from this peptide solution.
Congo Red Binding by Fibrils and Microcrystals.
Congo red binding was detected by using the method of Klunk et al. (22), with the modification of extending the incubation time to 16 h. For the microcrystals, a 7-μM filtered solution of Congo red was diluted 2:1 with GNNQQNY microcrystals prepared in water. After approximately 30 min incubation, microcrystals were mounted in a glass capillary tube and exposed to copper Kα X-radiation for 20 min with 2° oscillations (see above for additional experimental details). A radial profile of the resulting x-ray powder diffraction pattern was generated and, after scaling, was subtracted from the profile of a pattern collected before the addition of Congo red.
X-Ray Diffraction from Microcrystals.
X-ray diffraction data were collected from microcrystals at room temperature (as described for fibrils) and −180°C. For cryogenic data collection, microcrystals were transferred to 20% PEG 400 (EM Science) for 2–5 min and frozen on a cryostream of nitrogen at −180°C. Diffraction data from frozen crystals were collected on a Rigaku R-AxisIV detector. Synchrotron data were collected at the Advanced Photon Source at Argonne National Laboratories, Argonne, IL (beamline 19-ID, operating at 12 kV with a wavelength of 1.03321 Å), by using a charge-coupled device detector (SPC2-Oxford Instruments, Oxford, U.K.).
Crystal Density Determination.
The density of the GNNQQNY microcrystals was determined by floating the crystals in a bromobenzene/m-xylene density gradient and measuring their floating position along the gradient relative to cesium chloride standard solutions (23, 24). The densities of the cesium chloride standard solutions were pycnometrically determined.
Fourier Transform IR Spectrometry.
GNNQQNY microcrystals were lyophilized until the mass remained unchanged over three consecutive readings. Cesium iodide was dried for 2 h. The crystals and cesium iodide were mixed together, pulverized, and formed into a pellet. Data were collected on the solid pellet at room temperature on a Jasco Fourier transform IR 420A for wavelengths in the range of 4,000 to 220 cm-1.
GNNQQNY Microcrystalline Stability.
The following additives were tested for their ability to dissolve GNNQQNY microcrystals: (a) methanol, (b) 8 M guanidinium hydrochloride, (c) 8 M urea, (d) 0.5 M EDTA, pH 8, (e) 10% SDS, (f) 1 M acetic acid, (g) IM NaOH, (h) 0.24 M glutamine, (i) ammonium hydroxide, and (j) formic acid. Each additive was mixed with an equal volume of GNNQQNY microcrystals in water and sealed. The samples were observed by light microscopy immediately upon mixing and again after 5 h at room temperature.
Results
Fibrils, But No Crystals, Form from Proteins Containing the Sup35 PrD.
We tested various Sup35 constructs (Fig. 1 a and b) for their ability to form fibrils or crystals. All chimeras containing the full PrD aggregated either into fibrils or amorphous aggregates. Although GST was previously reported to block Sup35 aggregation (19), in our hands the GST-Sup35 (1–251) and the GST-Sup35 (1–251)-His chimeras aggregated. GST-Sup35 (1–251) fibrils produced the diagnostic cross-β x-ray diffraction pattern of amyloid, with meridional and equatorial reflections at 4.7 Å and approximately 10 Å, respectively, suggesting β-strands running perpendicular to the fiber axis and packed β-sheets parallel to the fiber axis. We also tested two PNM (psi-no-more) mutants, formed by deleting residues 22–69; these mutants block the [PSI+] phenotype in vivo (25, 26). Both the full-length protein with the 22–69 PNM deletion and the PrD with the same deletion still form fibrils in vitro, although at a slower rate than the unaltered Sup35 and PrD, respectively (16). No tested additive fully inhibited fibril formation, and no Sup35 chimera was found to form crystals under any of the hundreds of conditions tested.
Figure 1.

The Sup35 protein and constructs tested for crystallization. (a) Residues 1–123 make up the PrD or N region. Residues 124–253 are the highly charged M region, and residues 254–685 function in translation termination. (b) Sup35 constructs tested for crystallization. Δ22–69 indicates that residues 22–69, which confer the psi-no-more phenotype, have been deleted. Thx represents thioredoxin. (c) Peptidyl fragments from the Sup35 PrD. Underlined residues indicate non-native amino acid sequences. Superscripts give initial and final sequence positions. * indicate plasmids provided by Susan Lindquist.
Amyloid-Like Fibrils Form from Peptides of the Sup35 PrD.
Like the full Sup35 PrD, several peptidyl fragments of the PrD (Fig. 1c) also form amyloid fibrils. These peptides were selected from a 17-residue segment in the Sup35 sequence (residues 8–24) that is critical for maintenance of the [PSI+] phenotype in vivo (18). This segment also has been shown to create a species barrier among yeast Sup35 proteins (17). We chose peptides identical to this region of the Sup35 sequence and peptides with single amino acid substitutions (Gln to Arg). The altered peptides, GNNQQNYQR and SQNGNQQRG, correspond to mutants (underlined residues) of Sup35 that remain soluble in vivo and increase the solubility of wild-type Sup35 in vivo, respectively (18). Four of the five peptides form the unbranched fibrils typical of amyloid. Only the mutant peptide SQNGNQQRG remained soluble. Each peptide also was tested for its ability to inhibit Sup35 aggregation. Although some minor inhibition was observed, particularly with SQNGNQQRG, none of the peptides fully blocked Sup35 aggregation.
One of the fibril-forming peptides, GNNQQNY, which runs from residue 7 through residue 13 in the Sup35 sequence, displays the same amyloid properties as the Sup35 PrD, including the following:
Structural transition to β-sheet.
Amyloidogenic proteins, even if not rich in β-sheet structure in their native state, undergo a structural transition to adopt the mostly β-sheet structure in amyloid (27). The soluble form of the peptide, GNNQQNY, is in a coil conformation as determined by both CD and NMR spectroscopy (data not shown). The transition to β-sheet structure is demonstrated by the x-ray diffraction patterns obtained from GNNQQNY fibrils (discussed below).
Fibril formation.
After several hours at room temperature, GNNQQNY forms amyloid fibrils (Fig. 2a) with a diameter of approximately 75 Å, narrower than the roughly 100-Å diameter of the fibrils formed from whole Sup35. The outline of the GNNQQNY fibrils by negative staining EM is smooth. In contrast, whole Sup35 fibrils sometimes appear jagged, which has been attributed to a nonfibrillar region of the protein decorating a fibrillar core (16).
Figure 2.

Biophysical characterization of the Sup35 fragment GNNQQNY. (a) An electron micrograph of the GNNQQNY amyloid fibrils (Left) and x-ray diffraction pattern of partially aligned fibrils (Right). The meridional reflections at 4.7 Å and equatorial reflections at 10 Å are characteristic of the cross-β structure of amyloid. (b) Effect of Sup 35 on GNNQQNY turbidity. GNNQQNY aggregation was monitored in the presence (upper curve) and absence (middle curve) of Sup35. The turbidity of Sup35 alone also is shown (lower curve). (c) An electron micograph of the GNNQQNY microcrystals (Left) and their x-ray diffraction pattern (Right). An aggregate of these microcrystals diffracts x-rays to 0.9-Å resolution. The darkest ring in the powder pattern is the 4.7-Å reflection.
X-ray diffraction consistent with a cross-β structure.
The x-ray diffraction pattern produced from GNNQQNY fibrils is shown in Fig. 2a. Meridional diffraction peaks at 4.7-Å resolution and equatorial diffraction peaks at approximately 10-Å resolution correspond to interstrand separation in a β-sheet and intersheet separation, respectively. The strand orientation for the stacked sheets is perpendicular to the fiber axis. This is the cross-β structure characteristic of amyloid.
Congo red binding.
Protein fibrils traditionally have been characterized as amyloid by Congo red staining. Congo red binding by the GNNQQNY peptide was observed as a shift in the absorbance maximum of Congo red upon addition of peptide fibrils (data not shown). Although the peptide fibrils bind Congo red, we did not observe green birefringence by polarized light microscopy, a classic feature of amyloid. Neither whole Sup35 fibrils nor PrD fibrils display green birefringence, according to Glover et al. (16).
Cooperative aggregation kinetics.
The in vitro conversion of amyloidogenic proteins is cooperative with a concentration-dependent lag time, suggesting a nucleation-dependent process (28, 29). We observe a cooperative structural transition of GNNQQNY by its sigmoidal turbidity curve (Fig. 2b). At higher peptide concentrations, the lag time is diminished and the turbidity is increased (data not shown).
In short, the N-terminal GNNQQNY segment of Sup35 forms amyloid similar to that of full-length Sup35. Because GNNQQNY was selected from a 17-residue region in the Sup35 sequence that is critical for [PSI+] maintenance, we suggest that this fragment participates in the ordered aggregation of full-length Sup35.
Sup35 Seeds the Aggregation of GNNQQNY.
To test whether GNNQQNY interacts specifically with Sup35, we monitored the aggregation kinetics of the peptide alone and in the presence of Sup35 (Fig. 2b). The addition of Sup35 shortens the lag time for GNNQQNY aggregation and causes a higher total concentration of aggregated material. The increase in total aggregated material cannot be accounted for by Sup35 aggregation alone. It is therefore due, at least in part, to increased GNNQQNY aggregation. We conclude that the protein is effectively seeding the aggregation of the peptide. The peptide, however, slightly inhibits rather than seeds the aggregation of the full-length Sup35 protein (data not shown). These results suggest both that this seven-residue segment, and residues beyond this seven-residue segment, are involved in the aggregation of full-length Sup35.
Microcrystals of GNNQQNY Share Many of the Properties of Amyloid Fibrils.
GNNQQNY also forms well-ordered microcrystals (Fig. 2c) with amyloid properties, including the following:
Structural transition to β-sheet.
Fourier transform IR data collected from GNNQQNY microcrystals show a strong peak at 1633.41 cm−1 (data not shown), consistent with β-sheet structure. The diagnostic peak for an anti-parallel β-sheet (between 1675–1695 cm−1) is absent, suggesting that the strands within a β-sheet are parallel.
X-ray diffraction consistent with a cross-β structure.
X-ray diffraction data collected from an aggregate of microcrystals display over 30 prominent powder rings in the range of 23- to 0.9-Å resolution (Fig. 2c). Two of the strongest reflections in the pattern occur at 4.7-Å and 9.86-Å resolution, consistent with the strong reflections observed in the cross-β diffraction pattern obtained from GNNQQNY fibrils. Additional support for a cross-β structure is detailed below.
Possible Congo red binding.
The GNNQQNY microcrystals absorb Congo red from solution, leaving the surrounding solution clear. However, the resulting x-ray diffraction pattern from the Congo-red stained crystals is not measurably different from the diffraction pattern collected before adding Congo red.
Unusual structural stability.
Unlike typical protein aggregates, amyloid deposits are structurally very stable. We attempted to dissolve the GNNQQNY microcrystals in denaturants, detergents, and solvents. Of the 10 conditions tested, only three of them completely dissolved the crystals after 5 h: (i) 4 M guanidinium hydrochloride, (ii) 0.5 M sodium hydroxide, and (iii) 100% formic acid (Eastman). Neither 5% SDS nor 4 M urea dissolved the crystals.
Characterization of Microcrystals.
We indexed the x-ray powder diffraction data from GNNQQNY (Fig. 3), assigning the microcrystals to one of the orthorhombic space groups P22121 or P2212, with unit cell dimensions a = 22.63 ± 0.02 Å, b = 39.44 ± 0.02 Å, and c = 4.81 ± 0.05 Å, with one peptide in the asymmetric unit. Also possible is space group P21 with β very nearly 90°. A 2-fold screw axis parallel to b is indicated by the absence of reflections of the form 0k0 with k = odd for k = 3, 5, and 7. Reflection (0,1,0) is obscured by the beamstop and 0k0 reflections with k>7 are superimposed on other reflections. These systematic absences rule out other orthorhombic space groups and all hexagonal space groups.
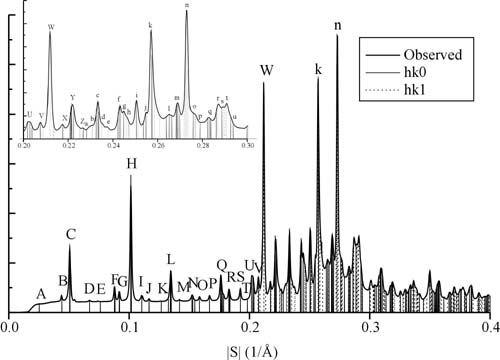
Figure 3.

Powder x-ray diffraction intensities obtained from GNNQQNY microcrystals shown as a radial distribution. Vertical lines superimposed on the pattern indicate calculated reflection positions. Peaks in the resolution range 0 to 0.227 Å−1 are labeled A–Z and those in the range 0.228 to 0.3 Å−1 are labeled a–u. See Table 1, which is published with an enlarged version of this figure as supplemental material on the PNAS web site, www.pnas.org.
Indexing of x-ray reflections with peak intensities (Fig. 3) began with hk0 reflections: A (0,1,0); B (1,0,0); D (1,2,0); E (0,3,0); G (2,1,0); I (1,4,0); J (2,3,0); K (0,5,0); M (3,2,0); S (4,3,0); and T (2,7,0). Because of overlapping reflections, indexing the strong peak at 4.7 Å (S = 0.21 Å−1) presents ambiguity. This uncertainty means the accuracy of the c unit cell dimension is less than that of the a and b dimensions. This uncertainty also leads to ambiguities in indexing hkl (l = one) reflections. Our best solution, based on minimizing residuals between calculated and observed peak positions, is shown in Fig. 3; in it we index the strong 4.72-Å reflection as (1,0,1). Our three next best solutions are obtained by indexing the 4.72-Å peak as (0,1,1) (0,2,1), and (1,1,1). The unit cell dimensions, and the assignment of the dominant component of observed peak H to reflection (0,4,0) are confirmed by electron diffraction patterns of the microcrystals. (D. L. D. Cospor and R. Diaz-Avalos, personal communication).
Density of Microcrystals and Unit Cell Contents.
The density of the GNNQQNY microcrystals was determined to be 1.39 ± 0.02 g/cm3. The number of water molecules per unit cell (14) follows directly from our measurements of the unit cell volume (V = 4.30 × 103 Å3) and crystal density (ρ = 1.39 ± 0.02 g/cm3), the molecular mass M (836.7 Da) and knowing that the liquid of crystallization is pure water. This follows from the well-known relationship, XP = (nM)/(NVρ), where XP is the weight fraction of protein in the crystals and N is Avogadro's number. Our measurements give a value for XP of 0.93, assuming initially that n = 4. The value n = 4 is consistent with the orthorhombic space groups P2212 and P22121, which demand some multiple of four peptides per unit cell. A value of n = 2 or n = 4 (but not 1, 3, or > 4) is compatible with monoclinic space groups. It can be shown that n = 2 is inconsistent with the crystal density.
Discussion
The Number of Side-Chain Hydrogen Bonds in GNNQQNY Crystals.
Our measurements lead directly to the conclusion that the number of peptide molecules per unit cell is 4 and the number of water molecules is 14 ± 3. Hence water molecules cannot saturate the many hydrogen bond-forming side chains of the peptide. Instead, side chains must form hydrogen bonds with other hydrogen bond-forming side chains of neighboring peptides, and perhaps their own peptide, creating a network of hydrogen bonds, as envisaged by Perutz et al. (30) in their proposal of the polar zipper. The striking extent to which this must be so emerges from an inventory of all hydrogen-bond donors and all acceptors that extend from the four peptides per unit cell: a total of 80 donors and 108 acceptors. The ≈14 water molecules per unit cell can hydrogen-bond to at most 28 donors and 28 acceptors. If we assume further that all backbone carbonyls and amide hydrogen bonds are engaged in forming β-sheets (see below), there remain roughly 16 each of nonhydrogen-bonded donors and acceptors. But all potential hydrogen bonds in protein crystals are found to form actual bonds (31). Thus there must be ≈16 hydrogen bonds between side chains in the unit cell, or roughly one per every other residue, in addition to backbone hydrogen bonds.
In short, the microcrystals of GNNQQNY contain no more than four water molecules per peptide, and probably fewer. The many potentially hydrogen-bonded chemical groups of the side chains of asparagine, glutamine, and tyrosine, rather than bonding to water, must form hydrogen bonds among themselves. The density of hydrogen bonds and the lack of water of solvation both probably contribute to the unusual density and cohesion of the crystals.
Analysis of the Packing of GNNQQNY in the Microcrystals.
Features of the atomic structure of the amyloid-like microcrystals (Fig. 4) emerge from the diffraction pattern, when considered in light of their high density and the known properties of β-sheets. Both our Fourier transform IR and x-ray measurements are consistent with an extended β-sheet structure for GNNQQNY in the microcrystals. The c unit cell dimension of 4.8 Å is the familiar spacing of β-strands forming a β-sheet and suggests that the strands of the sheet are roughly perpendicular to c. Furthermore, because all strands forming a β-sheet are related by the translation along c, the β-sheets must be parallel-stranded and have no twist. Such packing is permitted in each of the three possible space groups, P22121, P2212, and P21. Regardless of space group, the unit cell contains four GNNQQNY molecules, each of which participates in a separate β-sheet. In all three permitted space groups, there is a 2-fold screw axis parallel to b. This 2-fold screw requires that all four sheets pack roughly parallel to either the ac plane or the bc plane. For example, if the first two sheets were placed along the a-b diagonal, then the strands at the symmetry-generated interface would be roughly perpendicular to one another, leaving gaps between the sheets. There cannot be any such gaps in the crystal, because the solvent filling the gaps would have buoyancy inconsistent with the measured high density.
Figure 4.

Proposed packing of GNNQQNY β-strands (represented by arrows) in the unit cell of the microcrystals. The strands are extended and form parallel β-sheets. Each sheet packs against one neighboring sheet with its strands parallel to the first and one sheet with its strands antiparallel to the first. The registration of the sheets with one another along a and c is not yet known. The spacing between strands and sheets is ≈4.8 Å and ≈10 Å, respectively.
From the unit cell dimensions and diffraction data, we conclude that the β-sheets pack parallel to the ac plane, with the strands extended along a. The typical spacing between β-sheets is approximately 10 Å. With the sheets packed normal to b as shown in Fig. 4, four sheets each separated by 9.85 Å can be accommodated (4 × 9.85 Å = 39.4 Å = |b|). The x-ray powder pattern shows a strong reflection at 9.86-Å resolution, which can be indexed as (0,4,0), as expected for four nearly equally spaced sheets normal to b. Arguing for extended strands along a is the length of the a unit cell edge, essentially equal to the extended heptamer (|a| = 22.6 Å ≅ 7 × 3.25 Å = 22.7 Å, where 3.25 Å represents the repeat per residue distance expected in a parallel-stranded β-sheet) (32).
The main features of the model shown in Fig. 4 are consistent with all three possible space groups. These include: (i) parallel-stranded sheets; (ii) alternating sheet-sheet interfaces, one (the parallel interface) being between sheets with strands running the same direction and the next (the antiparallel interface) between sheets having strands running in opposite directions, and (iii) at the parallel interface, the backbone C⩵O groups in one residue point up (upward edge) and the C⩵O groups in that same residue in the other sheet point down (downward edge); at the antiparallel interface, both sheets have an upward edge or both have a downward edge.
Much of interest is not defined in the model of Fig. 4. This includes all atomic details of the sheet faces that pack against one another, including: the registration of the sheets against each other (along a and c); the precise angle of the strands to c; and the nature of the hydrogen-bonded networks between the side chains. Determining these details is now in progress by electron diffraction (R. Diaz-Avalos and D. L. D. Caspar, personal communication) and further x-ray diffraction.
What Do the GNNQQNY Amyloid Fibrils and Microcrystals Have in Common?
The GNNQQNY amyloid fibrils and microcrystals share the principal properties of amyloid, including: (i) a cooperative structural transition to β-rich structure, (ii) Congo red affinity, (iii) inordinate structural stability, and most notably (iv) an x-ray diffraction pattern consistent with the cross-β structure.
Implications of the Dry β-Sheet Model for the Amyloids, Prions, and Polyglutamine Aggregates.
A persistent puzzle about amyloids, as well as polyglutamine aggregates and the transformed or scrapie forms of prions has been their unusual stability. Our observation of the densely packed, nearly anhydrous, highly hydrogen-bonded structure for GNNQQNY, also a highly stable structure, offers an answer to this puzzle. Our proposed hydrogen-bonded sheet structure has essential features in common with Perutz et al.'s proposal of a polar zipper for polyglutamine aggregates (30), and indeed, GNNQQNY is glutamine and asparagine rich. This dry β-sheet model shares with the polar zipper model the hydrogen-bonding of backbone and side chains. But in the dry β-sheet model, there are fully formed β-sheets. This dry β-sheet model might be expected to be particularly stable because it expels most of the water molecules that ordinarily would be expected to hydrate the polar side chains of asparagine, glutamine, and tyrosine. Dunitz (33) noted that each water molecule that is localized within a solid aggregate has an associated entropy cost of up to 2 kcal/mole at 37°C. Because each Asn or Gln residue has the capacity to form hydrogen bonds with up to seven water molecules and the Tyr residues with five, the almost complete exclusion of water from the dry β-sheet could account for a major component of the stability of the dry β-sheet structure. The enthalpic contribution to water-residue hydrogen bonding would be compensated by interresidue hydrogen bonding. This dry β-sheet can accommodate polar sequences (e.g., Sup35) and apolar sequences (e.g., amyloid-β), both of which are known to form amyloid. Reopening the dry sheet must be a highly cooperative process of breaking many weak bonds, probably accounting for the unusual stability of amyloid.
Our model of the dry β-sheet also has implications for the transition to the amyloid state, including that for the prion form of Sup35 and perhaps for the scrapie form of other prions. The peptide GNNQQNY is only a small portion (seven of 685 residues) of the entire yeast prion Sup35, yet it is sufficient to form a fibrillar structure that closely resembles that for full Sup35. This result argues that the amyloid-forming nucleus of a protein need be only a short segment of the entire chain. Any in vivo or in vitro environmental condition that tends to open the native protein and expose this segment of the sequence could lead to amyloid formation and transition to the prion state. Thermal fluctuations at 37°C might well be sufficient to open the full protein and expose the segment, but for the dry sheet amyloid to form it would have to be stabilized by interaction with the same segment of other protein molecules, and dehydration of the sheet would have to take place. Our observation that full Sup35 promotes the transition to the dry sheet (Fig. 2b) confirms that interaction of the GNNQQNY segment with other Sup35 molecules is involved in the formation of amyloid. Conceivably the conversion of other prions to the transformed or scrapie state and the formation of polyglutamine aggregates in proteins such as huntingtin (34) involve the opening of the native structure to expose a peptide segment that can bond to similar segments of the same protein to form stable, dry β-sheets.
Supplementary Material
Acknowledgments
We thank Donald L. D. Caspar and Ruben Diaz-Avalos for stimulating collaboration; Maria Patino and Susan Lindquist for providing sup35 plasmid DNA; Mari Gingery for EM; Alf Bacher for Fourier transform IR data collection, Suzanna Horvath and Lynn Williams for peptide synthesis; Richard E. Marsh, Todd Yeates, Frank Rotella and Andrzej Joachimiak for help on x-ray diffraction; and the National Science Foundation, National Institutes of Health, and Department of Energy for support.
Abbreviations
- GST
glutathione S-transferase
- PrD
prion-determining domain
- EM
electron microscopy
References
- 1.Rochet J C, Lansbury P T., Jr Curr Opin Struct Biol. 2000;10:60–68. doi: 10.1016/s0959-440x(99)00049-4. [DOI] [PubMed] [Google Scholar]
- 2.Dobson C M. Trends Biochem Sci. 1999;24:329–332. doi: 10.1016/s0968-0004(99)01445-0. [DOI] [PubMed] [Google Scholar]
- 3.Koo E H, Lansbury P T, Jr, Kelly J W. Proc Natl Acad Sci USA. 1999;96:9989–9990. doi: 10.1073/pnas.96.18.9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelly J W. Curr Opin Struct Biol. 1996;6:11–17. doi: 10.1016/s0959-440x(96)80089-3. [DOI] [PubMed] [Google Scholar]
- 5.Prusiner S B. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sunde M, Serpell L C, Bartlam M, Fraser P E, Pepys M B, Blake C C. J Mol Biol. 1997;273:729–739. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- 7.Geddes A J, Parker K D, Atkins E D T, Beighton E. J Mol Biol. 1968;32:343–358. doi: 10.1016/0022-2836(68)90014-4. [DOI] [PubMed] [Google Scholar]
- 8.Sunde M, Blake C. Adv Protein Chem. 1997;50:123–159. doi: 10.1016/s0065-3233(08)60320-4. [DOI] [PubMed] [Google Scholar]
- 9.Wickner R B. Science. 1994;264:566–569. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 10.Sparrer H E, Santoso A, Szoka F C, Weissman J S. Science. 2000;289:595–599. doi: 10.1126/science.289.5479.595. [DOI] [PubMed] [Google Scholar]
- 11.Stansfield I, Tuite M F. Curr Genet. 1994;25:385–395. doi: 10.1007/BF00351776. [DOI] [PubMed] [Google Scholar]
- 12.Cox B S, Tuite M F, McLaughlin C S. Yeast. 1988;4:159–178. doi: 10.1002/yea.320040302. [DOI] [PubMed] [Google Scholar]
- 13.Patino M M, Liu J J, Glover J R, Lindquist S. Science. 1996;273:622–626. doi: 10.1126/science.273.5275.622. [DOI] [PubMed] [Google Scholar]
- 14.Ter-Avanesyan M D, Dagkesamanskaya A R, Kushnirov V V, Smirnov V N. Genetics. 1994;137:671–676. doi: 10.1093/genetics/137.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.King C Y, Tittmann P, Gross H, Gebert R, Aebi M, Wüthrich K. Proc Natl Acad Sci USA. 1997;94:6618–6622. doi: 10.1073/pnas.94.13.6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glover J R, Kowal A S, Schirmer E C, Patino M M, Liu J J, Lindquist S. Cell. 1997;89:811–819. doi: 10.1016/s0092-8674(00)80264-0. [DOI] [PubMed] [Google Scholar]
- 17.Santoso A, Chien P, Osherovich L Z, Weissman J S. Cell. 2000;100:277–288. doi: 10.1016/s0092-8674(00)81565-2. [DOI] [PubMed] [Google Scholar]
- 18.DePace A H, Santoso A, Hillner P, Weissman J S. Cell. 1998;93:1241–1252. doi: 10.1016/s0092-8674(00)81467-1. [DOI] [PubMed] [Google Scholar]
- 19.Paushkin S V, Kushnirov V V, Smirnov V N, Ter-Avanesyan M D. EMBO J. 1996;15:3127–3134. [PMC free article] [PubMed] [Google Scholar]
- 20.Clos J, Brandau S. Protein Expression Purifi. 1994;5:133–137. doi: 10.1006/prep.1994.1020. [DOI] [PubMed] [Google Scholar]
- 21.Schatz P J, Cull M G, Martin E L, Gates C M. Methods Enzymol. 1996;267:171–191. doi: 10.1016/s0076-6879(96)67012-8. [DOI] [PubMed] [Google Scholar]
- 22.Klunk W E, Pettegrew J W, Abraham D J. J Histochem Cytochem. 1989;37:1273–1281. doi: 10.1177/37.8.2666510. [DOI] [PubMed] [Google Scholar]
- 23.Low B W, Richards F M. J Am Chem Soc. 1952;74:1660–1666. [Google Scholar]
- 24.Low B W, Richards F M. J Am Chem Soc. 1954;76:2511–2518. [Google Scholar]
- 25.Ter-Avanesyan M D, Kushnirov V V, Dagkesamanskaya A R, Didichenko S A, Chernoff Y O, Inge-Vechtomov S G, Smirnov V N. Mol Microbiol. 1993;7:683–692. doi: 10.1111/j.1365-2958.1993.tb01159.x. [DOI] [PubMed] [Google Scholar]
- 26.Derkatch I L, Chernoff Y O, Kushnirov V V, Inge-Vechtomov S G, Liebman S W. Genetics. 1996;144:1375–1386. doi: 10.1093/genetics/144.4.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Booth D R, Sunde M, Bellotti V, Robinson C V, Hutchinson W L, Fraser P E, Hawkins P N, Dobson C M, Radford S E, Blake C C, Pepys M B. Nature (London) 1997;385:787–793. doi: 10.1038/385787a0. [DOI] [PubMed] [Google Scholar]
- 28.Jarrett J T, Lansbury P T., Jr Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 29.Harper J D, Lansbury P T. Annu Rev Biochem. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- 30.Perutz M F, Johnson T, Suzuki M, Finch J T. Proc Natl Acad Sci USA. 1994;92:6509–6513. [Google Scholar]
- 31.Baker E N, Hubbard R E. Progr Biophys Mol Biol. 1984;44:97–179. doi: 10.1016/0079-6107(84)90007-5. [DOI] [PubMed] [Google Scholar]
- 32.Pauling L, Corey R B. Proc Natl Acad Sci USA. 1953;39:253–256. doi: 10.1073/pnas.39.4.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dunitz J D. Science. 1994;264:670. doi: 10.1126/science.264.5159.670. [DOI] [PubMed] [Google Scholar]
- 34.Li X J. Mol Neurobiol. 2000;20:111–124. doi: 10.1007/BF02742437. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}