

Abstract
The paired-like homeodomain transcription factor Pitx2c has an essential role in patterning the left-right axis. However, neither its transcriptional targets nor the molecular mechanisms through which it exerts its patterning function are known. Here we provide evidence that the N-terminal domain of Pitx2c is important for this activity. Overexpression of the Pitx2c N-terminus in ovo randomizes the direction of heart looping, the first morphological asymmetry conserved in vertebrate embryos. In addition, the Pitx2c N-terminal domain blocks the ability of Pitx2c to synergize with Nkx2.5 to transactivate the procollagen lysyl hydroxylase (Plod-1) promoter in transient transfection assays. A five amino acid region containing leucine-41 is required for both of these effects. Our data suggest that the Pitx2c N-terminal domain competes with endogenous Pitx2c for binding to a protein interaction partner that is required for the activation of genes that direct asymmetric morphogenesis along the left-right axis.
Keywords: Pitx2c, left-right patterning, chick embryo, homeodomain, heart looping
Introduction
Over the past decade many of the molecules that direct patterning of the left-right axis have been identified and positioned with respect to one another in a developmental cascade (Levin, 2005; Hamada, 2008). Despite species-specific differences in the mechanisms that initiate left-right patterning, the downstream components of the left-right patterning cascade are evolutionarily conserved: in all species correct left-right patterning is dependent on the ability of Nodal, a member of the TGFβ superfamily of signaling molecules, to induce expression of the homeodomain transcription factor Pitx2c in the left lateral plate mesoderm. Asymmetric expression of Pitx2c on the left side of the embryo is required for the normal asymmetric organogenesis and positioning of the organs relative to the midline. Recently, several studies have identified specific morphogenetic processes required for asymmetric development that are dependent on Pitx2c, including cellular movements, changes in cellular organization and spindle orientation (Ai et al., 2006; Bajolle et al., 2006; Guioli and Lovell-Badge, 2007; Davis et al., 2008; Ishimaru et al., 2008; Rodriguez-Leon et al., 2008). However, no critical downstream transcriptional targets for left-right patterning have been identified in the lateral plate mesoderm.
Four different Pitx2 transcripts, derived from alternative promoter usage and splicing, have been described (Schweickert et al., 2000; Smidt et al., 2000; Yu et al., 2001; Cox et al., 2002). Pitx2a, Pitx2b and Pitx2c differ only in their N-termini and have been identified in chick, human, mice, zebrafish and Xenopus (Schweickert et al., 2000; Smidt et al., 2000; Yu et al., 2001; Cox et al., 2002), while the fourth isoform, PITX2D, which lacks the N-terminal domain and most of the homeodomain, has only been observed in humans (Cox et al., 2002). The unique N-termini of Pitx2a, 2b and 2c modify their ability to activate transcription in a cell-type and promoter-dependent manner (Tremblay et al., 2000a; Cox et al., 2002). In contrast, PITX2D inhibits the transactivation function of the other three isoforms (Cox et al., 2002).
During embryogenesis Pitx2 expression is observed in a large number of tissues, including the head mesenchyme, pituitary gland, heart, somites and lateral plate mesoderm on the left side of the embryo (Semina et al., 1996; Gage and Camper, 1997; Logan et al., 1998; Piedra et al., 1998; Ryan et al., 1998; St Amand et al., 1998; Yoshioka et al., 1998; Campione et al., 1999; Essner et al., 2000; Schweickert et al., 2000; Yu et al., 2001). Pitx2a transcripts are expressed only in regions exhibiting bilateral Pitx2 expression, including the head mesenchyme, somites and limb buds, while significant levels of Pitx2b have not been observed during embryogenesis. In contrast, the Pitx2c transcript is expressed asymmetrically in the left lateral plate mesoderm and its expression persists on the left side of many organs that are positioned asymmetrically with respect to the midline, including the lung, heart and gastrointestinal system (Logan et al., 1998; Piedra et al., 1998; Ryan et al., 1998; St Amand et al., 1998; Yoshioka et al., 1998; Campione et al., 1999; Essner et al., 2000; Schweickert et al., 2000; Yu et al., 2001).
In humans, mutations within the PITX2 locus lead to Axenfeld-Rieger syndrome, an autosomal dominant inherited disorder causing ocular anterior chamber, dental and umbilical anomalies (Semina et al., 1996; Amendt et al., 1998; Lines et al., 2002; Hjalt and Semina, 2005). Some individuals with Axenfeld-Rieger syndrome also have abnormal cardiac, limb and pituitary development. Similar defects are observed in a subset of mice that are heterozygous for a null allele at the Pitx2 locus (Gage et al., 1999; Kitamura et al., 1999; Lin et al., 1999; Lu et al., 1999; Suh et al., 2002; Liu et al., 2003). Mice that are homozygous null for the Pitx2 locus die in utero by embryonic day 12.5. In addition to tooth and pituitary defects, Pitx2 null mice exhibit a number of laterality defects that can be attributed to disruption of the left-right patterning cascade, including a delay in early embryonic rotation, right pulmonary isomerism, aberrant cardiac morphogenesis and failure of the ventral body wall to close (Gage et al., 1999; Kitamura et al., 1999; Lin et al., 1999; Lu et al., 1999; Liu et al., 2001; Liu et al., 2002).
Gain-of-function and loss-of-function experiments have shown that the asymmetric expression of Pitx2c in the left lateral plate mesoderm and in organ primordia is critical for asymmetric morphogenesis. Overexpression of Pitx2 on the right side of the embryo randomizes the direction of heart and gut looping (Semina et al., 1996; Logan et al., 1998; Piedra et al., 1998; Ryan et al., 1998; St Amand et al., 1998; Yoshioka et al., 1998; Campione et al., 1999; Essner et al., 2000; Schweickert et al., 2000; Yu et al., 2001). Studies with mice that were null for only the Pitx2c isoform demonstrate that asymmetric development of individual organs is dependent on distinct thresholds of Pitx2c activity (Gage et al., 1999; Liu et al., 2001), suggesting that Pitx2c-dependent morphogenetic events may be organ specific. In non-mammalian embryos, injection of antisense Pitx2c oligonucleotides in the left lateral plate mesoderm (Yu et al., 2001; Dagle et al., 2003) or overexpression of a dominant negative variant of Pitx2c (Yu et al., 2001) results in an increased incidence of leftward heart looping and/or reversed gut looping. A parallel dominant negative variant of Pitx2a was not able to affect the direction of heart looping. Given that the only difference between the Pitx2a and Pitx2c dominant negative variants is their N-terminal domains, the data suggest a specific function for the Pitx2c N-terminal domain in left-right patterning.
In this study, we have investigated the role of the Pitx2c N-terminal domain in patterning the left-right embryonic axis. We found that overexpression of the Pitx2c N-terminal domain on the left side of chick embryos led to a significant increase in the number of embryos with leftwardly looped heart tubes. Overexpression of the N-terminal domain did not affect the transactivation function of full-length wild type Pitx2c but did block its ability to cooperate with Nkx2.5 to synergistically activate the Plod-1 promoter in transient transfection assays. Using site-directed mutagenesis we determined that leucine-41 within the Pitx2c N-terminus is required for this function. Comparable mutations in the context of full-length Pitx2c did not affect its ability to transactivate target gene expression or to randomize the direction of heart looping when it was misexpressed on the right side of the embryo. Our data suggest that the overexpressed Pitx2c N-terminal inhibits the activity of endogenous Pitx2c in the left lateral plate mesoderm by competing for binding to a protein interaction partner that is critical for its left-right patterning function.
Results
Overexpression of the Pitx2c N-terminal Domain Randomizes the Direction of Heart Looping
Previous studies in chick using dominant negative variants of Pitx2 isoforms suggested that the N-terminus of Pitx2c has an important role in left-right patterning (Yu et al., 2001). In order to specifically explore this possibility, we ectopically expressed a truncated variant of Pitx2 that lacked an N-terminal domain (Pitx2ΔN-ter) or a variant that contained only the Pitx2c N-terminal domain (Pitx2cN-ter) in chick embryos (Figure 1). Pitx2ΔN-ter and Pitx2cN-ter retroviral particles were injected into the left or right side of HH stage 4 ex ovo cultured chick embryos and embryos were examined ∼24 hours later at HH stage 12 to determine if there were any effects on the direction of heart looping. As previously described, the experimental manipulations associated with ex ovo culturing of chick embryos resulted in 5-10% of the embryos having leftwardly looped hearts (Isaac et al., 1997, Linask et al., 2002, Simard et al., 2006). Ectopic expression of Pitx2ΔN-ter on the left side of the embryo did not alter the direction of heart looping (5%; n=1/18) as compared to the uninjected controls (10%; n= 11/110) (Figure1c,d; Table 2). In contrast, ectopic expression of the Pitx2c N-terminal domain on the left side of the embryo caused a significant increase in the number of embryos with abnormal leftward heart looping (27%; n=27/100; χ2 = 10.21; p<0.001) (Figure 1e,f; Table 2). Ectopic expression of the Pitx2c N-terminal domain on the right side of the embryo did not have any effect on the direction of heart looping (3% leftwardly looped hearts; n=1/28). Based on these data we hypothesized that the Pitx2c N-terminal domain randomized the direction of heart looping by antagonizing the function of the endogenous Pitx2c.
Figure 1. Ectopic expression of the Pitx2c N-terminal domain randomizes the direction of heart looping.
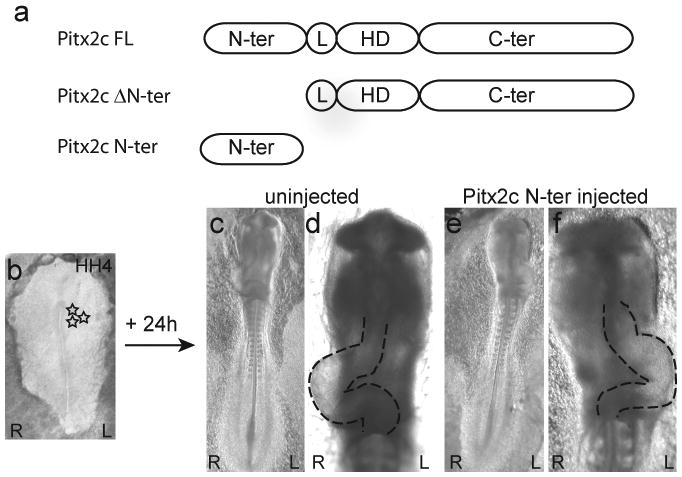
(a) Schematic representation of the full-length (Pitx2cFL) and truncated variants of Pitx2c (Pitx2c ΔN-ter and Pitx2c N-ter). The N-terminal domain is unique to each Pitx2 isoform, while the linker (L), homeodomain (HD) and C-terminal transactivation (C-ter) domains are identical in all Pitx2 isoforms. (b-f) Ventral views of embryos with left (L) and right (R) sides indicated. (b) An ex ovo cultured HH stage 4 chick embryo. The site of retroviral particle injection on the left side of the embryo is indicated by stars. (c,d) Ventral views of a HH stage 12 control embryo in which the heart is looping normally to the right side. (e,f) Ventral views of a HH stage 12 embryo that was injected with Pitx2c N-ter retroviral particles on its left side at HH stage 4. The heart has looped abnormally to the left.
Table 2. Expression of the Pitx2c N-terminus on the left side of the embryo randomizes the direction of heart looping.
| % Abnormal (Leftward) Heart Looping | |||
|---|---|---|---|
| Retroviral Construct | Injected on Left | Injected on Right | Uninjected |
| Pitx2c Δ N-ter | 5% (n=1/18) χ2 = 0.36 (N.S.) |
Not tested | 10% (n=11/110) |
| Pitx2c N-ter | 27% (n=27/100) χ2 = 10.21 (p<0.001) |
3% (n=1/28) χ2 = 1.16 (N.S.) |
|
Overexpression of the Pitx2c N-terminal Domain Does Not Affect Pitx2c's Transcriptional Activity
In order to assess the ability of the Pitx2c N-terminal domain to antagonize the transactivation function of Pitx2c we performed transient transfection assays. Pitx2 isoforms activate gene expression through binding of their homeodomains to consensus bicoid binding elements. We performed initial experiments using simple bicoid reporter constructs to ensure that the Pitx2c N-terminal domain did not have any effect on the transcriptional activity of the luciferase reporter genes in this assay system. pcDNA3.1 expression vectors expressing full-length Pitx2c or only the Pitx2c N-terminal domain were co-transfected with bicoid reporter constructs that contained one, two or three bicoid consensus binding elements cloned upstream of a luciferase reporter (Lamonerie et al., 1996) in human embryonic kidney (HEK) 293 cells. Full-length Pitx2c was able to transactivate all of the bicoid reporter constructs and showed the highest activity when 3 bicoid DNA response elements were present (Figure 2). As expected, the Pitx2c N-terminus, which does not contain DNA binding or transactivation domains, did not transactivate the bicoid reporter plasmid (Figure 2).
Figure 2. The Pitx2c N-terminal domain does not transactivate gene expression or inhibit transactivation of promoters containing bicoid response elements by full-length Pitx2c.

(a) HEK293 cells were seeded in 12-well plates and co-transfected with the Bicoid-response element (BRE) luciferase (Luc) reporter vector along with empty (pcDNA3.1-myc) or the Pitx2 N-terminal domain (N-ter) or full-length (FL) Pitx2c expression vectors. Data are derived from 2 experiments repeated in triplicate. Mean fold activation is shown. (b) CHO cells were co-transfected with the Plod-1 promoter-reporter vector and expression vectors containing full-length Pitx2c and/or the Pitx2c N-terminal domain. ‘-’ indicates empty vector. Data are derived from 3 independent experiments repeated in triplicate. Mean fold activation ± standard error is shown.
Although there are no known Pitx2c targets in the lateral plate mesoderm, transcriptional targets for Pitx2 have been identified in several tissues where it is required during embryonic development, including the developing tooth bud (Vadlamudi et al., 2005; Venugopalan et al., 2008), pituitary gland (Amendt et al., 1998; Tremblay et al., 2000b; Quentien et al., 2002) and heart (Ganga et al., 2003). We chose to use the Plod-1 promoter that contains bicoid response elements and is capable of being transactivated by the Pitx2c isoform (Hjalt et al., 2001; Ganga et al., 2003) to determine if the N-terminal domain could inhibit the transactivation function of full-length Pitx2c. The Plod-1 promoter reporter plasmid was cotransfected with expression vectors encoding full-length Pitx2c and/or the Pitx2c N-terminal domain in Chinese hamster ovary (CHO) cells. On its own, full-length Pitx2c caused an ∼20-fold increase in the luciferase activity of the Plod-1 reporter, while the Pitx2c N-terminal domain did not activate the Plod-1 reporter above background levels (Figure 2b). Co-transfection of the Pitx2c N-terminus expression vector at a 1:1 ratio did not inhibit the ability of full-length Pitx2c to transactivate the Plod-1 reporter.
Site-directed Mutagenesis of Amino Acids within the Pitx2c N-terminal Domain
The ability of the Pitx2c N-terminal domain to randomize the direction of heart looping when it is ectopically expressed on the left side of the embryo may be a consequence of its ability to compete with the endogenous Pitx2c for binding to an interaction partner that is critical for it to exert its role in left-right patterning. If this is true, we predicted that a mutating the amino acids involved in this interaction would eliminate the ability of the N-terminal domain to act as a competitive inhibitor.
To date, no mutations in the N-terminal domain of the human PITX2 locus have been identified in patients with Axenfeld-Rieger syndrome (Semina et al., 1996; Amendt et al., 2000; Saadi et al., 2001; Lines et al., 2002; Wang et al., 2003; Saadi et al., 2006; Weisschuh et al., 2006; Li et al., 2008). We assumed that since the role of Pitx2c in left-right patterning has been evolutionarily conserved, the amino acids that are critical for this function also would be conserved. Therefore, to identify the putative critical protein interaction domain within the N-terminal domain we first compared the amino acid sequence of the chick Pitx2c N-terminus to that of four other species (Figure 3a). Chick (accession # NP 990341), dog (accession # XM 545025), human (accession # NP 00316), mouse (accession # NP 001035967), zebrafish (accession # NP 571050) and Xenopus (accession # AAH73479) were aligned. The chick, dog, human and mouse sequences showed the highest degree of homology and were used primarily in selecting amino acid sequences for site-directed mutagenesis (Figure 3). Five conserved regions within the Pitx2c N-terminus were selected for mutagenesis analysis based on their degree of conservation and their location within the N-terminus. The amino acids selected for mutation were CMK (amino acids 3-5 of the chick Pitx2c isoform), CHH (amino acids 34-36), LAMAT (amino acids 41-45), KHRLE (amino acids 60-64) and SDTSS (amino acids 69-73). The codons for these blocks of 3-5 amino acids were mutated to codons for alanine. Mutations were made both in the context of expression constructs containing only the Pitx2c N-terminal domain (Pitx2cN-ter) and in the full-length Pitx2c protein (Pitx2c). For simplicity these mutations will be referred to as Pitx2ccmk, Pitx2cchh, Pitx2clamat, Pitx2ckhrle, and Pitx2csdtss, respectively. None of these mutations affected protein expression as assayed by Western blot analyses of transiently transfected HEK293 cells (data not shown).
Figure 3. Homology of vertebrate Pitx2c N-terminal domains.

(a) Alignment of Pitx2c amino acid sequences from chick, dog, human, mouse, zebrafish and Xenopus. Identities are indicated by a ‘.’ and deletions by ‘-’. Amino acids chosen for mutagenesis are shaded. (b) Table of amino acid identities in Pitx2c homologues.
Leucine-41 Is a Critical Residue for the Competitive Inhibitor Activity of the Pitx2c N-terminal Domain
Ex ovo cultured HH stage 4 embryos were injected with retroviral particles containing either wild type Pitx2cN-ter or the mutated variants on their left sides and examined 24 hours later to analyze the direction of heart looping (Table 3). The Pitx2ccmkN-ter, Pitx2cchhN-ter, Pitx2ckhrleN-ter and Pitx2csdtssN-ter mutants were able to randomize the direction of heart looping. Although the effect observed with Pitx2csdtssN-ter was not as strong as that observed with the wild type Pitx2c N-terminus, the number of embryos with leftwardly looped hearts was still significantly above background. In contrast, the Pitx2clamatN-ter retroviral particles did not randomize the direction of heart looping (Table 3): the percentage of embryos injected with Pitx2cNlamatN-ter that had leftwardly looped hearts was not significantly different from what was observed in uninjected embryos. The ability of the wild type or mutant N-terminal constructs to effect changes in the direction of heart looping did not correlate with differences in the titer of the retroviruses used for these studies. Thus, our data suggest that the inability of the Pitx2cNlamatN-ter protein to interfere with the normal process of leftward heart looping is directly due to the mutation of leucine-41, methionine-43 and threonine-45 and that the LAMAT sequence defines a critical interaction domain.
Table 3. Leucine-41 is required for the ability of ectopically expressed Pitx2c N-terminus to randomize the direction of heart looping.
| Injected Retroviral Construct | % Abnormal (Leftward) Heart Looping | χ2 (relative to uninjected) |
|---|---|---|
| Uninjected | 6.8% (n=20/291) | |
| Pitx2c N-ter | 24% (n=72/296) | 33.8 (p<0.0001) |
| Pitx2ccmk N-ter | 21.4% (n=18/84) | 15.2 (p<0.0001) |
| Pitx2cchh N-ter | 20% (n=13/65) | 10.9 (p<0.001) |
| Pitx2ckhrle N-ter | 22% (n=13/58) | 13.6 (p<0.0003) |
| Pitx2csdtss N-ter | 17% (n=25/147) | 10.9 (p<0.001) |
| Pitx2clamat N-ter | 10.7% (n=9/84) | 1.35 (N.S.) |
| Pitx2cM43A N-ter | 32% (n=10/31) | 21 (p<0.0001) |
| Pitx2cT45A N-ter | 18% (n=13/72) | 8.7 (p<0.005) |
| Pitx2cL41A N-ter | 12% (n=9/73) | 2.37 (N.S.) |
To further refine this domain, single amino acid mutations were made in which the leucine-41, methionine-43 or threonine-45 were converted to alanine (L41A, M43A and T45A, respectively). The effect of these single point mutations on left-right patterning was again assayed by injecting retroviral particles into the left side of ex ovo cultured embryos. Pitx2cM43AN-ter and Pitx2cT45AN-ter randomized the direction of heart looping in the injected embryos (Table 3). Pitx2cM43AN-ter behaved similarly to the wild type Pitx2c N-terminus while Pitx2cT45AN-ter had an intermediate effect but was not significantly different from the wild type N-terminus (χ2 = 1.3; N.S.) and was still above background (χ2 = 8.7; p<0.01). In contrast, the Pitx2cL41A mutation behaved similarly to the Pitx2clamat mutation: it significantly reduced the ability of the N-terminus to increase the incidence of leftwardly looped hearts (χ2 = 4.9; p<0.03). Thus, leucine-41 is required for the N-terminal domain to act as a competitive inhibitor of the endogenous Pitx2c's left-right patterning activity.
Pitx2c N-terminal Mutations Do Not Affect the Transactivation Function of Pitx2c
To determine if leucine-41 was essential for the transactivation function of Pitx2c, we generated expression vectors that harbored the same mutations in the context of full-length Pitx2c. We first compared the ability of all of the full-length Pitx2c mutant variants to transactivate the bicoid reporter construct with three bicoid response elements in HEK293 cells. No significant change in transactivation function was observed: all of the full-length Pitx2c mutants were able to activate the bicoid reporter at levels that were similar to wild type Pitx2c (Figure 4a).
Figure 4. Mutations within the Pitx2c N-terminal domain do not affect the ability of full-length Pitx2c transactivate Pitx2c responsive promoters.

(a) HEK293 cells were cotransfected with a bicoid promoter-reporter construct containing three bicoid response elements (BRE) cloned upstream of the pro-opiomelanocortin minimal promoter and empty vector or full-length wild type (wt) or mutated Pitx2c expression vectors. (b) CHO cells were cotransfected with the Plod-1 promoter-reporter construct and expression vectors encoding full-length wild type or mutated Pitx2c as indicated. ‘-’ indicates empty vector control. All data are derived from 3-5 independent experiments repeated in triplicate. Mean fold activation ± standard error is shown.
Although there are no known Pitx2c targets in the lateral plate mesoderm, transcriptional targets for Pitx2 have been identified in several tissues where it is required during embryonic development, including the developing tooth bud (Vadlamudi et al., 2005; Venugopalan et al., 2008), pituitary gland (Amendt et al., 1998; Tremblay et al., 2000b; Quentien et al., 2002) and heart (Ganga et al., 2003). We chose to use the Plod-1 promoter that contains bicoid response elements and is capable of being transactivated by the Pitx2c isoform (Hjalt et al., 2001; Ganga et al., 2003). The N-terminal mutations also did not affect the transactivation of the Plod-1 promoter (Figure 4b and data not shown). Only the Pitx2ckhrle mutant showed reduced ability to activate the Plod-1 promoter although co-transfection of the Pitx2ckhrle mutant with either wild-type Pitx2c or Pitx2clamat did not significantly affect their ability to transactivate the Plod-1 promoter. Thus, these data suggest that the mutations in Pitx2c N-terminus do not affect the general transactivation function of Pitx2c. However, we cannot rule out the possibility that the L41A mutation might have a specific effect in the context of a target gene(s) that is regulated by Pitx2c in the lateral plate mesoderm.
Full-length Pitx2cL41A Is Able to Randomize the Direction of Heart Looping
To determine if the L41A mutation would affect the ability of Pitx2c to randomize the direction of heart looping in gain-of-function assays, we injected ex ovo cultured HH stage 4 chick embryos with retroviral particles containing either wild type Pitx2c or the Pitx2cL41A mutation on their right sides. Misexpression of Pitx2c on the right side of the embryo randomized the direction of heart looping with 33% of the injected embryos having leftwardly looped hearts (Table 4). Misexpression of Pitx2cL41A also increased the incidence of leftward heart looping significantly (25%; χ2 = 4.98; p<0.03). The fact that Pitx2cL41A was able to randomize the direction of heart looping was not unexpected since the Pitx2a isoform, which has a completely different N-terminal domain than Pitx2c is also able to randomize the direction of heart looping when it is overexpressed on the right side of HH stage 4 chick embryos (Logan et al., 1998; Ryan et al., 1998; Yu et al., 2001). Thus, our data, in combination with previous gain- and loss-of-function studies suggest that the initial events induced by overexpression of Pitx2 isoforms on the right side of the body are distinct from the downstream molecular events that are regulated by endogenous Pitx2c in the left lateral plate mesoderm.
Table 4. Full-length Pitx2cL41A is able to randomize heart looping when overexpressed on the right side of HH stage 4 chick embryos.
| Injected Retrovirus (on right side) | % Abnormal (Leftward) Heart Looping |
|---|---|
| RCAS (empty vector) | 8% (n= 3/40) |
| Wild type Pitx2c | 33% (n=19/58) χ2 = 8.68 (p<0.005) |
| Pitx2cL41A | 25% (n=15/59) χ2 vs. RCAS = 5.15 (p<0.05) χ2 vs. WT = 0.76 (N.S.) |
Pitx2c N-terminal Domain Interferes with Synergistic Transactivation of the Plod-1 Promoter
Previous studies have shown that the Pitx2c isoform, but not the Pitx2a isoform, can synergize with Nkx2.5 to transactivate the atrial naturitic factor (ANF) and Plod-1 promoters (Ganga et al., 2003). We tested the ability of the Pitx2c N-terminal domain to antagonize the synergistic transactivation of the Plod-1 promoter by Nkx2.5 and Pitx2c. To determine if the Pitx2c N-terminal domain could inhibit the transactivation function of full-length Pitx2c and/or its ability to synergize with Nkx2.5, we cotransfected the Plod-1 promoter reporter plasmid with expression vectors encoding full-length Pitx2c and Nkx2.5 with or without the Pitx2c N-terminal domain in Chinese hamster ovary (CHO) cells. Individually, Nkx2.5 and Pitx2c caused a ∼7-fold and ∼30-fold increase in luciferase activity of the Plod-1 reporter, respectively, while the Pitx2c N-terminal domain did not activate the Plod-1 reporter above background levels (Figure 5a,b). Together Pitx2c and Nkx2.5 resulted in a ∼55-fold activation of the Plod-1 promoter (Figure 5a). Addition of the Pitx2c N-terminal domain at a 1:1 ratio had no effect on the level of transactivation by Nkx2.5. In this series of experiments we did observe a slight enhancement of the ability of Pitx2c to transactivate the Plod-1 promoter in the presence of the Pitx2c N-terminal domain. Importantly, the Pitx2c N-terminus was able to eliminate the synergistic activation of the Plod-1 promoter by Nkx2.5 and Pitx2c (Figure 5a; p<0.03). In contrast, the Pitx2clamat N-terminal domain caused only a slight but not significant decrease in the synergistic transactivation of the Plod-1 promoter by Nkx2.5 and Pitx2c (Figure 5b; p<0.05). Given that Nkx2.5 and Pitx2c do not directly interact on the Plod-1 promoter (Ganga et al., 2003), our data suggest that the amino acids leucine-41 to threonine-45 demarcate a critical interaction domain through which Pitx2c interacts with a ternary complex for synergistic gene activation. These data support previous findings that the role of the N-terminus in regulating the transactivation function of Pitx2c is promoter- and cell type-specific and suggests that the targets of Pitx2c in the lateral plate mesoderm may be uniquely regulated during embryogenesis.
Figure 5. The Pitx2c N-terminal domain eliminates synergistic transactivation of the Plod-1 promoter by Pitx2c and Nkx2.5.

CHO cells were cotransfected with the Plod-1 promoter-reporter construct, expression vectors encoding full-length wild type Pitx2c or Nkx2.5 and the wild type Pitx2c N-terminus (a) or the Pitx2clamat N-terminus (b) as indicated. All data are derived from 6 independent experiments repeated in triplicate. Mean fold activation ± standard error is shown.
Discussion
Gain-of-function and loss-of-function experiments in vertebrate embryos have demonstrated that Pitx2c is required downstream of Nodal signaling for patterning the left-right axis. However, we still have a limited understanding of how Pitx2c is exerting its effect. In the present study we have explored the role of the Pitx2c unique N-terminal domain in this process and found that misexpression of the Pitx2c N-terminus on the left side of the embryo had a biological impact. Rather than the normal rightward heart loop that was observed in uninjected embryos, a significant proportion of the injected embryos exhibited leftward heart looping without significant additional morphological features. We propose that the effect we observe is a result of the ectopically expressed Pitx2c N-terminal domain inhibiting the function of the endogenous Pitx2c by competing for binding to an essential protein interaction partner.
Data from in vitro transient transfection assays have shown that the Pitx2 N-terminus plays a role in modifying the transactivation function of the individual isoforms (Cox et al., 2002; Ganga et al., 2003; Vadlamudi et al., 2005; Amen et al., 2007). Although all three Pitx2 isoforms are capable of activating the same promoters, the degree of activation is isoform-specific (Tremblay et al., 2000a, Smidt et al., 2000). Similarly, both Pitx2a and Pitx2c were shown to synergize with MEF2A to activate the ANF promoter, but only in the LS8 oral epithelial cell line and not in NIH/3T3, Chinese hamster ovary or C2C12 cell lines (Toro et al., 2004). In contrast, Pitx2a, -2b and -2c behave quite differently with respect to their ability to cooperatively activate gene expression in conjunction with other transcription factors. For example, although each of the Pitx2 isoforms can activate the Plod-1 and ANF promoters, only Pitx2c acts synergistically with Nkx2.5 to activate these promoters (Ganga et al., 2003). Together these studies demonstrate that the N-terminal domains confer unique transactivation functions that are important for differentially regulating genes in a promoter- and cell-type specific context.
We and others have previously shown via gain-of-function experiments in chick and Xenopus that Pitx2a and Pitx2c behave equivalently when they are misexpressed on the right side of the embryo: bilateral expression of Pitx2 in the lateral plate mesoderm randomizes the direction of heart looping (Logan et al., 1998; Piedra et al., 1998; Ryan et al., 1998; St Amand et al., 1998; Yoshioka et al., 1998; Campione et al., 1999; Essner et al., 2000; Schweickert et al., 2000; Yu et al., 2001). Given the in vitro observations that the transcriptional differences in the Pitx2 isoform activity were largely qualitative rather than quantitative, it was generally accepted that ectopic expression of either Pitx2a and Pitx2c on the right side ‘simply’ activate patterns of gene expression that are normally activated by endogenous Pitx2c on the left side of the embryo. However, work in Xenopus demonstrated that ectopic expression of Pitx2c or Pitx2b on the right side of the embryo induces expression of endogenous Pitx2c (Schweickert et al., 2000) suggesting that the effect of Pitx2a or Pitx2b misexpression on left-right patterning is due to its induction of Pitx2c in the right lateral plate mesoderm. Therefore, the events that determine laterality are specifically regulated by and dependent on the Pitx2c isoform.
In contrast to gain-of-function studies, overexpression of dominant negative variants of Pitx2a and Pitx2c on the left side of the embryo do not have similar phenotypic effects (Yu et al., 2001). Dominant negative Pitx2c randomizes the direction of heart tube looping while dominant negative Pitx2a does not, despite the fact that both proteins should have identical DNA binding affinities. Thus, in the context of the left lateral plate mesoderm, Pitx2 isoforms do not behave equivalently, suggesting that the downstream events that direct left-right patterning are specifically dependent on the Pitx2c isoform.
Our current data provide further evidence that the N-terminal domain of the Pitx2c isoform plays a critical role in left-right patterning. The observation that overexpression of only its N-terminal domain was sufficient to disrupt normal patterning and randomize the direction of heart looping clearly indicates that this domain is critical for Pitx2c's left-right patterning function. In fact, our mutagenesis study allowed us to define a specific region within the Pitx2c N-terminal domain that is critical for this function. Of the five conserved regions that we chose to mutate, only the LAMAT→AAAAA significantly reduced the biological effect of overexpressing the N-terminal domain. Interestingly, the single amino acid mutation L41A behaved similarly to the three amino acid mutation indicating that the leucine must be an essential amino acid within this predicted protein interaction domain. A similarly positioned leucine was present in the other 5 species that we examined, albeit in Xenopus it was shifted one amino acid C-terminal, according to the most favorable alignment for the entire N-terminal domain. Recent data from Lamba et al., (2008) have demonstrated the existence of two variants of the Pitx2c isoform in a murine gonadotrope cell line. The shorter Pitx2c variant is proposed to arise from translation initiation at an internal methionine that is the equivalent of methionine-43 in the chick homologue. To date, we have not identified the two variants in the lateral plate mesoderm. However, the identification of leucine-41 as being essential for the competitive inhibitor activity of the Pitx2c N-terminal domain suggests that if multiple variants of the Pitx2c isoform are present in the lateral plate mesoderm, it is the longer full-length variant that is important for left-right patterning.
Overexpression of the Pitx2c N-terminal domain did not affect the transactivation function of full-length Pitx2c on either the bicoid reporter nor on the Plod-1 promoter. In fact, Pitx2c appears to activate the Plod-1 promoter with somewhat increased efficiency in the presence of the N-terminal domain. Previous studies demonstrated that there is an intramolecular interaction between the C-terminal tail and amino acids 16-38 of Pitx2a, which correspond to the linker region between the N-terminal domain and the homeodomain that is common to all isoforms (Amendt et al., 1999). Thus, it is possible that the presence of the N-terminal domain in these transfection studies allows Pitx2c to achieve a more active or stable conformation on the Plod-1 promoter by preventing the C-terminus from interacting with the linker domain.
Interestingly we found that overexpression of the Pitx2c N-terminus was able to completely abrogate the ability of full length Pitx2c to cooperate with Nkx2.5 to synergistically activate the Plod-1 promoter. Previous studies have shown that Pitx2c does not directly interact with Nkx2.5 (Ganga et al., 2003). Therefore, our data suggest that the Pitx2c N-terminal domain is competing for binding to a ‘bridging’ protein that acts to stabilize the ternary complex required for synergy between Pitx2c and Nkx2.5. The finding that overexpression of the Pitx2clamat mutant N-terminal domain did not significantly reduce the cooperative effects of Pitx2c and Nkx2.5 on the Plod-1 promoter further supports that leucine-41 to threonine-45 is part of a critical interaction domain within the N-terminus.
In the context of full length Pitx2c, neither the three amino acid mutation (LAMAT→AAAAA) nor the single point mutation significantly affected the ability of Pitx2c to transactivate the bicoid responsive promoters. Therefore, it was not surprising that misexpression of Pitx2L41A on the right side of the embryo was able to randomize the direction of heart looping in our gain-of-function study. We predict that as observed for Pitx2a and Pitx2b, ectopically expressed Pitx2cL41A induces the expression of endogenous Pitx2c in the right lateral plate mesoderm, which in turn mediates downstream events required for asymmetric morphogenesis.
Based on our data and that of previous studies, we propose that there are indeed two classes of genes that can be transcriptionally regulated by Pitx2 (Figure 6). The “isoform-independent” category can be induced by all three isoforms and would include Pitx2c itself since all Pitx2 isoforms can induce its expression in the right lateral plate mesoderm. The second class of genes that can be activated by Pitx2c are isoform-dependent and we would predict that downstream targets of Pitx2c in the left lateral plate mesoderm fall into this second category. The unique N-terminal domains may either confer a conformational change that affects the whole protein or directly interact with other transcription factors and/or cofactors in a promoter-specific manner. Our findings that overexpression of only the Pitx2c N-terminal domain can randomize the direction of heart looping and that we were able to map this ability to a specific amino acid strongly support that the Pitx2c N-terminus is directly involved in a ternary complex that regulates expression of genes that are required for patterning the left-right axis. Identification of Pitx2c-dependent protein interaction partners and transcriptional targets is the next step in understanding how it regulates development of the left-right axis and asymmetric organogenesis.
Figure 6. Model of Pitx2-dependent regulation of gene expression.

Pitx2 is proposed to regulate genes that are activated in an isoform-independent manner (shown on the left in blue) and genes that are isoform-dependent (shown in red in middle panel). Left-right patterning genes in the lateral plate mesoderm are proposed to be dependent on the Pitx2c isoform. Our model proposes that activation of genes important for left-right patterning requires interaction between the Pitx2c N-terminal domain and other transcription factors (purple molecule in middle diagram) either directly as shown or via an intermediate. Our data showed that these interactions can be blocked by overexpression of the wild type Pitx2c N-terminal domain (upper panel on right) but not by variants in which the predicted interaction domain has been disrupted as in Pitx2clamatN-ter or the Pitx2cL41AN-ter (L41A) mutants (lower panel on right).
Experimental Procedures
Isolation of Pitx2c cDNA, Site-directed Mutagenesis and Construction of Retroviral and Eukaryotic Expression Vectors
Full-length chick Pitx2c cDNA was obtained by RT-PCR of mRNA isolated from HH stage 9 to 12 chicken embryos (Hamburger and Hamilton, 1992) using the Qiagen OneStep RT-PCR kit. The sequences of the primers used for this study are listed in Table 1. The forward primer contained an EcoRI recognition site upstream of the initiation codon and the reverse primer contained a stop codon and a BamHI recognition site to permit directional cloning into the pBKS II vector. The Pitx2c N-terminal domain was obtained using the same forward primer and a unique reverse primer to generate a product that encoded the Pitx2c N-terminus upstream of the homeodomain. The PCR products were digested with EcoRI and BamHI and ligated into the pBKS II vector. The sequences of the inserts were confirmed using the Big Dye® Terminator v3.0 Cycle Sequencing Kit (Applied Biosystems) prior to subcloning them into the Slax adaptor plasmid (Hughes et al., 1987).
Table 1. Primer sequences used for RT-PCR, subcloning PCR and site-directed mutagenesis.
| Oligonucleotides for RT-PCR | |
| Pitx2c forward | 5′-CGAATTCCATATGAGTTGCATGAAGGACCC-3′ |
| Pitx2c reverse | 5′-AAGGGATCCTGCTCACACGGGCCTGTCCA-3′ |
| Pitx2c N-terminus forward | 5′-CGAATTCCATATGAGTTGCATGAAGGACCC-3′ |
| Pitx2c N-terminus reverse | 5′- AGGGATCCGCTTCTTCTTGGAGGGGT-3′ |
| Oligonucleotides for Subcloning PCR | |
| Pitx2c forward | 5′-GCGGGATCCCGCGCACTCCCTCGTCCATGAGTTGCATG-3′ |
| Pitx2c reverse | 5′-AGCTAGCGAAGCTTTCACGGGCCTGTCCACG3′ |
| Pitx2c N-terminus forward | 5′-GCGGGATCCCGCGCACTCCCTCGTCCATGAGTTGCATG-3′ |
| Pitx2c N-terminus reverse | 5′-AGCTAGCGAAGCTTTGCGGCCTCGGGGCTGG-3′ |
| Oligonucleotides for Mutagenesis (sense strand) | |
| Pitx2ccmk | 5′-CCGACCCATATGAGTGCCGCGGCGGACCCGTTAAGCCTGG-3′ |
| Pitx2cchh | 5′-CTCCTCCTCGTCCTCCGCCGCTGCCCAGCAGCCCGCG-3′ |
| Pitx2clamat | 5′-CCAGCAGCCCGCGGCGGCCGCAGCAGCAGCGCTGGCGCC-3′ |
| Pitx2ckhrle | 5′-CTGGAGGCCGCCGCAGCAGCAGCAGCAGTGCACACTATC-3′ |
| Pitx2csdtss | 5′-GTGCACACTATCGCTGCCGCCGCCGCCCCCGAGGCCGC-3′ |
| Pitx2cL41A | 5′-CCAGCAGCCCGCGGCGGCCATGGCGACG-3′ |
| Pitx2cM43A | 5′-GCCCGCGCTGGCCGCGGCGACGGCGCTG-3′ |
| Pitx2cT45A | 5′-CGCTGGCCATGGCGGCGGCGCTGGCGCCGGGG-3′ |
Site directed mutagenesis of the N-terminal domain and full-length Pitx2c inserts was performed using the QuickChange® Site-Directed Mutagenesis kit (Stratagene) in the context of the Slax adaptor plasmid. Oligonucleotides listed in Table 1 and their reverse complements were used to generate the eight mutations in the Pitx2c N-terminus that were used in this study. The mutations were confirmed by sequencing prior to subcloning the inserts into the RCASBP(A) vector (Hughes et al., 1987).
For transient transfection studies, inserts encoding full-length or N-terminal domains of wild type and mutant Pitx2c were PCR amplified using the Expand High Fidelity PCR system (Roche) and subcloned into the eukaryotic expression vector pcDNA3.1(-) (Invitrogen). Full-length Pitx2c inserts were amplified using a forward primer that contained the initiation codon and a BamHI recognition site and a reverse primer that eliminated the termination codon and permitted cloning of Pitx2c in-frame with the myc-tag (Table 1). The N-terminal domain inserts were amplified using the same forward primer and a reverse primer that contained a HindIII recognition site. Amplification products were digested with BamHI and HindIII and ligated into the pcDNA3.1(-) vector, which resulted in the addition of a myc-tag at the 3′ end of the Pitx2c insert. All clones were confirmed by sequencing and protein expression was confirmed by western blot analysis (data not shown).
In ovo Overexpression of the Pitx2c N-terminal domain (Pitx2cN-ter)
All eggs and embryos were handled according to McGill University and CCAC guidelines. Embryos were removed from the egg between HH stages 4 and 5 of embryogenesis using a filter paper carrier and cultured ventral side up on a soft agar-albumin medium (Chapman et al., 2001). Retroviral particles for full-length Pitx2c and for the Pitx2c N-terminal domain (Pitx2cN-ter) alone were prepared and titered using the chicken embryonic fibroblast cell line, CRL-12203 (ATCC), as previously described (Simard et al., 2006). Titers were as follows: Pitx2cN-ter = 7×108-1×108; Pitx2ccmkN-ter = 1×108; Pitx2cchhN-ter = 1×108-2×109; Pitx2clamatN-ter = 2×108; Pitx2ckhrleN-ter = 1×108-1×109; Pitx2csdtssN-ter = 1×109; Pitx2cL41AN-ter = 1×108-2×109; Pitx2cM43AN-ter = 2×108-1×109; and Pitx2cT45AN-ter = 1×108-1×109 retroviral particles per ml. Embryos were injected with retroviral particles in Dulbecco's Modified Eagle Medium (DMEM) high glucose media (Invitrogen) containing 1% Fast Green on their right or left side at a level that was parallel or slightly posterior to Hensen's node using a Narishige IM300 microinjector (3 times, 20 msec). On average 15-20 nl were injected per pulse. The embryos were then placed in a humidified chamber at 38.5°C for 24-30 hours until the embryos reached HH stage 11-12, at which point the status of heart looping was assessed. Embryos were photographed, dehydrated through a series of methanol washes and stored in methanol at -20°C.
Cell Culture, Transient Transfection, Luciferase and β-galactosidase Assays
HEK 293 cells were maintained in DMEM supplemented with 10% fetal bovine serum. Cells were plated at 1×105 cells/well in 12-well plates 24 hr prior to transfection. Transfections were carried out in triplicate with 0.25 μg bicoid-luciferase reporter plasmids which contained 1-3 copies of the bicoid response element cloned upstream of the pro-opiomelanocortin minimal promoter (Lamonerie et al., 1996), 0.25 μg of effector plasmid(s) and 10 ng CMV-β-galactosidase using Fugene reagent (Roche) according to the manufacturer's instructions. Transfected cells were harvested 48 hr after transfection, lysed and assayed for reporter activities. Luciferase was measured using luciferin (Pierce) in the presence of 1 mM Coenzyme A, 50 mM MgCl2 and 25 mM ATP. β-galactosidase was measured using the Galacto-Star™ reagents (Tropix). All luciferase activities were normalized to β-galactosidase activity.
CHO cells were cultured in DMEM supplemented with 5% fetal bovine serum (FBS) and penicillin/streptomycin in 60-mm dishes and fed 24 h prior to transfection. CHO cells were mixed with 2.5 μg of expression plasmid, 5 μg of reporter plasmid and 0.5 μg of CMV β-galactosidase plasmid, transfected by electroporation at 360V and 950μF, and then plated in 60-mm culture dishes and fed with 5% FBS and DMEM for 48 h as previously described (Amendt et al., 1998). Cells were then lysed and assayed for reporter activities and protein content by Bradford assay (Bio-Rad). Luciferase was measured using reagents from Promega. β-galactosidase was measured using the Galacto-Light Plus reagents (Tropix Inc.). All luciferase activities were normalized to β-galactosidase activity. The activities are shown as mean fold activation +/- SEM as compared to the Plod-1 promoter plasmid without Pitx2c FL, Pitx2c N-terminal domain (N-ter) and Nkx2.5 expression and normalized to β-galactosidase activity. All DNA was CsCl double banded.
Acknowledgments
We would like to thank Dr. J. Drouin for providing the bicoid-luciferase reporter constructs and Drs. C. Goodyer, I. Gupta and L. Jerome Majewska for helpful discussions and critical reading of the manuscript. This work was supported by grant MOP-84583 from the Canadian Institutes of Health Research to AKR and grant DE 13941 from the National Institute of Dental and Craniofacial Research to BAA. AKR is a member of the Research Institute of the McGill University Health Centre, which is supported in part by the FRSQ.
Grant Information: Canadian Institutes of Health Research, National Institutes of Health – National Institute of Dental and Craniofacial Research
References
- Ai D, Liu W, Ma L, Dong F, Lu MF, Wang D, Verzi MP, Cai C, Gage PJ, Evans S, Black BL, Brown NA, Martin JF. Pitx2 regulates cardiac left-right asymmetry by patterning second cardiac lineage-derived myocardium. Dev Biol. 2006;296:437–449. doi: 10.1016/j.ydbio.2006.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amen M, Liu X, Vadlamudi U, Elizondo G, Diamond E, Engelhardt JF, Amendt BA. PITX2 and beta-catenin interactions regulate Lef-1 isoform expression. Mol Cell Biol. 2007;27:7560–7573. doi: 10.1128/MCB.00315-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amendt BA, Semina EV, Alward WL. Rieger syndrome: a clinical, molecular, and biochemical analysis. Cell Mol Life Sci. 2000;57:1652–1666. doi: 10.1007/PL00000647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amendt BA, Sutherland LB, Russo AF. Multifunctional role of the Pitx2 homeodomain protein C-terminal tail. Mol Cell Biol. 1999;19:7001–7010. doi: 10.1128/mcb.19.10.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amendt BA, Sutherland LB, Semina EV, Russo AF. The molecular basis of Rieger syndrome. Analysis of Pitx2 homeodomain protein activities. J Biol Chem. 1998;273:20066–20072. doi: 10.1074/jbc.273.32.20066. [DOI] [PubMed] [Google Scholar]
- Bajolle F, Zaffran S, Kelly RG, Hadchouel J, Bonnet D, Brown NA, Buckingham ME. Rotation of the myocardial wall of the outflow tract is implicated in the normal positioning of the great arteries. Circ Res. 2006;98:421–428. doi: 10.1161/01.RES.0000202800.85341.6e. [DOI] [PubMed] [Google Scholar]
- Campione M, Steinbeisser H, Schweickert A, Deissler K, van Bebber F, Lowe LA, Nowotschin S, Viebahn C, Haffter P, Kuehn MR, Blum M. The homeobox gene Pitx2: mediator of asymmetric left-right signaling in vertebrate heart and gut looping. Development. 1999;126:1225–1234. doi: 10.1242/dev.126.6.1225. [DOI] [PubMed] [Google Scholar]
- Chapman SC, Collignon J, Schoenwolf GC, Lumsden A. Improved method for chick whole-embryo culture using a filter paper carrier. Dev Dyn. 2001;220:284–289. doi: 10.1002/1097-0177(20010301)220:3<284::AID-DVDY1102>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Cox CJ, Espinoza HM, McWilliams B, Chappell K, Morton L, Hjalt TA, Semina EV, Amendt BA. Differential regulation of gene expression by PITX2 isoforms. J Biol Chem. 2002;277:25001–25010. doi: 10.1074/jbc.M201737200. [DOI] [PubMed] [Google Scholar]
- Dagle JM, Sabel JL, Littig JL, Sutherland LB, Kolker SJ, Weeks DL. Pitx2c attenuation results in cardiac defects and abnormalities of intestinal orientation in developing Xenopus laevis. Dev Biol. 2003;262:268–281. doi: 10.1016/s0012-1606(03)00389-0. [DOI] [PubMed] [Google Scholar]
- Davis NM, Kurpios NA, Sun X, Gros J, Martin JF, Tabin CJ. The chirality of gut rotation derives from left-right asymmetric changes in the architecture of the dorsal mesentery. Dev Cell. 2008;15:134–145. doi: 10.1016/j.devcel.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essner JJ, Branford WW, Zhang J, Yost HJ. Mesendoderm and left-right brain, heart and gut development are differentially regulated by pitx2 isoforms. Development. 2000;127:1081–1093. doi: 10.1242/dev.127.5.1081. [DOI] [PubMed] [Google Scholar]
- Gage PJ, Camper SA. Pituitary homeobox 2, a novel member of the bicoid-related family of homeobox genes, is a potential regulator of anterior structure formation. Hum Mol Genet. 1997;6:457–464. doi: 10.1093/hmg/6.3.457. [DOI] [PubMed] [Google Scholar]
- Gage PJ, Suh H, Camper SA. Dosage requirement of Pitx2 for development of multiple organs. Development. 1999;126:4643–4651. doi: 10.1242/dev.126.20.4643. [DOI] [PubMed] [Google Scholar]
- Ganga M, Espinoza HM, Cox CJ, Morton L, Hjalt TA, Lee Y, Amendt BA. PITX2 isoform-specific regulation of atrial natriuretic factor expression: synergism and repression with Nkx2.5. J Biol Chem. 2003;278:22437–22445. doi: 10.1074/jbc.M210163200. [DOI] [PubMed] [Google Scholar]
- Guioli S, Lovell-Badge R. PITX2 controls asymmetric gonadal development in both sexes of the chick and can rescue the degeneration of the right ovary. Development. 2007;134:4199–4208. doi: 10.1242/dev.010249. [DOI] [PubMed] [Google Scholar]
- Hamada H. Breakthroughs and future challenges in left-right patterning. Dev Growth Differ. 2008;50 1:S71–78. doi: 10.1111/j.1440-169X.2008.01008.x. [DOI] [PubMed] [Google Scholar]
- Hamburger V, Hamilton HL. A series of normal stages in the development of the chick embryo. 1951. Dev Dyn. 1992;195:231–272. doi: 10.1002/aja.1001950404. [DOI] [PubMed] [Google Scholar]
- Hjalt TA, Amendt BA, Murray JC. PITX2 regulates procollagen lysyl hydroxylase (PLOD) gene expression: implications for the pathology of Rieger syndrome. J Cell Biol. 2001;152:545–552. doi: 10.1083/jcb.152.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjalt TA, Semina EV. Current molecular understanding of Axenfeld-Rieger syndrome. Expert Rev Mol Med. 2005;7:1–17. doi: 10.1017/S1462399405010082. [DOI] [PubMed] [Google Scholar]
- Hughes SH, Greenhouse JJ, Petropoulos CJ, Sutrave P. Adaptor plasmids simplify the insertion of foreign DNA into helper-independent retroviral vectors. J Virol. 1987;61:3004–3012. doi: 10.1128/jvi.61.10.3004-3012.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac A, Sargent MG, Cooke J. Control of vertebrate left-right asymmetry by a Snail-related zinc finger gene. Science. 1997;275:1301–1304. doi: 10.1126/science.275.5304.1301. [DOI] [PubMed] [Google Scholar]
- Ishimaru Y, Komatsu T, Kasahara M, Katoh-Fukui Y, Ogawa H, Toyama Y, Maekawa M, Toshimori K, Chandraratna RA, Morohashi K, Yoshioka H. Mechanism of asymmetric ovarian development in chick embryos. Development. 2008;135:677–685. doi: 10.1242/dev.012856. [DOI] [PubMed] [Google Scholar]
- Kitamura K, Miura H, Miyagawa-Tomita S, Yanazawa M, Katoh-Fukui Y, Suzuki R, Ohuchi H, Suehiro A, Motegi Y, Nakahara Y, Kondo S, Yokoyama M. Mouse Pitx2 deficiency leads to anomalies of the ventral body wall, heart, extra- and periocular mesoderm and right pulmonary isomerism. Development. 1999;126:5749–5758. doi: 10.1242/dev.126.24.5749. [DOI] [PubMed] [Google Scholar]
- Lamba P, Hjalt TA, Bernard DJ. Novel forms of Paired-like homeodomain transcription factor 2 (PITX2) generation by alternative translation initiation and mRNA splicing. BMC Mol Biol. 2008;9:31. doi: 10.1186/1471-2199-9-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamonerie T, Tremblay JJ, Lanctot C, Therrien M, Gauthier Y, Drouin J. Ptx1, a bicoid-related homeobox transcription factor involved in transcription of the pro-opiomelanocortin gene. Genes Dev. 1996;10:1284–1295. doi: 10.1101/gad.10.10.1284. [DOI] [PubMed] [Google Scholar]
- Levin M. Left-right asymmetry in embryonic development: a comprehensive review. Mech Dev. 2005;122:3–25. doi: 10.1016/j.mod.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Li D, Zhu Q, Lin H, Zhou N, Qi Y. A novel PITX2 mutation in a Chinese family with Axenfeld-Rieger syndrome. Mol Vis. 2008;14:2205–2210. [PMC free article] [PubMed] [Google Scholar]
- Lin CR, Kioussi C, O'Connell S, Briata P, Szeto D, Liu F, Izpisua-Belmonte JC, Rosenfeld MG. Pitx2 regulates lung asymmetry, cardiac positioning and pituitary and tooth morphogenesis. Nature. 1999;401:279–282. doi: 10.1038/45803. [DOI] [PubMed] [Google Scholar]
- Linask KK, Yu X, Chen Y, Han MD. Directionality of heart looping: Effects of Pitx2c misexpression on flectin asymmetry and midline structures. 2002 doi: 10.1006/dbio.2002.0661. [DOI] [PubMed] [Google Scholar]
- Lines MA, Kozlowski K, Walter MA. Molecular genetics of Axenfeld-Rieger malformations. Hum Mol Genet. 2002;11:1177–1184. doi: 10.1093/hmg/11.10.1177. [DOI] [PubMed] [Google Scholar]
- Liu C, Liu W, Lu MF, Brown NA, Martin JF. Regulation of left-right asymmetry by thresholds of Pitx2c activity. Development. 2001;128:2039–2048. doi: 10.1242/dev.128.11.2039. [DOI] [PubMed] [Google Scholar]
- Liu C, Liu W, Palie J, Lu MF, Brown NA, Martin JF. Pitx2c patterns anterior myocardium and aortic arch vessels and is required for local cell movement into atrioventricular cushions. Development. 2002;129:5081–5091. doi: 10.1242/dev.129.21.5081. [DOI] [PubMed] [Google Scholar]
- Liu W, Selever J, Lu MF, Martin JF. Genetic dissection of Pitx2 in craniofacial development uncovers new functions in branchial arch morphogenesis, late aspects of tooth morphogenesis and cell migration. Development. 2003;130:6375–6385. doi: 10.1242/dev.00849. [DOI] [PubMed] [Google Scholar]
- Logan M, Pagan-Westphal SM, Smith DM, Paganessi L, Tabin CJ. The transcription factor Pitx2 mediates situs-specific morphogenesis in response to left-right asymmetric signals. Cell. 1998;94:307–317. doi: 10.1016/s0092-8674(00)81474-9. [DOI] [PubMed] [Google Scholar]
- Lu MF, Pressman C, Dyer R, Johnson RL, Martin JF. Function of Rieger syndrome gene in left-right asymmetry and craniofacial development. Nature. 1999;401:276–278. doi: 10.1038/45797. [DOI] [PubMed] [Google Scholar]
- Piedra ME, Icardo JM, Albajar M, Rodriguez-Rey JC, Ros MA. Pitx2 participates in the late phase of the pathway controlling left-right asymmetry. Cell. 1998;94:319–324. doi: 10.1016/s0092-8674(00)81475-0. [DOI] [PubMed] [Google Scholar]
- Quentien MH, Pitoia F, Gunz G, Guillet MP, Enjalbert A, Pellegrini I. Regulation of prolactin, GH, and Pit-1 gene expression in anterior pituitary by Pitx2: An approach using Pitx2 mutants. Endocrinology. 2002;143:2839–2851. doi: 10.1210/endo.143.8.8962. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Leon J, Rodriguez Esteban C, Marti M, Santiago-Josefat B, Dubova I, Rubiralta X, Izpisua Belmonte JC. Pitx2 regulates gonad morphogenesis. Proc Natl Acad Sci U S A. 2008;105:11242–11247. doi: 10.1073/pnas.0804904105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan AK, Blumberg B, Rodriguez-Esteban C, Yonei-Tamura S, Tamura K, Tsukui T, de la Pena J, Sabbagh W, Greenwald J, Choe S, Norris DP, Robertson EJ, Evans RM, Rosenfeld MG, Izpisua Belmonte JC. Pitx2 determines left-right asymmetry of internal organs in vertebrates. Nature. 1998;394:545–551. doi: 10.1038/29004. [DOI] [PubMed] [Google Scholar]
- Saadi I, Semina EV, Amendt BA, Harris DJ, Murphy KP, Murray JC, Russo AF. Identification of a dominant negative homeodomain mutation in Rieger syndrome. J Biol Chem. 2001;276:23034–23041. doi: 10.1074/jbc.M008592200. [DOI] [PubMed] [Google Scholar]
- Saadi I, Toro R, Kuburas A, Semina E, Murray JC, Russo AF. An unusual class of PITX2 mutations in Axenfeld-Rieger syndrome. Birth Defects Res A Clin Mol Teratol. 2006;76:175–181. doi: 10.1002/bdra.20226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweickert A, Campione M, Steinbeisser H, Blum M. Pitx2 isoforms: involvement of Pitx2c but not Pitx2a or Pitx2b in vertebrate left-right asymmetry. Mech Dev. 2000;90:41–51. doi: 10.1016/s0925-4773(99)00227-0. [DOI] [PubMed] [Google Scholar]
- Semina EV, Reiter R, Leysens NJ, Alward WL, Small KW, Datson NA, Siegel-Bartelt J, Bierke-Nelson D, Bitoun P, Zabel BU, Carey JC, Murray JC. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat Genet. 1996;14:392–399. doi: 10.1038/ng1296-392. [DOI] [PubMed] [Google Scholar]
- Simard A, Di Pietro E, Young CR, Plaza S, Ryan AK. Alterations in heart looping induced by overexpression of the tight junction protein Claudin-1 are dependent on its C-terminal cytoplasmic tail. Mech Dev. 2006;123:210–227. doi: 10.1016/j.mod.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Smidt MP, Cox JJ, van Schaick HS, Coolen M, Schepers J, van der Kleij AM, Burbach JP. Analysis of three Ptx2 splice variants on transcriptional activity and differential expression pattern in the brain. J Neurochem. 2000;75:1818–1825. doi: 10.1046/j.1471-4159.2000.0751818.x. [DOI] [PubMed] [Google Scholar]
- St Amand TR, Ra J, Zhang Y, Hu Y, Baber SI, Qiu M, Chen Y. Cloning and expression pattern of chicken Pitx2: a new component in the SHH signaling pathway controlling embryonic heart looping. Biochem Biophys Res Commun. 1998;247:100–105. doi: 10.1006/bbrc.1998.8740. [DOI] [PubMed] [Google Scholar]
- Suh H, Gage PJ, Drouin J, Camper SA. Pitx2 is required at multiple stages of pituitary organogenesis: pituitary primordium formation and cell specification. Development. 2002;129:329–337. doi: 10.1242/dev.129.2.329. [DOI] [PubMed] [Google Scholar]
- Toro R, Saadi I, Kuburas A, Nemer M, Russo AF. Cell-specific activation of the atrial natriuretic factor promoter by PITX2 and MEF2A. J Biol Chem. 2004;279:52087–52094. doi: 10.1074/jbc.M404802200. [DOI] [PubMed] [Google Scholar]
- Tremblay JJ, Goodyer CG, Drouin J. Transcriptional properties of Ptx1 and Ptx2 isoforms. Neuroendocrinology. 2000a;71:277–286. doi: 10.1159/000054547. [DOI] [PubMed] [Google Scholar]
- Tremblay KD, Hoodless PA, Bikoff EK, Robertson EJ. Formation of the definitive endoderm in mouse is a Smad2-dependent process. Development. 2000b;127:3079–3090. doi: 10.1242/dev.127.14.3079. [DOI] [PubMed] [Google Scholar]
- Vadlamudi U, Espinoza HM, Ganga M, Martin DM, Liu X, Engelhardt JF, Amendt BA. PITX2, beta-catenin and LEF-1 interact to synergistically regulate the LEF-1 promoter. J Cell Sci. 2005;118:1129–1137. doi: 10.1242/jcs.01706. [DOI] [PubMed] [Google Scholar]
- Venugopalan SR, Amen MA, Wang J, Wong L, Cavender AC, D'Souza RN, Akerlund M, Brody SL, Hjalt TA, Amendt BA. Novel expression and transcriptional regulation of FoxJ1 during oro-facial morphogenesis. Hum Mol Genet. 2008;17:3643–3654. doi: 10.1093/hmg/ddn258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhao H, Zhang X, Feng H. Novel identification of a four-base-pair deletion mutation in PITX2 in a Rieger syndrome family. J Dent Res. 2003;82:1008–1012. doi: 10.1177/154405910308201214. [DOI] [PubMed] [Google Scholar]
- Weisschuh N, Dressler P, Schuettauf F, Wolf C, Wissinger B, Gramer E. Novel mutations of FOXC1 and PITX2 in patients with Axenfeld-Rieger malformations. Invest Ophthalmol Vis Sci. 2006;47:3846–3852. doi: 10.1167/iovs.06-0343. [DOI] [PubMed] [Google Scholar]
- Yoshioka H, Meno C, Koshiba K, Sugihara M, Itoh H, Ishimaru Y, Inoue T, Ohuchi H, Semina EV, Murray JC, Hamada H, Noji S. Pitx2, a bicoid-type homeobox gene, is involved in a lefty-signaling pathway in determination of left-right asymmetry. Cell. 1998;94:299–305. doi: 10.1016/s0092-8674(00)81473-7. [DOI] [PubMed] [Google Scholar]
- Yu X, St Amand TR, Wang S, Li G, Zhang Y, Hu YP, Nguyen L, Qiu MS, Chen YP. Differential expression and functional analysis of Pitx2 isoforms in regulation of heart looping in the chick. Development. 2001;128:1005–1013. doi: 10.1242/dev.128.6.1005. [DOI] [PubMed] [Google Scholar]