

Abstract
Due to the properties of silver as an antimicrobial, our research group has synthesized many different silver carbene complexes. Two new silver N-heterocyclic carbene complexes derived from 4,5-dichloroimidazole and theobromine bearing methyl benzoate substituents were synthesized by in situ carbene formation using silver acetate as the base in the reaction. The new compounds were fully characterized by several methods including NMR spectroscopy and X-ray crystallography. Preliminary antimicrobial efficacy studies against Pseudomonas aeruginosa, Staphylococcus aureus, and Escherichia coli were conducted. The results of this study demonstrated antimicrobial efficacy of the two complexes comparable to silver nitrate, showing their potential for use in the treatment of bacterial infections.
Keywords: Silver, N-heterocyclic carbene, antimicrobial, methyl benzoate, silver carbene complex
1. Introduction
Silver has a long standing use as an antimicrobial agent, particularly in modern medicine for the prevention and treatment of bacterial infections associated with severe burn wounds [1]. This is evidenced by the use of silver sulfadiazine, trademarked as Silvadene®, in burn wards worldwide since 1968 [1-3]. Silver has many properties that make it an attractive candidate for use as a medicinal agent. It is present in the human body in single digit μg/L concentrations despite no known biological function [4]. This fact indicates that the body can tolerate the presence of silver in low doses without toxic effect. In fact, its low toxicity is one of the greatest advantages of silver over other medicinally relevant metals. A cosmetic condition known as argyria, which is a blue-gray discoloration of the skin caused by extreme excess exposure of silver salts, is one of the only known side effects [5,6]. In general silver does not accumulate in the body, but rather is readily excreted, an important aspect for its use as a potential internalized therapeutic agent.
The first silver carbene was isolated by Arduengo in 1993 [7]. The synthesis involved first isolating the free carbene by using a strong base such as KH or KOtBu, followed by the reaction of the free carbene with silver triflate. Although it is possible to generate a stable free carbene in this manner, the conditions required resulted in decomposition when applied to complexes with methylene groups α to the nitrogen of the N-heterocyclic carbene (NHC) [8-10]. Due to this complication Lin and coworkers developed a method of creating metal carbenes in situ using silver oxide [11]. The synthesis began with an imidazolium cation and deprotonation was done using the silver salt as the base. These milder conditions lead to a more versatile method for making silver carbene complexes that remain stable under aerobic conditions.
Due to its promise as an antimicrobial, our research group has focused delivering silver by coupling silver (I) to NHCs to increase its stability in the body. The complexes developed by our research group have been synthesized by in situ carbene formation using silver acetate as a basic silver salt. The silver carbene complexes (SCCs) have utilized different backbones and substituents to allow for changes in water stability, water or organic solubility, and low toxicity of the degradation products. Most of these SCCs are stable to aerobic conditions, as well as in solution. They have been tested against a variety of bacterial species and have all shown effectiveness in vitro while some have also shown in vivo efficacy [12-20].
Herein we report the synthesis of two new SCCs utilizing the methyl-3-methylbenzoate group as a substituent on theobromine and 4,5-dichloroimidazole. Theobromine is a biocompatible natural product found in chocolate and 4,5-dichloroimidazole has been shown to give added stability to the silver complexes due to the electron withdrawing nature of the chloride groups of the backbone [15]. The benzoate group as a substituent serves as a site to complex targeting groups that can aid in the delivery of the compound to bacteria cells in the body. These two SCCs have been successfully synthesized, characterized, and tested for their stability and antimicrobial activity.
2. Experimental
2.1. Materials and General Methods
All reactions were carried out under aerobic conditions. 4,5-Dichloroimidazole, theobromine, iodomethane, methyl-3-bromomethyl benzoate, silver acetate, and silver nitrate were purchased from Alfa Aesar. All solvents were purchased from Fisher Scientific. All products and solvents were used as received without further purification. LB Brother Miller (DIFCO) and Bacto-agar (DIFCO) were prepared according to manufacturer’s instructions and sterilized before use. 1H and 13C NMR were collected on a Varian 500 MHz instrument. All spectra were referenced to deuterated solvents. Elemental analyses were performed by the University of Illinois microanalysis laboratory. Mass spectrometry was performed by the Ohio State University mass spectrometry and proteomics facility (compounds 1, 3, and 4) where exact mass was determined, as well as by the University of Akron mass spectrometry lab (compounds 2, 5, and 6) where nominal mass was determined. Crystals of the compounds were coated in paratone oil, mounted on a CryoLoop, and placed on a goniometer under a stream of nitrogen. Crystal structure data sets were collected on a Bruker SMART Apex CCD diffractometer with graphite-monochromated Mo Kα radiation (λ=0.71073 Å). The data was integrated using SAINT and a Bruker SHELXTL package was used for structure solution, refinement, and modeling of the crystals [21-23]. This data is represented in Table 1.
Table 1.
Crystallographic experimental data for compounds 1-6
| 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|
| C16H16N4O4 | 2(C17H19N4O4)·2(I)·O | C19H21AgN4O6·C2H4O2 | C12H10Cl2N2O2 | C13H13Cl2N2O2·I | C30H30Ag2Cl4N4O8 | |
| Lattice system | Triclinic | Triclinic | Monoclinic | Triclinic | Triclinic | Monoclinic |
| Space Group | P2(1)/c | C2/c | ||||
| a | 8.434(4) | 8.9713(15) | 6.718(4) | 7.836(4) | 5.980(3) | 47.430(20) |
| b | 8.897(4) | 11.6793(19) | 36.73(2) | 9.003(5) | 7.203(3) | 12.106(5) |
| c | 10.825(5) | 19.215(3) | 9.112(6) | 9.383(5) | 19.686(9) | 29.818(12) |
| α | 93.242(8) | 106.366(3) | 90.00 | 68.826(8) | 86.077(7) | 90.00 |
| β | 104.970(7) | 92.729(3) | 104.689(10) | 84.052(9) | 83.139(7) | 123.715(7) |
| γ | 108.245(7) | 93.658(3) | 90.00 | 81.241(8) | 66.480(7) | 90.00 |
| R1 (I>2σ(I)) | 0.0459 | 0.0824 | 0.0756 | 0.0367 | 0.0879 | 0.0770 |
| wR(F2) (I>2σ(I)) | 0.1164 | 0.1899 | 0.1673 | 0.0989 | .02260 | 0.1553 |
| R1 (all data) | 0.0554 | 0.0975 | 0.1238 | 0.0414 | 0.0925 | 0.1501 |
| wR(F2) (all data) | 0.1244 | 0.1961 | 0.1822 | 0.1036 | 0.2280 | 0.1862 |
| Goodness of fit | 1.034 | 1.233 | 0.925 | 1.054 | 1.267 | 1.013 |
2.2. Synthesis
Synthesis of 1. Theobromine (15.0 mmol, 2.70 g) was added to a 250 mL round bottom flask and 100 mL of dry DMF was added. Potassium carbonate (16.5 mmol, 2.28 g) was added and the mixture was stirred 30 minutes. Methyl-3-(bromomethyl)benzoate (16.0 mmol, 3.66 g) was added and the mixture was stirred at 120°C for 18 h. The white KBr precipitate was filtered and the volatiles were removed. The resulting oil was dissolved in 20 mL of methylene chloride and 200 mL of ether was added to yield the crude product as a cream colored solid. The solid was filtered and purified by stirring with 200 mL methanol for 30 minutes followed by vacuum filtration to isolate the desired product as soft white powder. Crystals were grown from methylene chloride. Yield: 2.35 g, 9.00 mmol, 60.0%. mp: >260°C. Anal. Calc. for C16H16N4O4: C, 58.53; H, 4.91; N, 17.06%. Found: C, 58.14; H, 4.83; N, 16.41%. ES-MS (m/z): calc., 351.1069 [M + Na]+; found, 351.1058. 1H NMR (500 MHz, d6-DMSO): δ ppm 3.42 (s, 3H, NCH3), 3.84 (s, 3H, OCH3), 3.89 (s, 3H, NCH3), 5.09 (s, 2H, NCH2N), 7.45 (t, J=7.76 Hz, 1H, CHbenzyl), 7.58 (d, J=1.51 Hz, 1H, CHbenzyl), 7.83 (d, J=7.57 Hz, 1H, CHbenzyl), 7.92 (s, 1H, CHbenzyl), 8.04 (s, 1H, NCHN). 13C NMR (500 MHz, d6-DMSO): δ 30.0 (NCH3), 33.7 (NCH3), 43.9 (NCH2), 52.2 (OCH3), 107.1 (COCN), 128.4 (Cbenzyl), 129.0 (Cbenzyl), 129.3 (Cbenzyl), 130.1 (Cbenzyl), 133.2 (Cbenzyl), 138.8 (Cbenzyl), 142.8 (NCHN), 143.3 (NCN), 151.4 (NCON), 154.8 (NCON), 166.6 (COOCH3). Crystal data for 1: C16H16N4O4, M = 328.33, triclinic, a = 8.434(4) Å, b = 8.897(4) Å, c = 10.825(5) Å, α = 93.242(8)°, β = 104.970(7)°, γ = 108.245(7)°, V = 736.9(6) Å3, T = 100(2) K, space group , Z = 2, μ(MoKα) = 0.109 mm−1, 6442 reflections measured, 3339 independent reflections (Rint = 0.0207). The final R1 values were 0.0459 (I > 2σ(I)). The final wR(F2) values were 0.1164 (I > 2σ(I)). The final R1 values were 0.0554 (all data). The final wR(F2) values were 0.1244 (all data). The goodness of fit on F2 was 1.034. Synthesis of 2. Compound 1 (8.00 mmol, 2.75 g) was placed in a pressure tube with 10 mL of DMF. Methyl iodide (15 mL) was added and the pressure tube was heated at 85 °C for 24 h. The volatiles were removed and methylene chloride was added. A small amount of white precipitate was filtered and the solution was concentrated to 10 mL. Ether was added and the sticky orange solid that remained on the flask and filter paper was dissolved in methanol. The volatiles were removed by rotary evaporation to yield the product as a shiny orange solid. Crystals were grown from acetonitrile/ethylacetate. Yield: 3.16 g, 6.51 mmol, 81.4%. mp: 146-147°C. Anal. Calc. for C17H19N4O4I: C, 43.42; H, 4.07; N, 11.91%. Found: C, 42.63; H, 4.04; N, 11.46%. ES-MS (m/z): calc., 343.1406 [M – I−]+; found, 343.2. 1H NMR (500 MHz, d6-DMSO): δ 3.76 (s, 3H, NCH3), 3.86 (s, 3H, OCH3), 4.09 (s, 3H, NCH3), 4.16 (s, 3H, NCH3), 5.16 (s, 2H, NCH2C), 7.51 (t, J=7.70 Hz, 1H, CHbenzyl), 7.63 (d, J=7.83 Hz, 1H, CHbenzyl), 7.88 (d, J=7.83 Hz, 1H, CHbenzyl), 7.98 (s, 1H, CHbenzyl), 9.34 (s, 1H, NCHN). 13C NMR (500 MHz, d6-DMSO): δ 32.0 (NCH3), 34.9 (NCH3), 36.2 (NCH3), 44.9 (NCH2), 52.7 (OCH3), 108.3 (COCN), 128.8 (Cbenzyl), 129.3(Cbenzyl), 129.4(Cbenzyl), 130.3 (Cbenzyl), 133.4 (Cbenzyl), 137.5(Cbenzyl), 140.2 (NCHN), 140.4 (NCN), 150.6 (NCON), 153.7 (NCON), 166.5 (COOCH3) ppm. Crystal data for 2: 2(C17H19N4O4)·2(I)·O, M = 956.52, triclinic, a = 8.9713(15) Å, b = 11.6793(19) Å, c = 19.215(3) Å, α = 106.366(3)°, β = 92.729(3)°, γ = 93.658(3)°, V = 1923.3(6) Å3, T = 100(2) K, space group , Z = 2, μ(MoKα) = 1.698 mm−1, 13814 reflections measured, 6729 independent reflections (Rint = 0.0363). The final R1 values were 0.0824 (I > 2σ(I)). The final wR(F2) values were 0.1899 (I > 2σ(I)). The final R1 values were 0.0975 (all data). The final wR(F2) values were 0.1961 (all data). The goodness of fit on F2 was 1.233.
Synthesis of 3. Compound 2 (1.82 mmol, 0.814 g) was dissolved in 30 mL methylene chloride. Silver acetate (3.64 mmol, 0.608 g) was added and the mixture was stirred at room temperature for 3 h. AgI precipitate was filtered and the solution was concentrated to 5 mL. Hexanes (100 ml) were added and the desired product was filtered as a white precipitate. The solid was stirred in 300 mL ether for 30 minutes to remove the acetic acid and the product was collected by vacuum filtration. Crystals were grown from methylene chloride. Yield: 0.829 g, 1.70 mmol, 93.4%. mp: 159-161°C. Anal. Calc. for AgC19H21N4O6: C, 44.72; H, 4.35; N, 10.98%. Found: C, 44.39; H, 4.40; N, 9.65%. ES-MS (m/z): calc., 343.1406 [M - AgOAc]+ and 531.0410 [M + Na]; found, 343.1272 and 531.0554. 1H NMR (500 MHz, d6-DMSO): δ 1.85 (s, 3H, OCCH3), 3.73 (s, 3H, NCH3), 3.84 (s, 3H, OCH3), 4.02 (s, 3H, NCH3), 4.15 (s, 3H, NCH3), 5.11 (s, 2H, NCH2C), 7.47 (t, J=7.80 Hz, 1H, CHbenzyl), 7.59 (d, J=6.65 Hz, 1H, CHbenzyl), 7.85 (d, J=7.76 Hz, 1H, CHbenzyl), 7.94 (s, 1H, CH benzyl). 13C NMR (500 MHz, d6-DMSO): δ 22.8 (COCH3), 32.1 (NCH3), 34.3 (NCH3), 38.8 (NCH3), 44.6 (NCH2), 52.7 (OCH3), 109.4 (COCN), 128.7 (Cbenzyl), 129.1 (Cbenzyl), 129.3 (Cbenzyl), 130.2 (Cbenzyl), 133.2 (Cbenzyl), 138.0 (Cbenzyl), 141.4 (NCN), 150.9 (NCON), 153.5 (NCON), 166.5 (COOCH3), 174.3 (NCN), 186.5 (COCH3) ppm. Crystal data for 3: C19H21AgN4O6·C2H4O2, M = 569.32, monoclinic, a = 6.718(4) Å, b = 36.73(2) Å, c = 9.112(6) Å, α = 90.00°, β = 104.689(10)°, γ = 90.00°, V = 2175(2) Å3, T = 100(2) K, space group P2(1)/c, Z = 4, μ(MoKα) = 0.985 mm−1, 18708 reflections measured, 5240 independent reflections (Rint = 0.1250). The final R1 values were 0.0756 (I > 2σ(I)). The final wR(F2) values were 0.1673 (I > 2σ(I)). The final R1 values were 0.1238 (all data). The final wR(F2) values were 0.1822 (all data). The goodness of fit on F2 was 0.925.
Synthesis of 4. 4,5-Dichloroimidazole (5.00 mmol, 0.685 g) was dissolved in 30 mL DMF in a 100 mL round bottom flask. K2CO3 (5.50 mmol, 0.760 g) was added and the mixture was stirred until dissolved. Methyl-3-(bromomethyl)benzoate (5.00 mmol, 1.14 g) was added and the mixture was stirred at reflux (120°C) overnight. The solid that was formed, presumably KBr, was filtered from the solution and the volatiles were removed from the filtrate by rotary evaporation. Dichloromethane was added and stirred resulting in a yellow solid that was filtered. The solid was dried under vacuum to yield clean product. Crystals were grown from acetonitrile. Yield: 1.25 g, 4.39 mmol, 87.7%. mp: 85-86°C. Anal. Calc. for C12H10N2O2Cl2: C, 50.55; H, 3.54; N, 9.82%. Found: C, 50.50; H, 3.53; N, 9.72%. ES-MS (m/z): calc., 307.0017 [M + Na]+; found, 306.9994. 1H NMR (500 MHz, d6-DMSO): δ 3.85 (s, 3H, OCH3), 5.34 (s, 2H, NCH2C), 7.51 (d, J=9.00 Hz, 1H, CHbenzyl), 7.55 (t, J=7.30 Hz, 1H, CHbenzyl), 7.83 (s, 1H, CHbenzyl), 7.90 (d, J=7.58 Hz, 1H, CHbenzyl), 8.03 (s, 1H, NCHN). 13C NMR (500 MHz, d6-DMSO): δ 49.0 (NCH2), 52.7 (OCH3), 112.8 (CCl), 125.3 (NCHN), 128.3 (Cbenzyl), 129.3 (Cbenzyl), 129.9(Cbenzyl), 131.0 (Cbenzyl), 132.6 (Cbenzyl), 137.0(Cbenzyl), 166.3 (COOCH3) ppm. Crystal data for 4: C12H10Cl2N2O2, M = 285.12, triclinic, a = 7.836(4) Å, b = 9.003(5) Å, c = 9.383(5) Å, α = 68.826(8)°, β = 84.052(9)°, γ = 81.241(8)°, V = 609.2(6) Å3, T = 100(2) K, space group , Z = 2, μ(MoKα) = 0.527 mm−1, 4879 reflections measured, 2438 independent reflections (Rint = 0.0238). The final R1 values were 0.0367 (I > 2σ(I)). The final wR(F2) values were 0.0989 (I > 2σ(I)). The final R1 values were 0.0414 (all data). The final wR(F2) values were 0.1036 (all data). The goodness of fit on F2 was 1.054.
Synthesis of 5. Compound 4 (1.20 mmol, 0.342 g) was placed in a 100 mL round bottom flask and dissolved in 25 mL acetonitrile. Methyl iodide (1.0 mL) was added and the mixture was stirred for 24 h at reflux. The volatiles were removed by rotary evaporation to yield a yellow solid. The yellow solid was washed with acetone and the off-white precipitate was filtered and collected. Crystals were grown from acetonitrile. Yield: 0.326 g, 0.750 mmol, 62.5%. mp: 159-160°C. Anal. Calc. for C13H13N2O2Cl2I: C, 36.56; H, 3.07; N, 6.56%. Found: C, 36.45; H, 3.11; N, 6.41%. ES-MS (m/z): calc., 299.0 [M - I]+; found, 299.0. 1H NMR (500 MHz, d6-DMSO): δ 3.84 (s, 3H, OCH3), 3.87 (s, 3H, NCH3), 5.60 (s, 2H, NCH2C), 7.61 (t, J=7.83 Hz, 1H, CHbenzyl), 7.69 (d, J=7.83 Hz, 1H, CHbenzyl), 8.00 (d, J=7.58 Hz, 1H, CHbenzyl), 8.08 (s, 1H, CHbenzyl), 9.45 (s, 1H, NCHN). 13C NMR (500 MHz, d6-DMSO): δ 35.4 (NCH3), 51.4 (NCH2), 52.9 (OCH3), 118.7 (CCl), 120.5 (CCl), 129.9 (Cbenzyl), 130.1 (Cbenzyl), 130.2 (Cbenzyl), 130.9 (Cbenzyl), 133.8 (Cbenzyl), 137.3 (Cbenzyl), 139.7 (NCHN), 166.2 (COOCH3) ppm. Crystal data for 5: C13H13Cl2N2O2·I, M = 427.05, triclinic, a = 5.980(3) Å, b = 7.203(3) Å, c = 19.686(9) Å, α = 86.077(7)°, β = 83.139(7)°, γ = 66.480(7)°, V = 771.7(6) Å3, T = 100(2) K, space group , Z = 2, μ(MoKα) = 2.423 mm−1, 6621 reflections measured, 3477 independent reflections (Rint = 0.0377). The final R1 values were 0.0879 (I > 2σ(I)). The final wR(F2) values were 0.2260 (I > 2σ(I)). The final R1 values were 0.0925 (all data). The final wR(F2) values were 0.2280 (all data). The goodness of fit on F2 was 1.264.
Synthesis of 6. Compound 5 (1.00 mmol, 0.427 g) was dissolved in 25 mL of methylene chloride in a 100 mL round bottom flask. Silver acetate (2.00 mmol, 0.338 g) was added and the mixture was stirred for 3 h at room temperature. The precipitate presumably AgI was filtered and the solution was concentrated to 2 mL. Ether was added and the product was crystallized in the freezer overnight. The resulting white fluffy solid was filtered and dried on a vacuum line to yield the clean, dry product. Crystals were grown from methylene chloride. Yield: 0.218 g, 0.470 mmol, 47.0%. Mp 92°C. Anal. Calc. for AgC15H15N2O2Cl2: C, 38.57; H, 3.45; N, 6.00%. Found: C, 38.07; H, 3.36; N, 5.81%. ES-MS (m/z): calc., 487.0 [M + Na]+ and 299.0 [M-AgOAc]+; found, 486.9 and 299.0. 1H NMR (500 MHz, d6-DMSO): δ ppm 1.79 (s, 3H, OCCH3), 3.84 (s, 3H, OCH3), 3.86 (s, 3H, NCH3), 5.55 (s, 2H, NCH2C), 7.54 (t, J=8.30 Hz, 1H, CHbenzyl), 7.59 (d, J=7.83 Hz, 1H, CHbenzyl), 7.92 (d, J=7.58, 1H, CHbenzyl), 7.95 (s, 1H, CHbenzyl). 13C NMR (500 MHz, d6-DMSO): δ 23.8 (COCH3, 38.3 (NCH3), 52.8 (COOCH3), 53.5 (NCH2), 116.9 (CCl), 118.4 (CCl), 128.5 (Cbenzyl), 129.4 (Cbenzyl), 129.9 (Cbenzyl), 130.6 (Cbenzyl), 132.6 (Cbenzyl), 136.7 (Cbenzyl), 166.3 (COOCH3), 176.2 (NCN), 181.2 (COCH3) ppm. Crystal data for 6: C30H30Ag2Cl4N4O8, M = 932.12, monoclinic, a = 47.430(20) Å, b = 12.106(5) Å, c = 29.818(12) Å, α = 90.00°, β = 123.715(7)°, γ = 90.00°, V = 14242(10) Å3, T = 100(2) K, space group C2/c, Z = 16, μ(MoKα) = 1.453 mm−1, 60807 reflections measured, 16796 independent reflections (Rint = 0.1181). The final R1 values were 0.0770 (I > 2σ(I)). The final wR(F2) values were 0.1553 (I > 2σ(I)). The final R1 values were 0.1501 (all data). The final wR(F2) values were 0.1862 (all data). The goodness of fit on F2 was 1.013.
2.3. MIC Studies
Single colonies of Pseudomonas aeruginosa, Staphylococcus aureus, and Escherichia coli were isolated from LB Agar plates and were diluted in 5.0 mL LB broth. The solutions were incubated at 37°C for 24 hours to yield bacteria solutions. Compounds 2, 3, 5, 6, and silver nitrate (0.01 mmol) were each dissolved separately in 4 mL of DMSO and diluted to 10 mL with LB broth. LB broth (2 mL) was added to each of 6 tubes. Serial dilutions were done for each compound by adding 1 mL of the dissolved compound to the first tube, taking 1 mL from tube 1 and adding to tube 2 and so on for all 6 tubes. Prepared bacteria solution (0.150 mL) was added to each tube and tubes were incubated at 37°C for 24 hours in a shaking incubator at 140 rpm. Bacteria growth was noted by turbidity of the solution in the tubes and MIC was determined by the lowest concentration lacking turbidity.
3. Results and Discussion
3.1. Synthesis and Characterization of 3
The synthesis of the theobromine derivative 3 was developed by adapting synthetic procedures previously established in our group based on the electronics of the theobromine building block [16]. The N1 nitrogen in theobromine (Figure 1a) is a good nucleophile when it is deprotonated making it a possible site for attachment of different substituents. The nucleophilicity of the N1 nitrogen is much greater than that of the N9 making it more likely to obtain single substituted products. Previous studies have taken advantage of this property where iodoethanol was reacted with theobromine to yield a hydrophilic silver complex 7 (Figure 2a). This complex has more water solubility than the methylated caffeine system 8 and has been shown to have antimicrobial properties against biosafety level 3 bacteria (Figure 2b) [16].
Figure 1.

Atom labeling of a) theobromine and b) 4,5-dichloroimidazole.
Figure 2.

Xanthene derivatives a) 7 and b) 8.
The synthesis of 3 was completed as shown in Scheme 1. The first step of the synthesis is the deprotonation of the N1 nitrogen with potassium carbonate in DMF followed by the addition of methyl-3-bromomethylbenzoate. The deprotonation produces KBr as a byproduct that falls out of solution and can be removed by filtration. The product 1 was isolated and N9 was methylated with excess CH3I. This imidazolium cation served as the carbene precursor for the synthesis of 3, which was accomplished by the addition of two equivalents of silver acetate. The first equivalent of silver acetate produces silver iodide and serves as the base to deprotonate C8 producing acetic acid and forming the carbene in situ. The second equivalent of silver acetate then reacts with the carbene to form 3.
Scheme 1.

Synthesis of 3 from theobromine.
The formation of each product was monitored by 1H and 13C NMR. The hydrogen on the N1 of theobromine has a 1H NMR signal at 11.12 ppm. After the reaction with methyl-3-bromomethylbenzoate, the signal is no longer present indicating that the substituent was attached. The signal for the C8 proton can also be monitored to assure that the substituent was not attached to N9. The theobromine proton signal for C8 is at 7.98 ppm. After the reaction with methyl-3-bromomethylbenzoate the signal only shifts slightly to 8.04 ppm, indicating that the reaction to form the imidazolium cation at N9 has not occurred. Once the methylation of 1 has taken place, we see a shift in the signal for the proton on C8 from 8.04 ppm to 9.34 ppm. This downfield shift is indicative of the imidazolium cation formation. Finally, the reaction of 2 with AgOAc causes the dissipation of the signal at 9.34 ppm and the appearance of a signal for the acetate methyl group at 1.85 ppm. The formation of 3 can further be confirmed by 13C NMR. The signal for C8 of 2 can be found at 140.4 ppm. After reaction with AgOAc the signal at 140.4 disappears and one appears further downfield at 186.5 ppm, a shift indicative of the formation of the carbene. A signal corresponding to the carbonyl carbon of the silver acetate portion can also be observed at 174.3 ppm. Further analysis of the products by elemental analysis and mass spectrometry confirmed the presence of compounds 1, 2, and 3. Crystals suitable for X-ray crystallography were grown of all the compounds and their X-ray structure was determined as another method of confirmation (Figures 3-5). The asymmetric units of compounds 2 and 3 show a molecule of water present in the crystal. This could be due to exposure to water in the atmosphere when growing the crystals or synthesizing the compounds. The Ag-C bond length in compound 3 is 2.023(6) Å and the Ag-O bond length was found to be 2.064(4) Å. The C-Ag-O bond angle of compound 3 only deviates slightly from linearity at 178.2(2) Å. Both the bond lengths and bond angles are consistent with xanthene based silver carbene complexes [16].
Figure 3.
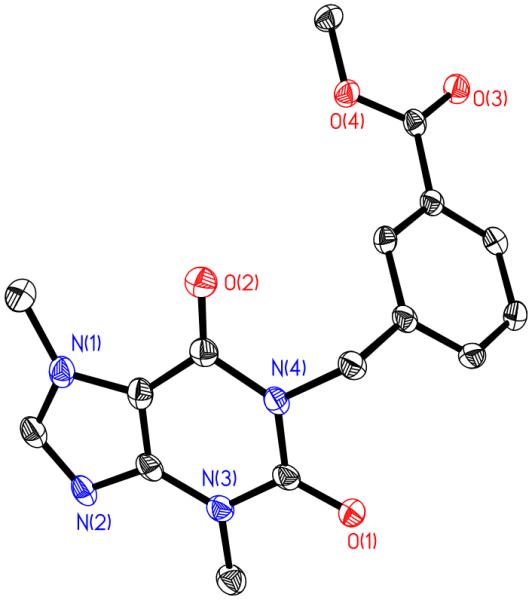
Crystal structure of 1 with thermal ellipsoids drawn at 50%.
Figure 5.

Crystal Structure of 3 with thermal ellipsoids drawn at 50%
3.2. Synthesis and Characterization of 6
One of the objectives in making different silver carbene complexes is finding ones that have enhanced stability in biological conditions. In an effort to accomplish this purpose, 4,5-dichloroimidazole was used as a starting material for several SCCs. The advantage of this precursor compared to others is the stability of its complexes in water and NaCl solutions. It has been demonstrated that compared to SCCs synthesized from 1-methylimidazole, those synthesized from 4,5-dichloroimidazole show increased stability due to the electron withdrawing chlorides in the 4 and 5 positions (Figure 1b). The electronic properties create a more stable silver complex by removing electron density from the carbene carbon, strengthening the silver-carbon bond in the complex. Due to the proven stability of these complexes, the methyl-3-methylbenzoate derivative was also synthesized. Synthesis of 6 was done by adapting previously reported procedures (Scheme 2) [12, 15]. 4,5-Dichloroimidazole was deprotonated with KOH and methyl-3-bromomethylbenzoate was added. The KBr byproduct was removed by filtration and compound 4 was isolated. This product was methylated with CH3I to obtain the imidazolium cation 5 and two equivalents of AgOAc were added to afford 6 in 87.7% yield.
Scheme 2.

Synthesis of 6 from 4,5-Dichloroimidazole.
Just as with 3, the formation of the products was monitored by 1H and 13C NMR. The hydrogen on N1 of 4,5-dichloroimidazole gives a signal at 13.33 ppm. After the addition of methyl-3-bromomethylbenzoate, that signal is no longer present indicating the complete transformation to compound 4. The signal for the proton on C2 shifts slightly from 7.70 ppm to 8.03 ppm. After methylation, the proton on C2 shifts downfield from 8.03 ppm to 9.45 ppm, indicative of imidazolium cation formation. The reaction of the cation with silver acetate causes the disappearance of the C2 proton peak and the resonance at 1.79 ppm appears representative of the acetate methyl group. The formation of 6 was also confirmed by elemental analysis, mass spectrometry, and X-ray crystallography (Figures 6-8). Crystals suitable for X-ray crystallography were grown from concentrated solvents. Just as with compounds 2 and 3, compounds 5 and 6 both have water in the asymmetric unit of the structure. This could be due to water absorbed from the atmosphere when growing the crystals or doing the reaction. The asymmetric unit of compound 6 also contained four molecules that are interacting through the silver on the silver acetate. It is possible that this could be due to the desire of silver to be 4 coordinate and the interaction with another soft silver atom makes it more stable in crystalline form. Similar to compound 3, the bond length of Ag-C in 6 was 2.060(8) Å. However, the Ag-O length of 6 was significantly larger at 2.127(7) Å than the Ag-O bond length of compound 3 (2.064(4) Å). The C-Ag-O bond angle in 6 is slightly less than linear at 177.9(3) Å.
Figure 6.

Crystal Structure of 4 with thermal ellipsoids drawn at 50%.
Figure 8.
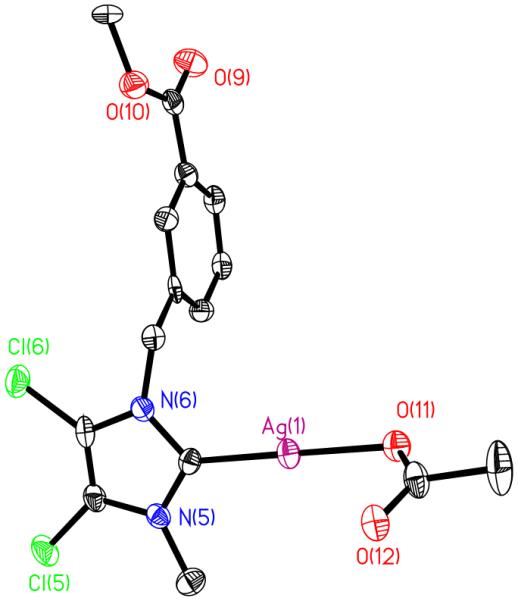
Crystal Structure of 6 with thermal ellipsoids drawn at 50%.
3.3. Antimicrobial Studies
An important characteristic of our SCCs is their antimicrobial properties. In order to determine the efficacy of the complexes, minimum inhibitory concentrations (MICs) are determined. The MIC is the minimum concentration of compound necessary to inhibit the growth of a bacterial strain. A preliminary study of silver complexes 3 and 6, and of the corresponding imidazolium cation, was done to determine their MIC. The results of these compounds were compared to that of silver nitrate which was used as a positive control based on its established antimicrobial properties [24]. The compounds were incubated for 24 hours with the non-virulent strains of Pseudomonas aeruginosa, Staphylococcus aureus, and Escherichia coli in growth media. The imidazolium cations 2 and 5 showed no antimicrobial activity at the concentrations tested (Table 2). Although there is evidence of imidazolium cations that have shown antimicrobial activity [25,26], the results from the study of 2 and 5 is consistent with studies on other SCCs synthesized in our research group in which no antimicrobial activity is observed [12,15,16]. The silver complexes, however, showed inhibition of bacterial growth with MICs of 64.0 μg/mL (126 nmol/mL) and 59.0 μg/mL (136 nmol/mL) for 3 and 6 respectively. These values were comparable to the MIC for silver nitrate (42.5 μg/mL, 250 nmol/mL), indicating that these new silver complexes are just as effective. Further studies are necessary to determine the efficacy of these compounds compared to other SCCs and their effectiveness against more virulent strains, but preliminary results demonstrate the potential of 3 and 6 in the treatment of bacterial infections.
Table 2.
MIC data (μg/mL and nmol/mL) for 3 and 6 and their corresponding imidazolium cations against bacteria strains
| Compound | P. Aerugionosa | S. aureus | E. coli | |||
|---|---|---|---|---|---|---|
| μg/mL | nmol/mL | μg/mL | nmol/mL | μg/mL | nmol/mL | |
| 2 | >235 | >500 | >235 | >500 | >235 | >500 |
| 3 | 64.0 | 126 | 64.0 | 126 | 64.0 | 126 |
| 5 | >215 | >503 | >215 | >503 | >215 | >503 |
| 6 | 59.0 | 136 | 59.0 | 136 | 59.0 | 136 |
| AgNO3 | 42.5 | 250 | 42.5 | 250 | 42.5 | 250 |
4. Conclusion
Two new silver carbene complexes were successfully synthesized and characterized. The results of the preliminary MIC studies conducted on these complexes showed concentrations comparable to silver nitrate, a proven antimicrobial agent. This demonstrates the potential of these complexes as agents in the treatment of bacterial infections. The benzoate substituent on these compounds could further be reacted with targeting moieties to allow for targeted delivery of the compounds to bacterial infections. Due to their low water solubility it may also be possible to encapsulate these compounds into nanoparticles for systemic delivery. Further studies are needed to investigate these two possibilities.
Figure 4.

Crystal Structure of the cation of 2 with thermal ellipsoids drawn at 50%.
Figure 7.

Crystal Structure of 5 with thermal ellipsoids drawn at 50%.
5. Acknowledgments
This work was supported by The University of Akron, The University of Akron Center for Silver Therapeutics Research, the National Institute of Allergies and Infectious Diseases (1 R01 A106785601) and The National Institute of General Medical Sciences (1 R01 GM86895-01). We wish to thank the Ohio Board of Regents and the National Science Foundation for funds used to purchase the 500 MHz NMR instrument (CHE-0341701 and DMR-0414599) and the Bruker-Nonius Apex CCD X-ray diffractometer (CHE-0116041) used in this research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Melaiye A, Youngs WJ. Expert Opin. Ther. Pat. 2005;14:125–130. [Google Scholar]
- [2].Fox CL. Arch. Surg. 1968;96:184–188. doi: 10.1001/archsurg.1968.01330200022004. [DOI] [PubMed] [Google Scholar]
- [3].Fox CL. U.S. Patent 3,761,590. 1970 May 18;
- [4].Davies RL, Etris SF. Catal. Today. 1997;36:107–114. [Google Scholar]
- [5].Fisher NM, Marsh E, Lazova R. J. Am. Acad. Dermatol. 2003;47:730–732. doi: 10.1067/s0190-9622(02)61574-9. [DOI] [PubMed] [Google Scholar]
- [6].White JML, Powell AM, Brady K, Russel-Jones R. Clin. Exp. Dermatol. 2003;28:254–256. doi: 10.1046/j.1365-2230.2003.01214.x. [DOI] [PubMed] [Google Scholar]
- [7].Arduengo AJ, Dias HVR, Calabrese JC, Davidson F. Organometallics. 1993;12:3405. [Google Scholar]
- [8].Guerret O, Sole S, Gornitzka H, Trinquier G, Bertrand G. J. Organomet. Chem. 2000;600:112. [Google Scholar]
- [9].Caballero A, Diez-Barra E, Jalon FA, Merino S, Tejeda J. J. Organomet. Chem. 2001;617-618:395. [Google Scholar]
- [10].Caballero A, Diez-Barra E, Jalon FA, Merino S, Rodriguez AM, Tejeda J. J. Organomet. Chem. 2001;627:263. [Google Scholar]
- [11].Wang HMJ, Lin IJB. Organometallics. 1998;17:972. [Google Scholar]
- [12].Kascatan-Nebioglu A, Melaiye A, Hindi KM, Durmus S, Panzner MJ, Hogue LA, Mallett RJ, Hovis CE, Coughenour M, Crosby SD, Milsted A, Ely DL, Tessier CA, Cannon CL, Youngs WJ. J. Med. Chem. 2006;49:6811–6818. doi: 10.1021/jm060711t. [DOI] [PubMed] [Google Scholar]
- [13].Nebioglu A, Panzner MJ, Tessier CA, Cannon CL, Youngs WJ. Coord. Chem. Rev. 2007;251:884–895. [Google Scholar]
- [14].Youngs WJ, Tessier CA, Garrison J, Quezada C, Melaiye A, Panzner M, Durmus S. U.S. Pat. Appl. Publ. 2007:45. [Google Scholar]
- [15].Hindi KM, Siciliano TJ, Durmus S, Panzner MJ, Medvetz DA, Reddy VD, Hogue LA, Hovis CE, Hilliard JK, Mallet RJ, Tessier CA, Cannon CL, Youngs WJ. J. Med. Chem. 2008;51:1577–1583. doi: 10.1021/jm0708679. [DOI] [PubMed] [Google Scholar]
- [16].Panzner MJ, Hindi KM, Wright BD, Taylor JB, Han DS, Youngs WJ, Cannon CL. Dalton Trans. 2009:7308–7313. doi: 10.1039/b907726j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Patil S, Claffey J, Deally A, Hogan M, Gleeson B, Méndez LMM, Muller-Bunz H, Paradisi F, Tacke M. Eur. J. Inorg. Chem. 2010;7:1020–1031. [Google Scholar]
- [18].Cannon CL, Hogue LA, Vajravelu RK, Capps GH, Ibricevic A, Hindi KM, Kascatan-Nebioglu A, Walter MJ, Brody SL, Youngs WJ. Antimicrob. Agents Chemother. 2009;53:3285–3293. doi: 10.1128/AAC.00314-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hindi KM, Panzner MJ, Tessier CA, Cannon CL, Youngs WJ. Chem. Rev. 2009;109:3859–3884. doi: 10.1021/cr800500u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ray S, Mohan R, Singh JK, Samantaray MK, Shaikh MM, Panda D, Ghosh P. J. Am. Chem. Soc. 2007;129:15042–15053. doi: 10.1021/ja075889z. [DOI] [PubMed] [Google Scholar]
- [21].Bruker . SMART, SAINT. Bruker AXS Inc.; Madison, Wisconsin, USA: 2007. [Google Scholar]
- [22].Bruker . SADABS. Bruker AXS Inc.; Madison, Wisconsin, USA: 2001. [Google Scholar]
- [23].Sheldrick GM. Acta. Cryst. 2008;A64:112–122. [Google Scholar]
- [24].Lansdown AB, Williams A, Chandler S, Benfield S. J. Wound Care. 2005;14:155–160. doi: 10.12968/jowc.2005.14.4.26762. [DOI] [PubMed] [Google Scholar]
- [25].Pernak J, Sobaszkiewicz K, Foksowicz-Flaczyk J. Chem. Eur. J. 2004;10:3579–3585. doi: 10.1002/chem.200400075. [DOI] [PubMed] [Google Scholar]
- [26].Pernak J, Kalewska J, Ksycinska H, Cybulski J. Eur. J. Med. Chem. 2001;36:899–907. [PubMed] [Google Scholar]