

Abstract
In caudal regions of the diencephalon, sonic hedgehog (Shh) is expressed in the ventral midline of prosomeres 1-3 (p1-p3), which underlie the pretectum, thalamus and prethalamus, respectively. Shh is also expressed in the zona limitans intrathalamica (zli), a dorsally projecting spike that forms at the p2-p3 boundary. The presence of two Shh signaling centers in the thalamus has made it difficult to determine the specific roles of either one in regional patterning and neuronal fate specification. To investigate the requirement of Shh from a focal source of expression in the ventral midline of the diencephalon, we used a newly generated mouse line carrying a targeted deletion of the 525 bp intronic sequence mediating Shh brain enhancer-1 (SBE1) activity. In SBE1 mutant mice, Shh transcription was initiated but not maintained in the ventral midline of the rostral midbrain and caudal diencephalon, yet expression in the zli was unaffected. In the absence of ventral midline Shh, rostral thalamic progenitors (pTH-R) adopted the molecular profile of a more caudal thalamic subtype (pTH-C). Surprisingly, despite their early mis-specification, neurons derived from the pTH-R domain continued to migrate to their proper thalamic nucleus, extended axons along their normal trajectory and expressed some, but not all, of their terminal differentiation markers. Our results, and those of others, suggest a model whereby Shh signaling from distinct spatial and temporal domains in the diencephalon exhibits unique and overlapping functions in the development of discrete classes of thalamic interneurons.
Keywords: Shh, Enhancer, Diencephalon, Thalamus, Mouse
INTRODUCTION
The vertebrate central nervous system is organized with great precision into discrete, yet interconnected, structural and functional units that control animal behavior. Early events in the regionalization of the CNS include its subdivision along the dorsoventral (d/v) and anteroposterior (a/p) axes. This is achieved, in large part, by the restricted exposure of neural progenitors to secreted ligands that are expressed initially from extrinsic signaling centers situated at key positions along the neural tube (Jessell, 2000). The axial mesoderm, including the notochord and prechordal plate, is one such polarizing tissue that serves as a primary source of the secreted morphogen Shh, which imparts identity to neurons in the ventral half of the neural tube. Functional studies in the mouse and chicken indicate that the notochord source of Shh is necessary and sufficient for the specification of the majority of ventral neuronal subtypes in the spinal cord, hindbrain and midbrain, whereas Shh emanating from the prechordal plate is required for ventral forebrain induction, including the separation of the cerebral hemispheres (Fucillo et al., 2006).
Shh is also a target of its own expression, as its activation in the ventral midline of the CNS is dependent on high levels of Shh signaling from the underlying axial mesoderm (Echelard et al., 1993; Ericson et al., 1996). However, owing to the severity of the CNS defects in Shh–/– mouse embryos, the full extent of Shh function from this secondary signaling source has yet to be fully realized (Chiang et al., 1996). The availability of mice carrying conditional mutations in Shh and downstream effectors of the Shh signaling cascade has been instrumental in uncovering some of the intrinsic roles of Shh within the ventral midline of the spinal cord, hindbrain and midbrain. These include well-characterized properties of Shh, such as the propagation and specification of distinct ventral neuronal and glial subtypes, as well as newly identified roles in neurogenic repression and axon guidance (Hynes et al., 1995; Ye et al., 1998; Ding et al., 1998; Matise et al., 1998; Charron et al., 2003; Bourikas et al., 2005; Andersson et al., 2006; Blaess et al., 2006; Bayly et al., 2007; Joksimovic et al., 2009).
Although significant advances have been made in elucidating the requirements of Shh signaling in posterior regions of the CNS, it is only recently that similar progress has been described for the diencephalon (Scholpp and Lumsden, 2010). In caudal regions of the diencephalon, Shh expression is localized to the ventral midline of prosomeres 1-3 (p1-p3), which underlie the pretectum, thalamus and prethalamus, respectively (Puelles and Rubenstein, 2003). Shh is also expressed in a dorsally projecting spike, the zona limitans intrathalamica (zli), that forms at the p2-p3 boundary. The exposure of the thalamus to two Shh signaling centers has made it difficult to reconcile the specific roles of either one in regional patterning and/or neuronal fate specification (Hashimoto-Torii et al., 2003; Kiecker and Lumsden, 2004; Vieira et al., 2005; Szabó et al., 2009; Vue et al., 2009).
The positioning of the thalamus between the mesencephalon and telencephalon is crucial to an important aspect of its principal function: to process and relay sensory and motor information to and from the cerebral cortex (Jones, 2007). Other functions of the thalamus include the regulation of consciousness, sleep and awareness. The thalamus is organized into five anatomical regions that are further divided into discrete nuclear subgroups (Jones, 2007). The neurons contributing to thalamic nuclei are derived from at least two distinct progenitor domains. The caudal population of thalamic progenitors, pTH-C, is the larger of the two groups and gives rise to all thalamic nuclei that relay sensory information from the periphery to primary sensory regions of the neocortex via thalamocortical axons (TCAs) (Vue et al., 2007). The rostral population of thalamic progenitors, pTH-R, consists of a narrow band of cells sandwiched between pTH-C and the zli. Thalamic neurons derived from pTH-R progenitors are thought to contribute to two dorsolaterally positioned thalamic nuclei with incompletely understood functions: the ventrolateral geniculate nucleus (vLG) and the intergeniculate leaflet (IGL), neither of which projects axons to the cortex (Horowitz et al., 2004; Morin and Blanchard, 2005; Vue et al., 2007; Vue et al., 2009; Jones, 2007).
The dependency of thalamic development on Shh appears to be regulated temporally. Early sources of Shh from the prechordal plate and/or ventral midline are required for the proliferation and survival of diencephalic precursors, whereas at later stages, a gradient of Shh signaling from the ventral midline and/or zli promotes thalamic regionalization, neuronal progenitor identity and nucleogenesis (Ishibashi and McMahon, 2002; Hashimoto-Torii et al., 2003; Kiecker and Lumsden, 2004; Vieira et al., 2005; Szabó et al., 2009; Vue et al., 2009). Conditional mouse mutants that either block the production of, or the response to, Shh throughout the thalamic primordium show a profound deficit in pTH-R and rostral pTH-C progenitor identities and their postmitotic derivatives (Szabó et al., 2009; Vue et al., 2009).
To investigate the requirement of Shh from a focal source of expression in the ventral midline of the diencephalon, we took advantage of a newly generated mouse line carrying a targeted deletion of the 525 bp intronic sequence mediating Shh brain enhancer-1 (SBE1) activity. Shh transcription was initiated, but not maintained, in the ventral midline of the rostral midbrain and caudal diencephalon of SBE1 mutants, yet expression in the zli remained intact. In the absence of ventral midline Shh, pTH-R progenitors adopted the molecular profile of pTH-C. Genetic labeling experiments indicated that neurons contributing to the vLG and IGL are dually derived from thalamic (pTH-R) and prethalamic progenitors. Surprisingly, despite their early mis-specification, pTH-R-derived neurons continued to migrate to the vLG, extended axons along their normal caudal trajectory and expressed a subset of their terminal differentiation markers. Our results, and those of others, suggest that Shh signaling from both the ventral midline and zli exhibits unique and overlapping functions in the development of thalamic interneurons.
MATERIALS AND METHODS
Generation of SBE1 mutant mice
A floxed SBE1 allele was generated by introducing loxP sites on either side of the SBE1 sequence by homologous recombination in embryonic stem (ES) cells. The design of the SBE1 targeting construct is depicted in Fig. S1A in the supplementary material. Upon successful germ line transmission, ShhfloxSBE1/+ mice were crossed with the EIIa-Cre line (Jackson Laboratory, Bar Harbor, ME, USA) in order to recombine out SBE1 and the Neo cassette. Deletion of SBE1 was confirmed by Southern blot (see Fig. S1B in the supplementary material). A PCR assay was subsequently developed to detect alleles corresponding to wild type (700 bp) and ΔSBE1 (186 bp) using primers 5′-TGGACCTTAAGTCTGCGCTGAAC-3′ and 5′-CTCCCTATTTGCTCAGGTGCAAAC-3′ under the following conditions: 94°C for 30 seconds, 60°C for 1 minute, 72°C for 1 minute for 35 cycles followed by a 72°C extension for 5 minutes.
Mouse lines
The Nkx2.2-cre and Tal1(Scl)-creER transgenic lines were kindly provided by M. Matise (UMDNJ, Piscataway, NJ, USA) and J. van Eekelen (University of Western Australia, Subiaco, Australia) (Bradley et al., 2007), respectively. The DLX-cre-IRES-eGFP and ccEGFP lines were generously provided by K. Campbell (CCHMC, Cincinnati, OH, USA) (Stenman et al., 2003; Nakamura et al., 2006). Shh+/– mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). ShhP1 mice were described previously (Riccomagno et al., 2002). Gsx1–/– (Li et al., 1996) and Ascl1–/– (Guillemot et al., 1993) embryos were provided by K. Campbell. Nkx2.2–/– embryos (Sussel et al., 1998) were provided by L. Sussel (Columbia University, New York, NY, USA). ShhΔSBE1/ΔSBE1 and Shh+/– mice were maintained on outbred (CD1) and inbred (C57BL/6J) backgrounds.
Whole-mount and section in situ hybridization
Whole-mount in situ hybridization was performed essentially as described (Matise et al., 1998). For gene expression studies at embryonic day (E) 12.5, heads were bisected sagitally along the midline prior to initiating the hybridization protocol. At least four brain halves were stained for any given probe. Section in situ hybridization was performed as described (Nissim et al., 2007). Embryos were fixed overnight in 4% paraformaldehyde at 4°C, washed three times in PBS, sunk in 30% sucrose in PBS at 4°C, embedded in O.C.T. (optimal cutting temperature embedding medium), quick frozen on dry ice and cryosectioned at 20 μm.
Immunohistochemistry
Primary antibodies used for immunohistochemistry were as follows: anti-GFP (1:1000; Molecular Probes); anti-Shh (5E1; 1:100; Developmental Studies Hybridoma Bank); anti-panDlx (1:250; Jhumku Kohtz, Children's Memorial Research Center, Northwestern University); anti-Nkx2.2 (74.5A5; 1:100; Developmental Studies Hybridoma Bank); anti-Sox2 (1:2000; Chemicon) and anti-Internexin (1:200; Millipore). Detection of primary antibodies was achieved using Cy3- (Jackson ImmunoResearch Laboratories) or Alexa 488- (Molecular Probes) conjugated goat anti-mouse or anti-rabbit secondary antibodies. For double labeling of mRNA and protein, antibody staining was performed on sections after completion of the in situ hybridization protocol.
Quantification of cell and axon number
At E12.5, Nkx2.2+ neurons were counted from every third coronal section of wild-type and SBE1 mutant brains. For quantification of vLG neurons at E16.5, every fourth section was double stained with pairs of antibodies against Sox2 and Nkx2.2, or GFP and Nkx2.2. Dlx+ cells were counted at a single axial level. For axon counting, a vertical line was overlaid onto the image and only those axons that crossed the vertical line were counted. To reduce sampling bias, the vertical line was positioned at the same distance from the dLG/vLG border on each section. Cell and axon counts were performed on three independent wild-type and ShhSBE1/– mutant embryos.
RESULTS
SBE1 is required for the maintenance of Shh expression in the ventral midline of the rostral midbrain and caudal diencephalon
We previously identified six enhancers distributed over 500 kb surrounding the Shh locus that regulate Shh-like expression in a regionalized manner along the a/p axis of the developing mouse CNS (Epstein et al., 1999; Jeong et al., 2006). Among these, Shh Brain Enhancer 1 (SBE1) was sufficient to direct the transcription of a reporter gene to the ventral midbrain, caudal diencephalon and zli in a pattern fitting with the spatial and temporal profile of the endogenous Shh gene. Furthermore, in the context of a Shh BAC transgene, SBE1 was deemed necessary for the elaboration of this pattern of expression (Jeong et al., 2006).
To evaluate the requirement of SBE1 in regulating the endogenous pattern of Shh transcription, we generated mice carrying a targeted deletion of the 525 bp DNA sequence in the Shh intron 2 that mediates SBE1 activity (see Fig. S1 in the supplementary material). Embryos homozygous for the SBE1 deletion (ShhΔSBE1/ΔSBE1) showed a normal pattern of Shh expression in the presumptive midbrain at E8.5 (8-10 somites), coinciding with the initial site of Shh transcription in the CNS (Fig. 1A,E). However, at E10.0, Shh expression started to fade in ShhΔSBE1/ΔSBE1 embryos and by E10.5 there was a highly reproducible gap in Shh staining in the rostral midbrain and caudal diencephalon, but not other Shh-expressing tissues, including the zli (Fig. 1B,F; data not shown). The reduction in Shh transcription persisted throughout later developmental stages in ShhΔSBE1/ΔSBE1 embryos and differed both temporally and spatially from the domain of Shh expression within the ventral midbrain that normally becomes repressed after E11.5 (Fig. 1C,D,G,H). Despite the lower levels of Shh, no gross morphological defects were observed in the brains of ShhΔSBE1/ΔSBE1 mice, which were born in normal Mendelian ratios and showed no differences in viability, fertility or overt behavior, compared to wild-type littermates when raised on either outbred (CD1) or inbred (C57BL/6J) genetic backgrounds (Fig. 2; data not shown). Thus, SBE1 is required for the maintenance, but not the initiation of, Shh expression in the ventral midline of the rostral midbrain and caudal diencephalon. As the expression of Shh was not lost from all cell types (e.g. zli) and stages (E8.5-9.5) in which SBE1 is active, an additional Shh enhancer must be compensating for the loss of SBE1 in ShhΔSBE1/ΔSBE1 embryos.
Fig. 1.

Maintenance of Shh expression is dependent on SBE1. (A-L) Whole-mount in situ hybridization for Shh in wild-type (WT; A-D), ShhΔSBE1/ΔSBE1 (E-H), and ShhΔSBE1/– (I-L) mouse embryos at various stages of development (E8.5-12.5). The initiation of Shh expression within the presumptive ventral midbrain (arrowhead) was equivalent in all three genotypes (A,E,I). Brackets highlight the site of Shh downregulation at E10.5 (F,J). At E11.5 and E12.5 whole-mount in situ hybridization for Shh on bisected heads (medial view) is shown (C,D,G,H,K,L). Arrows indicate the residual Shh staining in ShhΔSBE1/ΔSBE1 embryos (G,H) that is absent in ShhΔSBE1/– embryos (K,L). Scale bars: 500 μm. hb, hindbrain; hyp, hypothalamus; mge, medial ganglionic eminence; mb, midbrain; pret, pretectum; preth, prethalamus; thal, thalamus; zli, zona limitans intrathalamica.
Fig. 2.
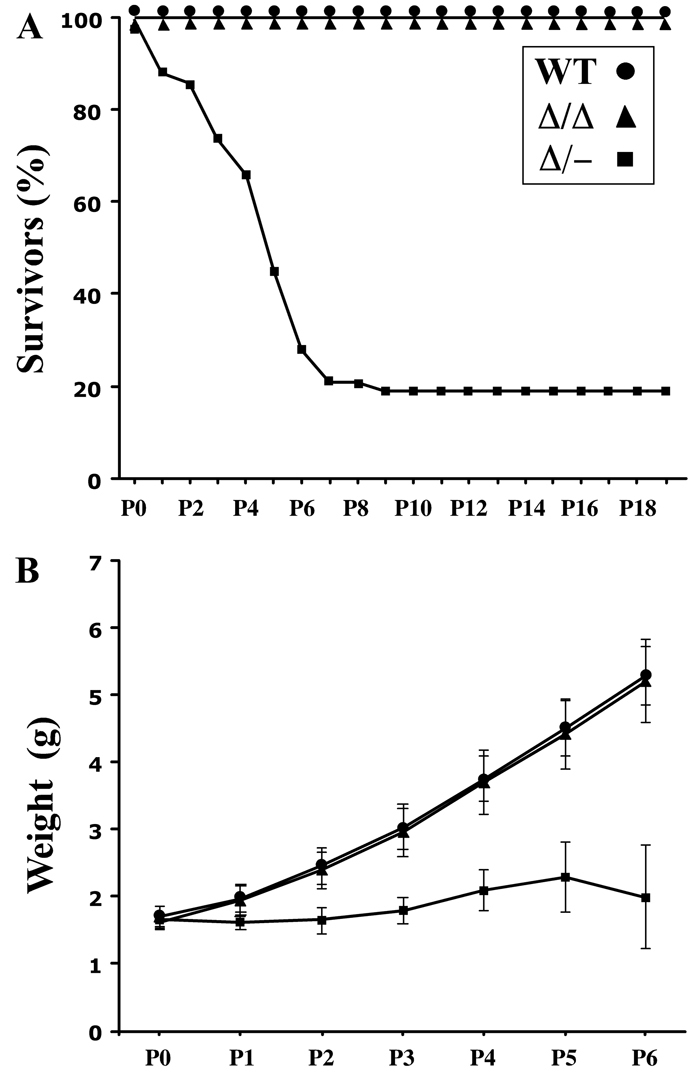
Postnatal lethality and growth retardation in ShhΔSBE1/– pups. (A) The percentage of ShhΔSBE1/– mouse pups (Δ/–; square points) surviving the first postnatal week is significantly reduced compared with wild-type (WT; round points) and ShhΔSBE1/ΔSBE1 mutants (Δ/Δ; triangle points). (B) Body weight is significantly reduced in ShhΔSBE1/– pups in the first postnatal week compared with wild-type and ShhΔSBE1/ΔSBE1 mutants. Data are expressed as mean ± s.e.m.
To evaluate further the impact of SBE1 on the regulation of Shh expression, we generated ShhΔSBE1/– embryos by crossing ShhΔSBE1/ΔSBE1 and Shh+/– mouse lines. We reasoned that the greater attenuation of Shh expression brought about by the removal of a wild-type Shh allele would partially circumvent the redundant mechanisms governing Shh transcription and, hence, reveal additional SBE1-dependent functions. The reduction in Shh expression in the ventral midbrain and caudal diencephalon was even more pronounced in ShhΔSBE1– than in ShhΔSBE1/ΔSBE1 embryos when evaluated between E10.5 and E12.5 (Fig. 1F-H,J-L). The small amount of residual Shh staining in the ventral midline of ShhΔSBE1/ΔSBE1 embryos was consistently absent in ShhΔSBE1– mutants. However, other sites of SBE1-mediated expression, including the presumptive midbrain (E8.5) and zli (E10.5), failed to show discernible differences in Shh expression between the mutant and wild-type genotypes (Fig. 1A-D,I-L).
ShhΔSBE1/– mice fail to thrive and exhibit impaired Shh signaling activity
The further reduction in Shh expression in the brains of ShhΔSBE1/– embryos correlated with a poor postnatal outcome. ShhΔSBE1/– pups were born in expected Mendelian ratios with a normal birth weight; however, ∼80% of these mutant pups (n=117) failed to gain weight and died by postnatal day (P) 7 (Fig. 2A,B). The 20% of ShhΔSBE1/– pups (n=26) that survived to adulthood never recovered their lost body weight and remained one-third the size of their wild-type littermates (data not shown). Although the cause of death of ShhΔSBE1/– pups is unknown, it is unlikely to be due to their inability to feed as the mice were often found dead with milk in their stomachs. Moreover, the ShhΔSBE1/– pups exhibited a proper suckling response upon oral stimulation, demonstrated coordinated motor control (they were able to turn over when placed on their backs) and displayed blood glucose levels in the normal range when measured at P3, a stage when the mutants were already showing signs of distress (data not shown).
We attribute the failure-to-thrive phenotype to the loss of Shh in the ventral midline of the rostral midbrain and/or caudal diencephalon for several reasons. First, these are the only known sites of SBE1 activity (Epstein et al., 1999; Jeong et al., 2006). Second, no other tissues showed a discernable reduction in Shh expression when examined at multiple stages between E8.5 and E18.5 (including lung, gut, kidney and other regions of the gastrointestinal tract; data not shown). Third, neonatal lethality was prevented in ShhΔSBE1/– mice by restoring Shh expression in the ventral midbrain and diencephalon with a ShhP1 transgene (see Fig. S2A-F in the supplementary material). Although our data point to the downregulation in Shh expression in the midbrain and diencephalon as the most likely cause of the failure-to-thrive phenotype in ShhΔSBE1/– pups, the possibility that Shh expression is disrupted in other vital organs that went undetected in our analysis cannot be ruled out.
Given that the failure-to-thrive phenotype was observed in ShhΔSBE1/– but not ShhΔSBE1/ΔSBE1 neonates, we wondered whether the subtle differences in their expression of Shh could account for the disparities in their phenotypes. We examined first whether differences in Shh signaling activity could be detected in mutant embryos by evaluating Gli1 and Ptc1 (Ptch1 – Mouse Genome Informatics) expression, two transcriptional targets of Shh signaling and effective readouts of pathway activation. Gli1 and Ptc1 displayed graded patterns of expression in the diencephalon of wild-type embryos at E12.5 (Fig. 3A, only the results for Gli1 are shown). The highest levels of Gli1 and Ptc1 were detected in close proximity to the sources of Shh in the ventral midline and zli (white brackets in Fig. 3A). Lower levels of Gli1 and Ptc1 were observed in more dorsocaudal regions of the thalamic primordium (red brackets in Fig. 3A). In ShhΔSBE1/– embryos, the high levels of Gli1 and Ptc1 in the basal plate of the rostral midbrain and caudal diencephalon were consistently reduced compared with wild-type littermates (Fig. 3A,C). This contrasts with other Shh responsive regions, including the domains on either side of the zli, which showed relatively normal Gli1 and Ptc1 expression in ShhΔSBE1/– mutants. The perturbation in Shh signaling activity was less apparent in ShhΔSBE1/ΔSBE1 embryos (compare Fig. 3B and 3C). The expression of Nkx2.2, another direct target of Shh signaling, was also downregulated in the basal plate of the midbrain and diencephalon (Fig. 3D-F). Taken together, these results indicate that the failure to maintain Shh transcription in the ventral midline of the rostral midbrain and caudal diencephalon results in a downregulation in Shh signaling activity that is more severe in ShhΔSBE1/– compared with ShhΔSBE1/ΔSBE1 embryos.
Fig. 3.

Shh signaling is impaired in SBE1 mutant embryos. (A-F) Gli1 (A-C) and Nkx2.2 (D-F), were downregulated to varying degrees in the basal plate of the caudal diencephalon in SBE1 mutant mice. The white, pink and red brackets indicate strong, moderate and weak domains of Gli1 expression, respectively. Arrowheads in D-F indicate affected domain of Nkx2.2 expression. (G-L) Foxa2 (G-I) and GFP expression from the Shh-BAC reporter line 429M20-GFP (Shh-GFP; J-L) were unaffected by the loss of SBE1.
The loss of Shh signaling did not affect the integrity of ventral midline cells in the midbrain and caudal diencephalon, as determined by the expression of Foxa2 and a Shh reporter transgene (Fig. 3G-L). The 429M20-GFP transgenic line harbors a BAC reporter construct that expresses GFP under the transcriptional control of Shh regulatory sequences, including SBE1 (Jeong et al., 2006). Additional zli markers (Sim1, Sim2, Barhl1, Pitx2) were also properly expressed in the two mutants (data not shown). From this analysis, we conclude that Shh is not required to maintain ventral midline integrity, or its own expression, in the rostral midbrain and caudal diencephalon.
Midbrain dopaminergic neurons are correctly specified in ShhΔSBE1/– embryos
Midbrain dopaminergic neurons (mDA) are dependent on Shh signaling for multiple aspects of their formation and are vital for postnatal survival (reviewed by Abeliovich and Hammond, 2007). Upon surveying a panel of genes required for mDA neuron formation [Th, Nurr1 (Nr4a2 – Mouse Genome Informatics), Lmx1b and Pitx3], we observed no differences in their expression in either ShhΔSBE1/– or ShhΔSBE1/ΔSBE1 embryos, compared with wild-type littermates (see Fig. S3 in the supplementary material). Moreover, the number of mature mDA neurons in the substantia nigra and ventral tegmental area of ShhΔSBE1/– embryos (E12.5) or neonatal pups (P2) as assessed by tyrosine hydroxylase (TH) immunostaining, was not significantly different from wild-type littermates (data not shown). These findings indicate that the premature termination of Shh signaling in the basal plate of the rostral midbrain of ShhΔSBE1/– embryos does not compromise the development of mDA neurons.
Thalamic progenitors are mis-specified in ShhΔSBE1/– embryos
Thalamic neurons are derived from at least two distinct progenitor domains. The caudal population of thalamic progenitors (pTH-C) is the larger of the two groups, expresses the basic helix-loop-helix (bHLH) transcription factors Ngn1 (Neurog1 – Mouse Genome Informatics) and Ngn2 (Neurog2 – Mouse Genome Informatics), and is fated to give rise to all thalamic nuclei with projections to the cerebral cortex (Fig. 4A, green) (Vue et al., 2007). The pTH-R domain consists of a narrow band, sandwiched between pTH-C and the zli, comprising cells that express Nkx2.2, Gsx1, Ascl1 and several other genes (Fig. 4A) (Vue et al., 2007). Thalamic neurons derived from pTH-R progenitors are thought to contribute to two dorsolaterally positioned thalamic nuclei, the ventrolateral geniculate nucleus (vLG) and the intergeniculate leaflet (IGL), neither of which projects axons to the cerebral cortex (Horowitz et al., 2004; Morin and Blanchard, 2005; Vue et al., 2007; Jones, 2007).
Fig. 4.

Rostral thalamic progenitor identity is dependent on Shh signaling from the ventral midline. (A) Schematic representation of a sagittal view through the mouse diencephalon, color coded as follows: gray, Shh-expressing cells; green, pTH-C domain; blue, pTH-R domain; yellow, prethalamic region; red line, border between the basal and alar plates. (B-M) Whole-mount in situ hybridization of genes expressed in pTH-R (Gsx1, Ascl1 and Tal1) and pTH-C (Ngn2) of wild-type (WT) and SBE1 mutant embryos at E12.5. The expression of genes in the pTH-R domain is missing (black arrowheads) or reduced (red arrowheads) to varying degrees in ShhΔSBE1/ΔSBE1 (C,F,I), and ShhΔSBE1/– (D,G,J) mutants. The prethalamic expression of Gsx1 and Ascl1 is not affected by the loss of SBE1 (asterisk in D,G). The gap in Ngn2 expression between pTH-C and the zli, corresponding to pTH-R (white arrowhead in K), is reduced in ShhΔSBE1/ΔSBE1 embryos (L) and absent in ShhΔSBE1/– embryos (M). pTH-C, caudal thalamic progenitors; pTH-R, rostral thalamic progenitors.
We examined first the expression of thalamic progenitor markers at E12.5 and observed that genes expressed in the pTH-R domain (Nkx2.2, Gsx1, Ascl1, Tal1) were partially reduced in ShhΔSBE1/ΔSBE1 embryos and almost completely downregulated in ShhΔSBE1/– mutants (Fig. 3D-F; Fig. 4B-J). None of these markers was altered in Shh+/– or ShhΔSBE1/+embryos compared to wild-type littermates. Concurrent with the reduction in pTH-R markers, the size of the Ngn2-expressing pTH-C domain appeared to expand. Normally, the expression of Ngn2 in the pTH-C and zli is separated by a gap, corresponding to pTH-R (Fig. 4K, white arrow). This gap in Ngn2 expression was narrower in ShhΔSBE1/ΔSBE1 embryos and eliminated in ShhΔSBE1/– embryos (Fig. 4K-M). These data suggest that Shh signaling from the ventral midline of the caudal diencephalon is required for the specification of pTH-R progenitors. It is worth noting that the expression of genes in the prethalamus of ShhΔSBE1/– embryos was unaltered, further indicating that Shh signaling from the zli is intact in these mutants (Fig. 3A-C; Fig. 4D,G).
Next, we addressed whether the differentiation of thalamic neurons was also compromised in ShhΔSBE1/– embryos by evaluating markers of postmitotic neurons derived from pTH-R (Sox14, Tal1) and pTH-C (Gbx2) (Hashimoto-Torii et al., 2003; Bradley et al., 2007; Vue et al., 2007). The expression of Sox14 and Tal1 was significantly diminished in the thalamus of ShhΔSBE1/– embryos, simultaneous with a shift in the rostral border of Gbx2 towards the zli (Fig. 5B-G).
Fig. 5.

Maturation of rostral thalamic neurons is dependent on the ventral midline source of Shh. (A) Schematic representation of a sagittal view through the mouse diencephalon (see Fig. 4A for details) showing the plane of sections in L-S. (B-G) In situ hybridization of coronal sections through wild-type (WT) and ShhΔSBE1/– mutants at E12.5 stained for markers of postmitotic neurons derived from pTH-R (Sox14 and Tal1) and pTH-C (Gbx2). The red dashed lines mark the borders between caudal (c), rostral (r) and zli (z) regions of the thalamus. The Shh-GFP reporter line was used to mark the zli in D and G. (H-K) Gad67 (pTH-R neurons) and Vglut2 (pTH-C neurons) expression in WT and ShhΔSBE1/– embryos. The black arrowhead in J indicates the reduction in Gad67 expression. The gap in Vglut2 expression (white arrowhead in I) is filled in K. (L-S) Nkx2.2 (pTH-R) and GFP (zli) immunostaining on coronal sections through WT and ShhΔSBE1/ embryos at E12.5. Nkx2.2 expression was reduced in both pTH-R progenitors (white arrows) and postmitotic neurons (blue arrows). (T) The number of Nkx2.2+ neurons was significantly reduced in ShhΔSBE1/– embryos (*P<0.005). Data are expressed as mean ± s.e.m. pTH-C, caudal thalamic progenitors; pTH-R, rostral thalamic progenitors.
Another distinguishing feature of neurons derived from pTH-R and pTH-C is their neurotransmitter profile. Neurons derived from pTH-R are GABAergic and express genes encoding the glutamate decarboxylase enzymes Gad65 (Gad2 – Mouse Genome Informatics) and Gad67 (Gad1 – Mouse Genome Informatics) (Fig. 5H; data not shown). By contrast, pTH-C neurons are glutamatergic and express the vesicular glutamate transporter Vglut2 (Slc17a6 – Mouse Genome Informatics) (Fig. 5I). There was a significant reduction in Gad65 and Gad67 staining in pTH-R-derived neurons from ShhΔSBE1/– embryos at E12.5, and a corresponding expansion of Vglut2 expression (Fig. 5H-K). These results suggest that the failure to maintain Shh signaling in the basal plate of the caudal diencephalon of ShhΔSBE1/– embryos compromises the differentiation of newly born pTH-R derived neurons at E12.5.
Given the dynamic nature of Sox14 and Gbx2 expression, these markers were not fully informative for evaluating the contribution of pTH-R derived neurons to specific thalamic nuclei at later aspects of their development. Fortunately, two additional markers, Nkx2.2 and Tal1, retained their expression in neurons derived from this domain. In wild-type embryos examined at E12.5, a stream of Nkx2.2+ neurons appeared to migrate radially from the pTH-R ventricular zone along the zli to a lateral position within the mantle layer, at which point they turned in a caudal direction (Fig. 5L-O). The number of these Nkx2.2+ neurons was diminished by ∼70% in ShhΔSBE1/– embryos (Fig. 5P-T).
At E16.5, the Nkx2.2+ neurons in the thalamus were observed in the vLG nucleus of wild-type embryos (Fig. 6). As with the earlier time point, the number of neurons that expressed Nkx2.2 was significantly reduced in ShhΔSBE1/– embryos (Fig. 6D,E). This effect was more pronounced in caudal regions of the vLG. The reduction in Nkx2.2 expression appeared to be limited to those neurons co-expressing Tal1 (Fig. 6D-I). Despite the reduced number of Nkx2.2+ Tal1+ neurons in the vLG nucleus of ShhΔSBE1/– embryos, the total number of cells was unchanged (Fig. 6J). Furthermore, no differences were observed in the mitotic index or number of apoptotic cells in pTH-R progenitors or vLG neurons when comparing ShhΔSBE1/– and wild-type embryos at E10.5, E12.5 and E16.5 (data not shown). These results suggest that the altered expression of genes in the pTH-R domain and its derivatives in ShhΔSBE1/– embryos is not due to a loss in cell number per se, but might be due to a change in their identity.
Fig. 6.

vLG neurons are dependent on ventral midline Shh for specific aspects of their differentiation. (A) Sox2 immunostaining shows a sharp boundary between the dorsal lateral geniculate nucleus (dLG, black arrowhead) and ventral lateral geniculate nucleus (vLG, white arrowhead) in a wild-type mouse embryo at E16.5. (B-I) Triple labeling for Sox2, Nkx2.2 and Tal1 on coronal sections through wild-type (WT) and ShhΔSBE1/– embryos at E16.5. Merge of triple images shows overlap of the expression of Nkx2.2 and Tal1 in vLG neurons from wild-type (H) but not ShhΔSBE1/– embryos (I). (J) Quantification of Nkx2.2 (red) and DAPI (white) positive cells in the vLG of WT and ShhΔSBE1/– embryos. A significant reduction in the number of Nkx2.2+ neurons was observed in ShhΔSBE1/– embryos (*P<0.005) whereas the total number of vLG neurons was unaffected. Data are expressed as mean ± s.e.m.
Heterogeneous origin of vLG neurons
Results from recent lineage-tracing experiments suggest that pTH-R progenitors contribute to the vLG (Vue et al., 2007). However, these findings were not definitive, as the genetic labeling strategy employed did not make use of genes that were exclusively expressed in the pTH-R domain. Also, these results contradicted previously held views that vLG and IGL nuclei were of prethalamic origin (Jones, 2007). To reconcile this apparent discrepancy, we performed fate-mapping experiments of pTH-R and prethalamic neurons using cre-expressing mouse lines unique to each domain. The expression of a cre-inducible eGFP reporter gene (ccEGFP) was induced in the pTH-R domain using the tamoxifen-regulated Tal1(Scl)-creER transgenic line (Nakamura et al., 2006; Bradley et al., 2007). Upon tamoxifen administration at E11.5, a subset of Tal1-expressing progenitors in the pTH-R domain activated GFP. When examined at E16.5, the vast majority of GFP-expressing cells were confined to the region of the vLG and IGL, as shown by the high degree of co-expression with Nkx2.2 (Fig. 7A-C). Not all of the Nkx2.2-expressing cells co-labeled with GFP, probably because only a subset of Tal1+ progenitors activated cre at E11.5, and not all Nkx2.2+ vLG neurons co-express Tal1. These results conclusively demonstrate that pTH-R progenitors contribute to the vLG and IGL.
Fig. 7.

Dual origin of vLG neurons in mouse. (A-C) Tal1creER;ccEGFP embryos were exposed to tamoxifen at E11.5, harvested at E16.5 and stained for Nkx2.2 and GFP. (D-F) Dlx5/6Cre;ccEGFP embryos were immunostained for Nkx2.2 and GFP at E16.5. (G-I) Nkx2.2 and panDlx antibodies mark distinct neurons in the vLG at E16.5. (J,K) The number of Dlx+ cells in the vLG in ShhΔSBE1/– embryos (J,K) is comparable with that observed in wild type (H). Data are expressed as mean ± s.e.m. ZI, zona incerta.
To mark the fate of prethalamic neurons, we crossed the Dlx5/6-cre and ccEGFP lines (Stenman et al., 2003). As expected, eGFP expression was activated in prethalamic nuclei including the reticular nucleus and the zona incerta (data not shown). Interestingly, eGFP-positive neurons were also found in the location of the vLG (Fig. 7D-F). These eGFP-positive neurons were intermingled with Nkx2.2+ neurons but did not co-express the two markers (Fig. 7F). The same result was observed when vLG neurons were co-labeled with antibodies against Nkx2.2 and pan-Dlx proteins (Fig. 7G-I). Therefore, the vLG nucleus is composed of a heterogeneous group of neurons originating from both thalamic (pTH-R) and prethalamic progenitors.
Having identified a dual origin of vLG neurons, we next addressed whether the prethalamic population of vLG neurons was affected by the loss of Shh in ShhΔSBE1/– embryos. No significant differences in the number of Dlx+ cells in the vLG were observed between wild-type and ShhΔSBE1/– embryos at E16.5 (Fig. 7H,J,K).
Mapping the fate of pTH-R progenitors in wild-type and ShhΔSBE1/– embryos
The reduced expression of Nkx2.2 and Tal1 in vLG and IGL neurons of ShhΔSBE1/– embryos raised several questions regarding the fate of pTH-R progenitors in ShhΔSBE1/– embryos. Did the mis-specified pTH-R progenitors still migrate to the vLG and IGL in the absence of Shh, or did they intermingle with more caudal thalamic nuclei? Did they send axonal projections to their proper targets in the brainstem? Did neurons derived from mis-specified pTH-R progenitors express differentiation markers appropriate for their thalamic nucleus? To address these questions, we once again used a recombination-based lineage tracing approach to permanently label pTH-R progenitors. However, because the expression of Tal1-creER was downregulated in ShhΔSBE1/– embryos prior to its ability to activate ccEGFP, it could not be used for these experiments.
Fortunately, we had previously identified an Nkx2.2 enhancer that recapitulates endogenous Nkx2.2 expression in the developing CNS, including neuronal progenitors in the pTH-R domain (data not shown) (Lei et al., 2006). This enhancer was used to generate an Nkx2.2-cre transgenic mouse line, in which cre recombinase was placed under the transcriptional control of the Nkx2.2 CNS regulatory element. The Nkx2.2 CNS enhancer is active in the ventral diencephalon at least 30 hours before Shh expression is extinguished in ShhΔSBE1/– embryos (Lei et al., 2006). We predicted that this temporal window would be sufficient for Nkx2.2-cre to activate ccEGFP in most, if not all, pTH-R progenitors prior to its downregulation in ShhΔSBE1/– mutants.
To map the fate of pTH-R progenitors on wild-type and mutant backgrounds, we bred Nkx2.2-cre; Shh+/– males with ccEGFP/ccEGFP; ShhΔSBE1/ΔSBE1 females and harvested wild-type (Nkx2.2-cre; ShhΔSBE1/+; ccEGFP) and mutant (Nkx2.2-cre; ShhΔSBE1/–; ccEGFP) embryos at E16.5. The added benefit of the ccEGFP line is that eGFP fills both cell bodies and neuronal processes, allowing for a convenient method to visualize axonal projections.
Wild-type embryos harvested at E16.5 showed a pattern of GFP expression in a stream of cells extending radially from the pTH-R ventricular zone along the zli, terminating in a triangular cluster consistent with the vLG nucleus (Fig. 8A). The distribution of the GFP+ neurons within the vLG was confirmed by Nkx2.2 co-labeling (Fig. 8C-E). Notably, GFP+ immunostaining was detected in caudally projecting axons that extended from the cell bodies of vLG neurons (Fig. 8J-L). Surprisingly, a similar pattern of GFP staining was also observed in ShhΔSBE1/– mutant embryos. In particular, GFP+ neurons remained confined to the vLG and were not intermingled with caudal thalamic nuclei derived from pTH-C progenitors (Fig. 8B,F-H). In addition, vLG-labeled axons maintained their caudal trajectory in ShhΔSBE1/– embryos and showed no evidence of erroneous projections to the cerebral cortex (Fig. 8M-O).
Fig. 8.

Tracing the fate of pTH-R neurons in wild-type and ShhΔSBE1/– embryos. (A,B) Nkx2.2cre; ccEGFP embryos at E16.5 stained for GFP on coronal sections through wild-type (WT; A) and ShhΔSBE1/– (B) embryos. GFP-positive cell bodies (arrow) and axons (arrowheads) are shown. Boxed area indicates magnified region in C-H. (C-H) Colocalization of Nkx2.2 and GFP-expressing neurons in the vLG of WT (C-E) and ShhΔSBE1/– (F-H) embryos. (I) Quantification of data from E and H. Data are expressed as mean ± s.e.m. *P<0.05, **P<0.005. (J-O) Colocalization of GFP and α-internexin (intermediate filament marker) on caudally projecting vLG axons from WT (J-L) and ShhΔSBE1/– (M-O) embryos. The majority of GFP+ neurons and caudally projecting axons are present in the vLG of ShhΔSBE1/– mutants compared with wild-type littermates despite the reduction in Nkx2.2 expression. Arrows in K and N point to the optic tract (OT). (P) Quantification of data from J and M. Data are expressed as mean ± s.e.m. P<0.05. dLG, dorsolateral geniculate; Th, thalamus; vLG, ventrolateral geniculate nucleus; zli, zona limitans intrathalamica.
We quantified the number of GFP+ neurons contributing to the vLG and detected a slight reduction (20%) in ShhΔSBE1/– compared with wild-type embryos (Fig. 8I). A similar deficit in the number of axons extending from vLG neurons was also observed (Fig. 8P). These differences can be attributed to either a smaller number of cre-expressing cells in ShhΔSBE1/– embryos or a minimal loss of pTH-R progenitors that went undetected in our analysis. Nonetheless, of the vLG and IGL neurons that did label with GFP, the number that co-expressed Nkx2.2 was significantly reduced in ShhΔSBE1/– embryos compared with wild-type littermates. These results clearly indicate that the majority of pTH-R progenitors are present in ShhΔSBE1/– embryos and that the loss of their identity at earlier stages can probably be attributed to a mis-specification defect rather than their failure to proliferate or survive.
When examining the neurotransmitter profile of thalamic neurons at E16.5, we observed that Vglut2 was no longer ectopically expressed in pTH-R derivatives of ShhΔSBE1/– embryos, but was now appropriately restricted to pTH-C neurons. Moreover, Gad67 was properly expressed in vLG neurons. By contrast, IGL neurons failed to express neuropeptide Y (NPY; see Fig. S4 in the supplementary material). Thus, the apparent pTH-R to pTH-C fate switch observed in ShhΔSBE1/– mutants at E12.5 was partially reverted by E16.5. Recent studies demonstrated that embryos lacking Shh (or the ability to respond to Shh) from both the diencephalic ventral midline and zli showed a more profound thalamic phenotype, including the complete loss of pTH-R and rostral pTH-C neuronal derivatives (Vue et al., 2009; Szabó et al., 2009). Therefore, in ShhΔSBE1/– embryos, it is conceivable that the exposure of mis-specified pTH-R neurons to the zli source of Shh during their migration to the vLG resulted in their partial recovery.
Determinants of GABA identity in pTH-R-derived neurons
Several of the genes expressed in the pTH-R domain were previously shown to function as determinants of neurotransmitter phenotypes in other regions of the CNS. For instance, the bHLH protein Helt (also known as megane or Heslike) promotes GABAergic and suppresses glutamatergic fates in the mesencephalon, in part by repressing Ngn2 transcription (Miyoshi et al., 2004; Guimera et al., 2006; Nakatani et al., 2007). Gsx1, Gsx2 and Ascl1 are also necessary and sufficient determinants of GABA identity in the ventral telencephalon (Casarosa et al., 1999; Fode et al., 2000; Corbin et al., 2000; Toresson and Campbell, 2001; Yun et al., 2001; Parras et al., 2002; Wang et al., 2009). By contrast, Ngn1 and Ngn2 promote glutamatergic fates in the dorsal telencephalon (Parras et al., 2002).
Surprisingly, GABA expression was not impaired in neurons derived from the pTH-R domain of Helt–/– mutants (Guimera et al., 2006). We also failed to detect alterations in the rostral thalamic expression of Gad67 in Gsx1–/– and Ascl1–/– mutants (data not shown). However, a partial reduction in Gad67 expression was observed in pTH-R derivatives from Nkx2.2–/– embryos, concomitant with an expansion in Vglut2 staining, as indicated by the smaller gap in expression between the caudal thalamus and zli (Fig. 9A-D). A comparable loss and gain of Ascl1 and Ngn2 expression, respectively, was also observed in Nkx2.2 mutants (Fig. 9E-H). The switch in thalamic progenitor identity in Nkx2.2–/– embryos was not as extensive as that seen in ShhΔSBE1/– mutants. This is likely to be due to the partial compensation by Nkx2.9, which continues to be expressed in the pTH-R domain of Nkx2.2 but not ShhΔSBE1/– embryos (data not shown). Thus, Nkx2.2 (and probably Nkx2.9) mediates GABA identity in pTH-R derivatives downstream of Shh.
Fig. 9.
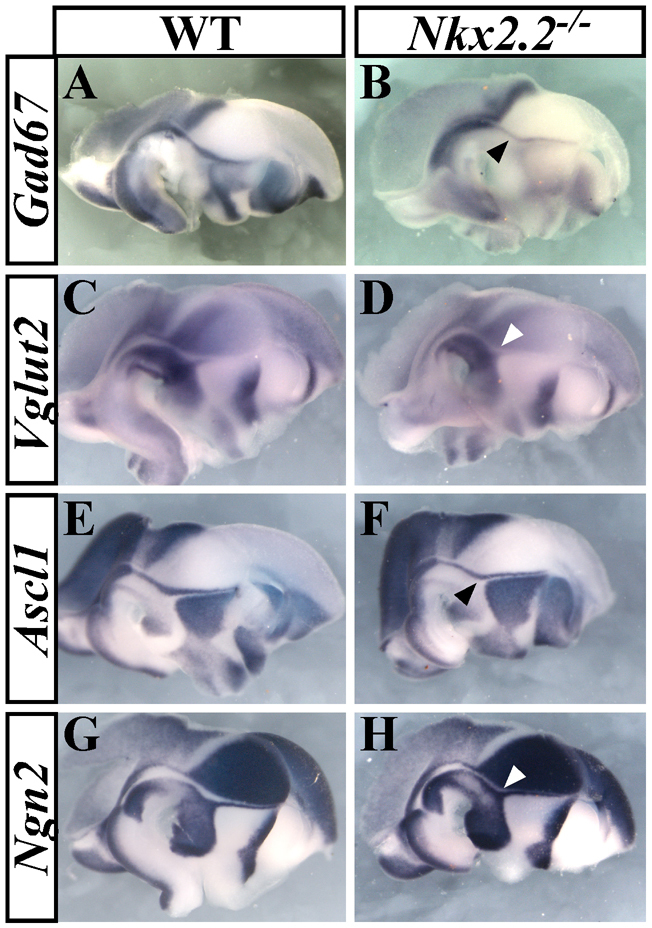
pTH-R identity is partially dependent on Nkx2.2. (A-H) The expression of Gad67 (A,B) and Ascl1 (E,F) are reduced (black arrowheads), whereas Vglut2 (C,D) and Ngn2 (G,H) are upregulated (white arrowheads) in the pTH-R (rostral thalamic progenitors) domain of Nkx2.2–/– embryos versus wild type (WT) at E12.5.
In summary, ShhΔSBE1/– mutants showed a selective loss of Shh expression in the ventral midline of the rostral midbrain and caudal diencephalon that resulted in the mis-specification of pTH-R progenitors. Notwithstanding their transient switch in identity at E12.5, pTH-R-derived neurons from ShhΔSBE1/– embryos recovered some of their differentiation properties by E16.5 (migration to the vLG nucleus; guidance of axons along a caudal trajectory; GABA expression), whereas others were permanently lost (expression of Nkx2.2 and Tal1 in the vLG; expression of NPY in the IGL). From these and previously published data, we conclude that the two sources of Shh adjacent to the thalamic primordium, the ventral midline and zli, have unique and partially overlapping functions in the specification and differentiation of thalamic interneurons.
DISCUSSION
Initiation versus maintenance of Shh expression
Our results demonstrate a requirement for SBE1, a cis-acting regulatory element located in the second intron of the Shh gene, in the spatiotemporal control of Shh transcription in the mouse CNS. In embryos lacking SBE1, Shh expression in the ventral midline of the midbrain and caudal diencephalon initiated properly at E8.5 but was not maintained beyond E10.0. Because SBE1 is sufficient to direct reporter activity to these sites of expression at both stages of development (Epstein et al., 1999), we surmise that SBE1 functions redundantly with an unidentified enhancer(s) to control Shh transcription at the earlier time point. Our finding exemplifies a clear case of gene regulation that is partitioned into initiation and maintenance phases. Identification of the redundant enhancer should help to elaborate on the mechanistic differences in the temporal regulation of Shh expression in the ventral midline of the mid- and forebrain.
Spatiotemporal and threshold models of Shh signaling in the thalamus
Previous studies have implicated Shh signaling in the growth, regionalization and specification of thalamic progenitors (Scholpp and Lumsden, 2010). Early sources of Shh in the prechordal plate and/or ventral midline of the diencephalon promote the proliferation, survival and patterning of the thalamic primordia (Ishibashi et al., 2002). At slightly later stages, the source of Shh in the zli is required to establish regional identity on either side of this diencephalic boundary (Kiecker and Lumsden, 2004; Vieira et al., 2005; Scholpp et al., 2006; Vieira and Martinez, 2006). In addition to these early patterning events, Shh signaling is also required to specify neuronal subtypes within thalamic nuclei. The prevailing model stipulates that graded Shh signaling from the zli is necessary and sufficient to promote distinct classes of thalamic progenitor subtypes (Hashimoto-Torii et al., 2003; Szabó et al., 2009; Vue et al., 2009; Scholpp et al., 2009). The pTH-R domain, which develops closest to the zli, is dependent on the highest concentration of Shh, whereas the rostral population of pTH-C progenitors, developing several cell diameters away from the zli, is dependent on a lower concentration of Shh. The conditional inactivation of either Shh or Smo throughout the diencephalon results in a loss of pTH-R progenitors and their postmitotic derivatives in the vLG and IGL, as well as rostral pTH-C progenitors and the formation of cortex-projecting thalamic nuclei (Szabó et al., 2009; Vue et al., 2009).
Although these studies have mainly focused on the role of Shh emanating from the zli, the contribution of the ventral midline source of Shh in the diencephalon to specification of thalamic neural fates has received considerably less attention, aside from a disputed function in regulating zli outgrowth (Kiecker and Lumsden, 2004; Zeltser, 2005; Scholpp et al., 2006; Vieira and Martinez. 2006). As Shh signaling activity from the zli was not impaired in either ShhΔSBE1/ΔSBE1 or ShhΔSBE1/– mutants, we have been able to take advantage of a unique opportunity to uncover the specific contribution of Shh from the diencephalic ventral midline on thalamic development.
The extent of Shh downregulation in the ventral midline of the caudal diencephalon correlated with the severity of the thalamic defects observed in SBE1 deficient embryos. ShhΔSBE1/ΔSBE1 mutants showed residual Shh expression in the basal plate of p1 and p2, and only a slight alteration in pTH-R progenitor identity. However, a more pronounced loss of Shh expression was observed in ShhΔSBE1/– embryos resulting in the mis-specification of the majority of pTH-R progenitors. Given our data, and those recently reported by others (Szabó et al., 2009; Vue et al., 2009), we propose that the threshold model of Shh signaling in the thalamus should be revised to include spatial and temporal components. Rather than a simple concentration gradient of Shh signaling from the zli, we suggest that the two classes of Shh-dependent thalamic progenitors, pTH-R and rostral pTH-C, are specified by two spatially distinct sources of Shh, the ventral midline and zli, respectively. The zli source of Shh is not sufficient to activate genes in the pTH-R domain in the absence of ventral midline Shh, but is capable of activating lower threshold targets in pTh-C and its derivatives. Our results do not exclude the possibility that pTH-R progenitors are specified by the high levels of Shh signaling that result from the convergence of both sources of Shh in the caudal diencephalon. Indeed, the blockade of Shh signaling from the zli in chick results in a loss of Nkx2.2 expression, consistent with the argument that both sources of Shh surrounding the thalamus are necessary for pTH-R identity (Kiecker and Lumsden, 2004). It might also be the case that the pTH-R domain is dependent on high levels of Shh signaling for a prolonged period of time, as was described for Nkx2.2-expressing p3 progenitors in the spinal cord (Dessaud et al., 2007).
Our data indicating that Shh signaling from the ventral midline is required to establish pTH-R progenitor identity in the mouse thalamus differs from that described in zebrafish. In zebrafish oep mutants, which lack a nodal co-receptor, Shh expression was absent from the ventral midline of the diencephalon but was retained in the zli. Nevertheless, the thalamus was reported to develop normally in oep mutants (Scholpp et al., 2006). One explanation for this apparent species difference might have to do with the earlier onset of Shh expression in the zli of zebrafish compared with mouse (Barth and Wilson, 1995; Epstein et al., 1999). Owing to its slower development and increased brain complexity, the mouse might have co-opted the two sources of Shh in the diencephalon to increase neuronal diversity in the thalamus, whereas the zebrafish relies on just one source of Shh.
The pTH-R to pTH-C fate switch in ShhΔSBE1/– embryos is partially reverted in postmitotic neurons
Our lineage tracing experiments yielded the surprising finding that despite their altered progenitor status from pTH-R to pTH-C, the fate of mis-specified pTH-R neurons in ShhΔSBE1/– mutants was partially restored at later stages of their differentiation. GFP-labeled neurons derived from the mis-specified pTH-R domain of ShhΔSBE1/– mutants still managed to migrate to the vLG and IGL, instead of segregating with the caudal population of Gbx2-expressing neurons. This result was not anticipated given our observation that Gbx2 was ectopically expressed in postmitotic pTH-R derivatives of ShhΔSBE1/– mutants at E12.5 and that Gbx2 controls lineage-restriction boundaries in the thalamus (Chen et al., 2009). Additional indications of the partial fate restoration of pTH-R neurons in ShhΔSBE1/– mutants included their projection of axons towards the brainstem rather than the cortex and their re-employment of the GABA neurotransmitter over glutamate. However, not all aspects of their differentiation profile were restored as pTH-R derivatives in the vLG and IGL of ShhΔSBE1/– mutants failed to express Nkx2.2, Tal1 and NPY (IGL only).
We also considered an alternative explanation for the partial recovery of vLG neurons in ShhΔSBE1/– mutants. As vLG neurons are derived from both thalamic (pTH-R) and prethalamic sources that are dually marked by Nkx2.2cre;ccEGFP, it was possible that the loss of pTH-R progenitors was compensated for by an expansion of pre-thalamic (Dlx+) neurons. However, as no increase in the number of Dlx+ neurons was observed in the vLG of ShhΔSBE1/– mutants, we feel confident that this hypothesis can be ruled out.
The partial recovery of thalamic neurons from the pTH-R to pTH-C fate switch in ShhΔSBE1/– mutants suggests that these neurons continue to rely on cell-fate determinants to maintain their identity at postmitotic stages of their differentiation. One intriguing candidate that might be fulfilling this role is Shh from the zli. The pTH-R neurons originating in the basal plate of p1 and p2 remained in close proximity to the zli as they migrate dorsally towards the vLG and IGL (Fig. 8A). Experiments that selectively eliminate Shh signaling in postmitotic pTH-R-derived neurons and/or conditionally remove Shh from the zli after E12.5 will be needed to address whether the maturation of vLG and IGL neurons is dependent on later aspects of Shh function. Fgf8 is another candidate signal that might be regulating the differentiation of vLG and IGL neurons. Fgf8 is expressed in the dorsal diencephalon and signals to cells in the region of the vLG and IGL to regulate aspects of their identity (Kataoka and Shimogori, 2008).
How the mis-specification of vLG and IGL neurons contributes to the failure-to-thrive phenotype in ShhΔSBE1/– mutants is unclear. The restoration of pTH-R progenitor identity and postnatal viability in ShhP1; ShhΔSBE1/– embryos is consistent with our hypothesis that the mutant phenotype stems from the loss of Shh in the SBE1-dependent region of the diencephalon. Interestingly, mice carrying a conditional deletion of Tal1 in the CNS show early postnatal lethality and growth retardation similar to ShhΔSBE1/– mutants (Bradley et al., 2006). Determining which aspect of the vLG and/or IGL circuitry is altered in ShhΔSBE1/– will be a challenge for the future.
Acknowledgments
The authors would like to thank Dr Hong Fu and Dr Jean Richa and his staff at the University of Pennsylvania Transgenic and Mouse Chimeric Facility for their assistance in generating the targeted SBE1 mouse line. The Nkx2.2-Cre transgenic line was generated by the Transgenic and Knockout Mouse Core Facility at the UMDNJ Cancer Institute of New Jersey and was supported by grants from the NSF and NIH to M.P.M. Funding sources for work described in this study include grants from the NIH/NINDS (R01 NS39421) to D.J.E., NIH/NIDDK (R01 DK082590) to L.S. and the National Research Foundation of Korea (2009-0087420) to Y.J. Deposited in PMC for release after 12 months.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.058917/-/DC1
References
- Abeliovich A., Hammond R. (2007). Midbrain dopamine neuron differentiation: factors and fates. Dev. Biol. 304, 447-454 [DOI] [PubMed] [Google Scholar]
- Andersson E., Tryggvason U., Deng Q., Friling S., Alekseenko Z., Robert B., Perlmann T., Ericson J. (2006). Identification of intrinsic determinants of midbrain dopamine neurons. Cell 124, 393-405 [DOI] [PubMed] [Google Scholar]
- Barth K. A., Wilson S. W. (1995). Expression of zebrafish nk2.2 is influenced by sonic hedgehog/vertebrate hedgehog-1 and demarcates a zone of neuronal differentiation in the embryonic forebrain. Development 121, 1755-1768 [DOI] [PubMed] [Google Scholar]
- Bayly R. D., Ngo M., Aglyamova G. V., Agarwala S. (2007). Regulation of ventral midbrain patterning by Hedgehog signaling. Development 134, 2115-2124 [DOI] [PubMed] [Google Scholar]
- Blaess S., Corrales J. D., Joyner A. L. (2006). Sonic hedgehog regulates Gli activator and repressor functions with spatial and temporal precision in the mid/hindbrain region. Development 133, 1799-1809 [DOI] [PubMed] [Google Scholar]
- Bourikas D., Pekarik V., Baeriswyl T., Grunditz A., Sadhu R., Nardó M., Stoeckli E. T. (2005). Sonic hedgehog guides commissural axons along the longitudinal axis of the spinal cord. Nat. Neurosci. 8, 297-304 [DOI] [PubMed] [Google Scholar]
- Bradley C. K., Takano E. A., Hall M. A., Göthert J. R., Harvey A. R., Begley C. G., van Eekelen J. A. (2006). The essential haematopoietic transcription factor Scl is also critical for neuronal development. Eur. J. Neurosci. 23, 1677-1689 [DOI] [PubMed] [Google Scholar]
- Bradley C. K., Takano E. A., Göthert J. R., Göttgens B., Green A. R., Begley C. G., van Eekelen J. A. (2007). Temporal regulation of Cre-recombinase activity in Scl-positive neurons of the central nervous system. Genesis 45, 145-151 [DOI] [PubMed] [Google Scholar]
- Casarosa S., Fode C., Guillemot F. (1999). Mash1 regulates neurogenesis in the ventral telencephalon. Development 126, 525-534 [DOI] [PubMed] [Google Scholar]
- Charron F., Stein E., Jeong J., McMahon A. P., Tessier-Lavigne M. (2003). The morphogen Sonic Hedgehog is an axonal chemoattractant that collaborates with Netrin-1 in midline axon guidance. Cell 113, 11-23 [DOI] [PubMed] [Google Scholar]
- Chen L., Guo Q., Li J. Y. (2009). Transcription factor Gbx2 acts cell-nonautonomously to regulate the formation of lineage-restriction boundaries of the thalamus. Development 136, 1317-1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C., Litingtung Y., Lee E., Young K. E., Corden J. L., Westphal H., Beachy P. A. (1996). Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 383, 407-413 [DOI] [PubMed] [Google Scholar]
- Corbin J. G., Gaiano N., Machold R. P., Langston A., Fishell G. (2000). The Gsh2 homeodomain gene controls multiple aspects of telencephalic development. Development 127, 5007-5020 [DOI] [PubMed] [Google Scholar]
- Dessaud E., Yang L. L., Hill K., Cox B., Ulloa F., Ribeiro A., Mynett A., Novitch B. G., Briscoe J. (2007). Interpretation of the sonic hedgehog morphogen gradient by a temporal adaptation mechanism. Nature 450, 717-720 [DOI] [PubMed] [Google Scholar]
- Ding Q., Motoyama J., Gasca S., Mo R., Sasaki H., Rossant J., Hui C. C. (1998). Diminished Sonic Hedgehog signaling and lack of floor plate differentiation in Gli2 mutant mice. Development 125, 2533-2543 [DOI] [PubMed] [Google Scholar]
- Echelard Y., Epstein D. J., St-Jacques B., Shen L., Mohler J., McMahon J. A., McMahon A. P. (1993). Sonic hedgehog, a member of a family of putative signaling molecules is implicated in the regulation of CNS and limb polarity. Cell 75, 1417-1430 [DOI] [PubMed] [Google Scholar]
- Epstein D. J., McMahon A. P., Joyner A. L. (1999). Regionalization of Sonic hedgehog transcription along the anteroposterior axis of the mouse central nervous system is regulated by Hnf3-dependent and -independent mechanisms. Development 126, 281-292 [DOI] [PubMed] [Google Scholar]
- Ericson J., Morton S., Kawakami A., Roelink H., Jessell T. M. (1996). Two critical periods of sonic hedgehog signaling required for the specification of motor neuron identity. Cell 87, 661-673 [DOI] [PubMed] [Google Scholar]
- Fode C., Ma Q., Casarosa S., Ang S. L., Anderson D. J., Guillemot F. (2000). A role for neural determination genes in specifying the dorsoventral identity of telencephalic neurons. Genes Dev. 14, 67-80 [PMC free article] [PubMed] [Google Scholar]
- Fuccillo M., Joyner A. L., Fishell G. (2006). Morphogen to mitogen: the multiple roles of hedgehog signalling in vertebrate neural development. Nat. Rev. Neurosci. 7, 772-783 [DOI] [PubMed] [Google Scholar]
- Guillemot F., Lo L. C., Johnson J. E., Auerbach A., Anderson D. J., Joyner A. L. (1993). Mammalian achaete-scute homolog 1 is required for the early development of olfactory and autonomic neurons. Cell 75, 463-476 [DOI] [PubMed] [Google Scholar]
- Guimera J., Weisenhorn D. V., Wurst W. (2006). Megane/Heslike is required for normal GABAergic differentiation in the mouse superior colliculus. Development 133, 3847-3857 [DOI] [PubMed] [Google Scholar]
- Hashimoto-Torii K., Motoyama J., Hui C. C., Kuroiwa A., Nakafuku M., Shimamura K. (2003). Differential activities of Sonic hedgehog mediated by Gli transcription factors define distinct neuronal subtypes in the dorsal thalamus. Mech. Dev. 120, 1097-1111 [DOI] [PubMed] [Google Scholar]
- Horowitz S. S., Blanchard J. H., Morin L. P. (2004). Intergeniculate leaflet and ventral lateral geniculate nucleus afferent connections: An anatomical substrate for functional input from the vestibulo-visuomotor system. J. Comp. Neurol. 474, 227-245 [DOI] [PubMed] [Google Scholar]
- Hynes M., Poulsen K., Tessier-Lavigne M., Rosenthal A. (1995). Control of neuronal diversity by the floor plate: contact-mediated induction of midbrain dopaminergic neurons. Cell 80, 95-101 [DOI] [PubMed] [Google Scholar]
- Ishibashi M., McMahon A. P. (2002). A sonic hedgehog-dependent signaling relay regulates growth of diencephalic and mesencephalic primordia in the early mouse embryo. Development 129, 4807-4819 [DOI] [PubMed] [Google Scholar]
- Jeong Y., El-Jaick K., Roessler E., Muenke M., Epstein D. J. (2006). A functional screen for Sonic hedgehog regulatory elements across a 1 Mb interval identifies long range ventral forebrain enhancers. Development 133, 761-772 [DOI] [PubMed] [Google Scholar]
- Jessell T. M. (2000). Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nat. Rev. Genet. 1, 20-29 [DOI] [PubMed] [Google Scholar]
- Joksimovic M., Yun B. A., Kittappa R., Anderegg A. M., Chang W. W., Taketo M. M., McKay R. D., Awatramani R. B. (2009). Wnt antagonism of Shh facilitates midbrain floor plate neurogenesis. Nat. Neurosci. 12, 125-131 [DOI] [PubMed] [Google Scholar]
- Jones E. G. (2007) The Thalamus. Cambridge, UK: Cambridge University Press; [Google Scholar]
- Kataoka A., Shimogori T. (2008). Fgf8 controls regional identity in the developing thalamus. Development 135, 2873-2881 [DOI] [PubMed] [Google Scholar]
- Kiecker C., Lumsden A. (2004). Hedgehog signaling from the ZLI regulates diencephalic regional identity. Nat. Neurosci. 7, 1242-1249 [DOI] [PubMed] [Google Scholar]
- Lei Q., Jeong Y., Misra K., Li S., Zelman A. K., Epstein D. J., Matise M. P. (2006). Wnt signaling inhibitors regulate the transcriptional response to morphogenetic Shh-Gli signaling in the neural tube. Dev. Cell 11, 325-337 [DOI] [PubMed] [Google Scholar]
- Li H., Zeitler P. S., Valerius M. T., Small K., Potter S. S. (1996). Gsh-1, an orphan Hox gene, is required for normal pituitary development. EMBO J. 15, 714-724 [PMC free article] [PubMed] [Google Scholar]
- Matise M. P., Epstein D. J., Park H. L., Platt K. P., Joyner A. L. (1998). Gli2 is required for induction of floor plate but not most ventral neurons in the mouse central nervous system. Development 125, 2759-2770 [DOI] [PubMed] [Google Scholar]
- Miyoshi G., Bessho Y., Yamada S., Kageyama R. (2004). Identification of a novel basic helix-loop-helix gene, Heslike, and its role in GABAergic neurogenesis. J. Neurosci. 24, 3672-3682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin L. P., Blanchard J. H. (2005). Descending projections of the hamster intergeniculate leaflet: relationship to the sleep/arousal and visuomotor systems. J. Comp. Neurol. 487, 204-216 [DOI] [PubMed] [Google Scholar]
- Nakamura T., Colbert M. C., Robbins J. (2006). Neural crest cells retain multipotential characteristics in the developing valves and label the cardiac conduction system. Circ. Res. 98, 1547-1554 [DOI] [PubMed] [Google Scholar]
- Nakatani T., Minaki Y., Kumai M., Ono Y. (2007). Helt determines GABAergic over glutamatergic neuronal fate by repressing Ngn genes in the developing mesencephalon. Development 134, 2783-2793 [DOI] [PubMed] [Google Scholar]
- Nissim S., Allard P., Bandyopadhyay A., Harfe B. D., Tabin C. J. (2007). Characterization of a novel ectodermal signaling center regulating Tbx2 and Shh in the vertebrate limb. Dev. Biol. 304, 9-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parras C. M., Schuurmans C., Scardigli R., Kim J., Anderson D. J., Guillemot F. (2002). Divergent functions of the proneural genes Mash1 and Ngn2 in the specification of neuronal subtype identity. Genes Dev. 16, 324-338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puelles L., Rubenstein J. L. (2003). Forebrain gene expression domains and the evolving prosomeric model. Trends Neurosci. 26, 469-476 [DOI] [PubMed] [Google Scholar]
- Riccomagno M. M., Martinu L., Mulheisen M., Wu D. K., Epstein D. J. (2002). Specification of the mammalian cochlea is dependent on Sonic hedgehog. Genes Dev. 16, 2365-2378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholpp S., Lumsden A. (2010). Building a bridal chamber: development of the thalamus. Trends Neurosci. 33, 373-380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholpp S., Wolf O., Brand M., Lumsden A. (2006). Hedgehog signalling from the zona limitans intrathalamica orchestrates patterning of the zebrafish diencephalon. Development 133, 855-864 [DOI] [PubMed] [Google Scholar]
- Scholpp S., Delogu A., Gilthorpe J., Peukert D., Schindler S., Lumsden A. (2009). Her6 regulates the neurogenetic gradient and neuronal identity in the thalamus. Proc. Natl. Acad. Sci USA. 106, 19895-19900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenman J., Toresson H., Campbell K. (2003). Identification of two distinct progenitor populations in the lateral ganglionic eminence: implications for striatal and olfactory bulb neurogenesis. J. Neurosci. 23, 167-174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussel L., Kalamaras J., Hartigan-O'Connor D. J., Meneses J. J., Pedersen R. A., Rubenstein J. L., German M. S. (1998). Mice lacking the homeodomain transcription factor Nkx2.2 have diabetes due to arrested differentiation of pancreatic beta cells. Development 125, 2213-2221 [DOI] [PubMed] [Google Scholar]
- Szabó N. E., Zhao T., Zhou X., Alvarez-Bolado G. (2009). The role of Sonic hedgehog of neural origin in thalamic differentiation in the mouse. J. Neurosci. 29, 2453-2466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toresson H., Campbell K. (2001). A role for Gsh1 in the developing striatum and olfactory bulb of Gsh2 mutant mice. Development 128, 4769-4780 [DOI] [PubMed] [Google Scholar]
- Vieira C., Martinez S. (2006). Sonic hedgehog from the basal plate and the zona limitans intrathalamica exhibits differential activity on diencephalic molecular regionalization and nuclear structure. Neuroscience 143, 129-140 [DOI] [PubMed] [Google Scholar]
- Vieira C., Garda A. L., Shimamura K., Martinez S. (2005). Thalamic development induced by Shh in the chick embryo. Dev. Biol. 284, 351-363 [DOI] [PubMed] [Google Scholar]
- Vue T. Y., Aaker J., Taniguchi A., Kazemzadeh C., Skidmore J. M., Martin D. M., Martin J. F., Treier M., Nakagawa Y. (2007). Characterization of progenitor domains in the developing mouse thalamus. J. Comp. Neurol. 505, 73-91 [DOI] [PubMed] [Google Scholar]
- Vue T. Y., Bluske K., Alishahi A., Yang L. L., Koyano-Nakagawa N., Novitch B., Nakagawa Y. (2009). Sonic hedgehog signaling controls thalamic progenitor identity and nuclei specification in mice. J. Neurosci. 29, 4484-4497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B., Waclaw R. R., Allen Z. J., 2nd, Guillemot F., Campbell K. (2009). Ascl1 is a required downstream effector of Gsx gene function in the embryonic mouse telencephalon. Neural Dev. 4, 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye W., Shimamura K., Rubenstein J. L. R., Hynes M. A., Rosenthal A. (1998). FGF and Shh signals control dopaminergic and serotonergic cell fate in the anterior neural plate. Cell 93, 755-766 [DOI] [PubMed] [Google Scholar]
- Yun K., Potter S., Rubenstein J. L. (2001). Gsh2 and Pax6 play complementary roles in dorsoventral patterning of the mammalian telencephalon. Development 128, 193-205 [DOI] [PubMed] [Google Scholar]
- Zeltser L. M. (2005). Shh-dependent formation of the ZLI is opposed by signals from the dorsal diencephalon. Development 132, 2023-2033 [DOI] [PubMed] [Google Scholar]