

INTRODUCTION
The fact that plasma contains one system designed to clot blood and another to dissolve thrombi is one of the teleologic wonders of vascular biology (Ratnoff and Forbes 1984). Following vessel injury, the coagulation cascade is activated and generates thrombin, which converts soluble fibrinogen into insoluble fibrin. Upon vessel healing, the process of fibrinolysis begins, whereby, the sequential activation of specific proteases culminates in production of plasmin, which cleaves fibrin into soluble, defined degradation products, and restores vascular patency.
In the last century, the principal fibrinolytic molecules, plasminogen, tissue plasminogen, and urokinase, were identified, cloned, and studied in genetic mouse models (Hajjar 2009). From the work of many investigators, it is now clear that plasminogen, an inactive zymogen, circulates in blood and can be converted to plasmin by either of two physiologic activators, tissue plasminogen activator (tPA) or urokinase (uPA). tPA is a secretory product of endothelial cells, while uPA is produced by renal epithelial cells or by endothelial cells stimulated by inflammatory cytokines (Wojta et al. 1989). Upon resolution of vascular injury, these agents dissolve mature blood clots or thrombi renewing the flow of blood.
Until the middle of the 20th century, fibrinolysis was thought to be confined to the surface of fibrin-containing thrombi. In 1964, however, Todd observed that fibrin gels applied directly to fresh tissue sections displayed zones of protein lysis that were specifically localized to blood vessels, and required the integrity of the endothelium (Todd 1964). Our research group has focused on vascular fibrinolysis, the generation of plasmin activity in the vicinity of the blood vessel wall, postulating that small amounts of this activity may protect the blood vessel from fibrin accumulation resulting from subliminal injury. We have hypothesized that cell surface receptors enable vascular fibrinolysis by providing a micro-environment that localizes and protects low-level fibrinolytic activity. The relevant receptors include the urokinase receptor (uPAR), which appears to be preferentially expressed on migrating endothelial cells (Pepper et al. 1993), and the annexin A2 complex, which is expressed on both resting and activated endothelial cells (Cesarman et al. 1994; Hajjar et al. 1994).
THE ENDOTHELIUM AND VASCULAR FIBRINOLYSIS
Collectively, endothelial cells (ECs) constitute the vascular endothelium, a “distributed organ” whose functional properties are versatile and specific to individual organ systems (Gimbrone 2007). The endothelium's intimate association with flowing blood allows it to participate dynamically in modulated secreted activity, selective permeability, and thromboresistance (Altschul 1954; Gimbrone 1980; Gimbrone 2007). ECs are heterogeneous in terms of their structure, protein synthetic repertoire, and response to environmental stimuli (Augustin et al. 1994; Cines et al. 1998; Durr et al. 2004; Furchgott and Zawadzki 1980; Garlanda and Dejana 1997; Gimbrone 1995; Gimbrone et al. 1993; Hajjar 2006; Oh et al. 2004). In regulating hemostasis, ECs possess both antiplatelet properties, through production of nitric oxide (NO) and prostacyclin (PGI2), and anticoagulant properties, through expression of thrombomodulin and the endothelial protein C receptor (Hajjar et al. 2001).
The fibrinolytic properties of endothelial cells are complex. Although the endothelium appears to be the major source of tPA in blood, tPA expression in vivo appears to be restricted to the microvasculature in specific anatomic locations (Hajjar et al. 2001), and is subject to stimulation by desmopressin (1-deamino-8-D-arginine vasopressin, DDAVP), bradykinin, endothelin, hyperoxia, and thrombin (Diamond et al. 1989; Dichek and Quertermous 1989; Kooistra et al. 1987; Levin et al. 1989). uPA and its receptor, on the other hand, are not highly expressed by resting endothelium, but are stimulated by inflammatory cytokines, during wound repair, and in physiologic angiogenesis (Bacharach et al. 1992; Pepper et al. 1993). Similarly, the physiologic inhibitor of both tPA and uPA, plasminogen activator inhibitor-1 (PAI-1), is expressed minimally by quiescent ECs, but increases in response to angiogenic or inflammatory cytokines. Finally, thrombin bound to the integral endothelial membrane protein, thrombomodulin, can activate a protein called thrombin-activatable fibrinolysis inhibitor (TAFI), which, as a carboxypeptidase, removes C-terminal Lys and Arg residues, the primary PLG and tPA binding sites on fibrin and many receptors (Bajzar et al. 1996). Thus, it seems that the endothelial cell is equipped with a number of mechanisms for the regulation of vessel associated fibrinolytic activity.
ENDOTHELIAL CELL RECEPTORS IN VASCULAR FIBRINOLYSIS
Plasminogen Receptors
Plasminogen (PLG) receptors are a diverse group of proteins expressed on a wide array of cell types (Miles et al. 2005). The most widely recognized PLG receptors include α-enolase and histone H2B on monocytoid cells, glycoprotein IIb/IIIa complex on platelets, the Heymann nephritis antigen on renal epithelial cells, amphoterin on neuroblastoma cells, and annexin A2 (A2)/protein p11 on ECs (Barnathan et al. 1988; Das et al. 2007; Hajjar 1991; Hajjar and Hamel 1990; Kanalas and Makker 1991; Miles et al. 1991; Miles et al. 1986; Parkkinen and Rauvala 1991). These binding proteins commonly possess a C-terminal Lys residue, either in the native state, or upon proteolytic processing, which allows them to interact with the lysine-binding “kringle” domains of PLG (Das et al. 2007; Miles et al. 1991). Receptor mediated binding of plasminogen serves first to protect plasmin (PN), once it has been activated, from its principal circulating inhibitor, α2-antiplasmin, and secondly to concentrate PN activity at the cell surface (Hajjar and Francis 2006).
UPAR
uPA is expressed by ECs, as well as monocytes, macrophages, fibroblasts, and a variety of tumor cells (Cines et al. 1998). While uPA appears to be absent in quiescent endothelium (Cines et al. 1998; Wojta et al. 1989), it is present in ECs involved in wound repair or angiogenesis. This is consistent with the hypothesized importance of uPA in cell migration and tissue remodeling (Cines et al. 1998; Wojta et al. 1989). uPAR, a three-domain protein linked to cell surfaces by a glycerophosphatidyl inositol (GPI) anchor (Cines et al. 1998), binds uPA and activates PLG. While uPA appears to contribute to vascular hemostasis, since mice genetically deficient in uPA show fibrin deposition in tissues (Carmeliet et al. 1994), uPAR has yet to be shown to participate in baseline hemostasis, since mice lacking uPAR develop normally and do not exhibit spontaneous vascular occlusion. uPAR may be important for vascular repair, however, since it may be expressed mostly on the surface of migrating ECs participating in angiogenesis, rather than on quiescent ECs lining normal vessels (Bugge et al. 1996; Pepper et al. 1993).
The (A2•p11)2 receptor complex
The annexins are a family of Ca2+ regulated, phopholipid-binding proteins, characterized by the unique architecture of their Ca2+ binding sites (Gerke et al. 2005). These structures enable the annexins to interact with anionic membrane phospholipids in their Ca2+-bound conformation, a property that appears to link many annexins to a range of membrane-related events, such as exocytosis, endocytosis, and the regulation of ion fluxes across membranes. Some annexins pair with members of the S100 family of Ca2+-binding proteins, which appear to modulate their function.
Protein p11, a member of the S100 family (S100A10), contains two EF hand motifs, structures consisting of two nearly perpendicular α-helices flanking a roughly 12-residue loop. EF-hand proteins usually bind to cellular targets, in response to changes in Ca2+ concentration (Gerke et al. 2005). p11, however, is an exception to the Ca2+ activation rule, since it permanently assumes a “Ca2+-on” conformation and can readily target A2 subunits. Consequently, partnering of p11 appears to be regulated mainly by the concentration of each binding partner, rather than by Ca2+ ion fluxes (He et al. 2008). In ECs, an increase in intracellular Ca2+ is predicted to mobilize (A2•p11)2 complexes to the inner surface of the plasma membrane (Thiel et al. 1992).
The function of A2 in fibrinolysis was discovered by members of our lab while seeking the EC surface receptor for PLG and tPA (Hajjar et al. 1994). In 1986, we reported for the first time that PLG could bind directly to cultured ECs with high affinity (Kd 300 nM) and specificity (Hajjar et al. 1986). We later found that the circulating form of PLG, N-terminal glutamic acid-plasminogen (Glu-PLG), was converted to a more readily activated form (N-terminal Lys-PLG) upon binding to ECs (Hajjar et al. 1986). These findings identified the EC surface as a profibrinolytic microenvironment. We subsequently discovered that PLG binding to human ECs could be inhibited in the presence of lipoprotein(a) (Lp(a)), a highly atherogenic lipoprotein particle whose apoprotein, apoprotein(a), is structurally homologous to PLG (Hajjar et al. 1989); these data implicated Lp(a) in atherogenesis through inhibition of EC surface fibrinolysis.
In 1987, we reported the first demonstration that tPA could bind to cultured human ECs at two independent sites with Kd's of 29 pM and 18 nM, respectively (Hajjar et al. 1987). The higher affinity site had characteristics of the physiologic plasminogen activator inhibitor type 1 (PAI-1), and could be blocked by uPA. The lower affinity site, on the other hand, appeared to be novel. Ligand blotting of an EC plasma membrane protein fraction revealed an approximately 40-kDa protein that bound tPA, but not uPA, thus distinguishing it from PAI-1 and uPAR (Hajjar and Hamel 1990). The protein was subsequently isolated from human placental membranes and shown, in both this preparation and in an EC membrane fraction, to interact specifically with both tPA and its substrate, PLG (Hajjar 1991). These findings suggested a common EC surface binding protein for tPA and PLG, somewhat reminiscent of fibrin.
Our identification of the tPA-PLG binding protein emanated from amino acid sequence analysis, which revealed a 100% match with residues 29-46 of annexin A2, then known as “annexin II” (Hajjar et al. 1994). In further investigations, antibody directed at authentic A2 blocked ~50% PLG and tPA binding to human ECs, and A2 antisense oligonucleotides blocked 50 and 40% of tPA and PLG binding to these cells, respectively. In addition, treatment of A2 with carboxypeptidase B (CPB) eliminated its ability to bind PLG, thus implicating a C-terminal Lys or Arg residue. Mutation of Lys307 to Ala specifically eliminated PLG binding as well, suggesting that PLG binding to A2 required a proteolytic processing event to liberate Lys307 at a new C-terminus.
In a companion study, native annexin A2 purified from human placenta bound tPA, PLG, and PN saturably (Cesarman et al. 1994). This A2 preparation increased the catalytic efficiency (kcat/Km) of tPA-dependent PLG activation by ~60-fold, but had no effect on uPA activation of PLG. The catalytic effect of A2 disappeared in the presence of the Lys analog ε-aminocaproic acid, or by treatment of A2 with CPB, again implicating an interaction between a C-terminal Lys or Arg of A2 and a Lys binding domain of PLG. These experiments raised the possibility of a C-terminal Lys residue for PLG binding, and demonstrated the fibrin-like cofactor-like behavior of A2 with respect to tPA-dependent PLG activation.
In follow-up studies, we explored the mechanism by which A2 interacted with the EC surface (Hajjar et al. 1996). A2 was biosynthetically labeled by cultured human ECs incubated with 35S-radiolabeled methionine, indicating direct synthesis by these cells. Moreover, in the presence of Ca2+, but not other divalent cations, both recombinant and native A2 showed high affinity, equilibrium binding to cultured ECs (Kd ~ 50 nM). This binding could be out-competed by phosphatidylserine (Ptd-L-Ser)-containing vesicles, by peptides mimicking the annexin A2 repeat 2, or upon mutation of the Asp161 residue that coordinates with the annexin repeat (Lys-Gly-Leu-Gly-Thr) sequence. Together, this study showed that the interaction of A2 with the EC surface involved both the Ca2+-dependent, anionic phospholipid-binding repeat 2 of A2, and Ptd-L-Ser moieties within the plasma membrane.
The tPA-binding domain of A2 was examined separately (Hajjar et al. 1998). While PLG bound only to native A2, suggesting the need for a proteolytic processing event to reveal a C-terminal Lys or Arg, tPA bound to both native and recombinant forms. Unlike full-length A2, however, the core fragment of A2 failed to compete for binding of tPA to full-length A2, thus implicating the N-terminal tail domain in this interaction. Indeed, residues Leu7-Cys8-Lys9-Leu10-Ser11-Leu12, and larger peptides containing this fragment, specifically blocked binding of tPA to A2. Mutation of Cys8, but neither Cys133, Cys262, nor Cys335 prevented binding of tPA to A2; further implicating the N-terminal region. Interestingly, when A2 was incubated in a purified protein system with the atherothrombotic amino acid, homocysteine (HC), A2 was modified by HC. This treatment blocked the ability of tPA to bind to A2. Finally, incubation of cultured ECs with 35S-HC led to metabolic labeling of A2 that was sensitive to protein reduction, suggesting a disulfide-mediated association between Cys8 and HC. These data revealed a binding domain for tPA in the N-terminal tail of A2, and showed its susceptibility to modification by HC, an agent highly associated with atherothrombotic vascular disease.
Based upon these data, our working model has postulated that both PLG and tPA interact with A2 within the (A2•p11)2 heterotetrameric complex at the EC surface. Our model suggests further that this assembly augments the catalytic efficiency of plasmin activation by 1-to 2-log orders-of-magnitude. The model predicts that the A2•p11 system contributes to blood vessel patency, i.e. that gain-of-function would lead to hemorrhage, and that loss-of-function would lead to vascular thrombosis. To test this hypothesis, we have carried out a series of studies both in vitro and in vivo. Using homologous recombination, we created a mouse globally deficient in A2, and discovered that it displayed both fibrin accumulation, mainly in microvessels, and defects in angiogenesis (Ling et al. 2004). Interestingly, these animals, in retrospect, also expressed very low levels of p11. In humans, overexpression of A2 in blast cells from patients with acute promyelocytic leukemia correlated with hyperfibrinolysis and hemorrhage (Menell et al. 1999). In patients with antiphospholipid syndrome, on the other hand, we found high titers of anti-A2 IgG that were associated with major thrombotic episodes. Additionally, in vascular injury models, A2-/- mice exhibited an increase in the degree of thrombotic vascular occlusion, compared to basal conditions, with an equivalent decrease in blood flow recovery. In rats, treatment with recombinant A2 counteracted the onset of carotid artery thrombosis (Ishii et al. 2001), and reduced cerebral infarct size secondary to increased cerebral blood flow after induced embolic stroke (Tanaka et al. 2007). Taken together, these findings support our hypothesis that the annexin A2 system functions to promote vascular fibrinolysis.
Additional studies have suggested that the proenzymatic activity of (A2•p11)2 is not limited to ECs. Annexin A2 supports macrophage matrix invasion and degradation (Falcone et al. 2001). Moreover, monocytes, the major A2-expressing cells in circulating blood, express cell surface A2, and augment its expression upon differentiation into macrophages (Brownstein et al. 2004). Annexin A2 appears to support neuritogenesis of PC12 cell in vitro (Jacovina et al. 2001). In chick embryo, A2 promotes epithelial mesenchymal transformation during heart development by enabling plasmin-mediated activation of transforming growth factor ß3 (Krishnan et al. 2004). These data, therefore further suggest that the A2•p11 system facilitates the directed migration and/or remodeling of a spectrum of cell types.
PRIMARY BINDING SITES FOR PLASMIN GENERATION
With regard to alternative models for Plg and tPA binding to the (A2•p11)2 complex, another group has investigated these interactions. In 1998, it was reported that, while recombinant A2 tetramer (A2t) stimulated tPA-dependent PLG activation, it inhibited plasmin-mediated fibrinolysis in a purified system (Choi et al. 1998). This latter inhibitory effect was attributed to the in vitro observation of PN inactivation by autoproteolysis (Fitzpatrick et al. 2000). The same group reported that A2 binds PN, but not PLG (MacLeod et al. 2003), suggesting that p11, rather than A2, is the true binding site relevant to PN generation. It was further reported that, in a purified protein system, recombinant protein p11, which has a Lys-Gly-Lys-Lys sequence at its C-terminus, binds both tPA and PLG through Lys binding sites, whereas recombinant A2 monomer does not (Kassam et al. 1998).
While our group has not examined interactions between PLG and p11 directly, it seems quite possible that p11 could serve as a cellular PLG binding site under some circumstances. With regard to A2 binding of PLG, however, it is important to note that the studies cited above do not take into account our previous work showing that proteolytic processing of A2 appears to expose C-terminal Lys residues, converting A2 into a PLG-binding protein. This possibility has been reiterated in a study from a third group in which the authors found that, in addition to α-enolase and histone 2B, both A2 and p11 could serve as PLG receptors in murine and human macrophage-like cells (Das et al. 2007). While binding of PLG to purified, recombinant A2, required cleavage of the recombinant protein by trypsin, intact cells showed colocalization of PLG and A2 by confocal microscopy. These authors demonstrated further that this association was sensitive to carboxypeptidase B, indicating that proteolytic processing of A2 had already occurred. These data emphasize that A2 is processed to a PLG-binding form at the surface of monocytoid cells and perhaps other cells as well, and that data generated from recombinant proteins in purified systems may not reflect their full behavior in the cellular context.
REGULATION OF (A2•p11)2 ENDOTHELIAL CELL SURFACE EXPRESSION
Though there is a spectrum of evidence supporting the central role of the A2 system in fibrinolysis, the mechanisms governing A2 cell surface expression have only recently been addressed. It is now clear that the regulation of heterotetrameric (A2•p11)2 cell surface expression is governed by intracellular signaling pathways that respond to changes in the extracellular environment. Key factors that influence EC surface expression and function of (A2•p11)2 are oxidative stress (Rowan et al. 2002; Sullivan et al. 2000), heat stress (Deora et al. 2004), and thrombin stimulation (Bhattacharjee et al. 2008; Peterson et al. 2003).
Under oxidative stress, A2 becomes more susceptible to the incorporation of prothrombic amino acid homocysteine (HC) (Hajjar and Jacovina 1998). In the presence of mid-to-high micromolar HC, there appears to be reduced EC surface potential for tPA-dependent PLG activation due to blockade of tPA binding to A2 by derivatization Cys8 within the N-tail domain of annexin A2 (Hajjar 1993; Hajjar et al. 1998). In this situation, the stress-associated modification of A2 alters the properties of the protein (Gerke et al. 2005) and impairs the function of the (A2•p11)2 complex.
On the other hand, our group has shown, that temperature-stress stimulates translocation of A2 to the EC surface, increasing the potential for plasmin generation (Deora et al. 2004). In recent studies, Deora et al. injected GFP-tagged A2 cDNAs into human ECs, and noted that when the injected cells were exposed to a brief increase in temperature (42° C), there was a doubling or tripling of the cell surface expression of A2. This process required both tyrosine phosphorylation of A2 and the presence of protein p11. Interestingly, ECs treated with the protein synthesis inhibitor, cyclohexamide, exhibited enhanced, rather than repressed, translocation at both 37° C and 42° C. There was no change in steady-state mRNA levels during heat stress (Deora et al. 2004). In an additional experiment, HUVECs treated with brefeldin A, a drug that disrupts the endoplasmic reticulum (ER)-Golgi complex did not decrease the ability for A2 to be expressed on the cell surface. It was therefore concluded that A2 translocation occurs independently of the classical ER-Golgi pathway, and does not require de novo protein synthesis (Deora et al. 2004).
At the intracellular surface of the plasma membrane, (A2•p11)2 is susceptible to phosphorylation (Bellagamba et al. 1997; Okuse et al. 2002). A2 is a known substrate for both serine/threonine and tyrosine protein kinases (Beaton et al. 2002). Previous studies in HEK 293 cells have shown that Tyr23 of A2 is a target for phosphorylation by the src-family kinase, pp60-c-src kinase (Glenney and Tack 1985). Src-kinase is activated when Tyr527 in the C-terminus kinase domain of pp60-c-src is dephosphorylated, and Tyr416 remains phosphorylated (Cooper and King 1986). When Tyr23 in the tail domain of A2 is mutated, preventing phosphorylation, neither translocation nor phosphorylation of A2 occurs. Once phosphorylated, the new phospho-(A2•p11)2 heterotetramer is predicted to couple more tightly to inner leaflet anionic Ptd-L-Ser (Montaville et al. 2002) and possibly undergo conformational changes that enhance its ability to be translocated to the outer membrane surface.
In a recent study focusing on p11, our group showed that p11 is unstable and rapidly degraded in the EC in the absence of A2 (He et al. 2008). When HEK 293 cells, which express very low levels of endogenous p11 and A2, were transfected with FLAG-p11, unbound p11 monomers were observed to be rapidly ubiquitinated and degraded through a proteasome-dependent mechanism (He et al. 2008). Co-transfection of A2, however, rescued p11, which bound to the 13 N-terminal amino acids in the A2 tail peptide, thus masking an autonomous p11 ubiquitination signal. Ubiquitination of p11 required residues Tyr86-Gly95, near its A2-binding C-terminal motif. These studies suggest that A2 directly regulates intracellular levels of p11 in endothelial cells.
CONCLUDING REMARKS
Partnering of A2 and p11 to form the (A2•p11)2 complex is a cooperative process: A2 binds and stabilizes p11, thereby regulating the intracellular concentration of p11. Intracellular p11 levels, on the other hand, are likely to determine how much (A2•p11)2 translocates to the cell surface. Stress-induced stimuli, such as heat shock or the action of thrombin, can activate a cascade of events that enhance translocation to the cell surface through an ER-Golgi independent pathway (Figure 1). Further understanding of the regulation of the (A2•p11)2 system will be important for evaluation of the proposed functions for A2 and p11 in a wide range of cell and tissue types (Foulkes et al. 2006; Liu et al. 2003; Sharma and Sharma 2007; Tsai 2007). For instance, the reported role of A2 in angiogenesis and fibrin homeostasis (Ling et al. 2004) may contribute to our understanding of cancer cell biology, retinal disease, and vascular occlusion. Forthcoming investigative studies should be not only exciting, but also useful for developing future therapies.
Figure 1.
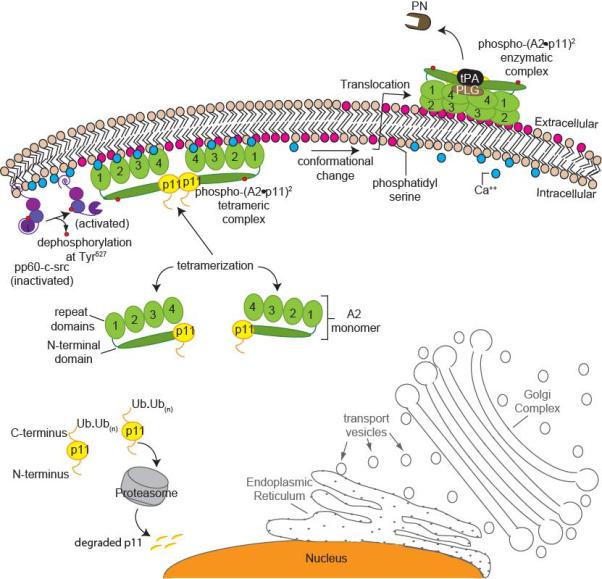
A stress-related working model for heterotetrameric (A2•p11)2 complex formation and translocation to the cell surface. (A2•p11)2 cell surface translocation and activation is independent of the endoplasmic reticulum (ER)-Golgi pathway (outlined in grey). Unpartnered p11 monomers (yellow) within the cell are polyubiquitinated and degraded by the proteasome (grey). Annexin A2 (A2) monomers (green) are bound to p11 subunits. Under stress, intracellular calcium levels increase in response to various stimuli (blue), and this increases the affinity of the (A2•p11)2 complex for binding to anionic phospholipid (shown in dark pink) at the inner membrane surface. There, pp60-c-src kinase (purple) becomes activated following dephosphorylation of Tyr527 within its kinase domain, while Tyr416 within its SH2 domain remains phosphorylated. Once the (A2•p11)2 complex is phosphorylated (red circles) by pp60-c-src, it becomes more tightly associated with inner leaflet phosphatidyl serine (Ptd-L-Ser). The newly phospho-(A2•p11)2 complex may undergo a conformational change, making it more susceptible to translocation to the outer membrane surface. On the outer membrane surface, phospho-(A2•p11)2 associates with plasminogen (PLG) and tissue plasminogen activator (tPA), giving rise to active plasmin (PN).
REFERENCES
- Altschul R. Endothelium: its development, morphology, function, and pathology. Macmillan; New York: 1954. [Google Scholar]
- Augustin HG, Kozian DH, Johnson RC. Differentiation of endothelial cells: analysis of the constitutive and activated endothelial cell phenotypes. Bioessays. 1994;16:901–906. doi: 10.1002/bies.950161208. [DOI] [PubMed] [Google Scholar]
- Bacharach E, Itin A, Keshet E. In vivo patterns of expression of urokinase and its inhibitor PAI-1 suggest a concerted role in regulating physiological angiogenesis. Proc Natl Acad Sci U S A. 1992;89:10686–10690. doi: 10.1073/pnas.89.22.10686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajzar L, Morser J, Nesheim M. TAFI, or plasma procarboxypeptidase B, couples the coagulation and fibrinolytic cascades through the thrombin-thrombomodulin complex. J Biol Chem. 1996;271:16603–16608. doi: 10.1074/jbc.271.28.16603. [DOI] [PubMed] [Google Scholar]
- Barnathan ES, Kuo A, Van der Keyl H, McCrae KR, Larsen GR, et al. Tissue-type plasminogen activator binding to human endothelial cells. Evidence for two distinct binding sites. J Biol Chem. 1988;263:7792–7799. [PubMed] [Google Scholar]
- Beaton AR, Rodriguez J, Reddy YK, Roy P. The membrane trafficking protein calpactin forms a complex with bluetongue virus protein NS3 and mediates virus release. Proc Natl Acad Sci U S A. 2002;99:13154–13159. doi: 10.1073/pnas.192432299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellagamba C, Hubaishy I, Bjorge JD, Fitzpatrick SL, Fujita DJ, et al. Tyrosine phosphorylation of annexin II tetramer is stimulated by membrane binding. J Biol Chem. 1997;272:3195–3199. doi: 10.1074/jbc.272.6.3195. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee G, Ahamed J, Pawlinski R, Liu C, Mackman N, et al. Factor Xa binding to annexin 2 mediates signal transduction via protease-activated receptor 1. Circ Res. 2008;102:457–464. doi: 10.1161/CIRCRESAHA.107.167759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownstein C, Deora AB, Jacovina AT, Weintraub R, Gertler M, et al. Annexin II mediates plasminogen-dependent matrix invasion by human monocytes: enhanced expression by macrophages. Blood. 2004;103:317–324. doi: 10.1182/blood-2003-04-1304. [DOI] [PubMed] [Google Scholar]
- Bugge TH, Flick MJ, Danton MJ, Daugherty CC, Romer J, et al. Urokinase-type plasminogen activator is effective in fibrin clearance in the absence of its receptor or tissue-type plasminogen activator. Proc Natl Acad Sci U S A. 1996;93:5899–5904. doi: 10.1073/pnas.93.12.5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Schoonjans L, Kieckens L, Ream B, Degen J, et al. Physiological consequences of loss of plasminogen activator gene function in mice. Nature. 1994;368:419–424. doi: 10.1038/368419a0. [DOI] [PubMed] [Google Scholar]
- Cesarman-Maus G, Hajjar KA. Molecular mechanisms of fibrinolysis. Br J Haematol. 2005;129:307–321. doi: 10.1111/j.1365-2141.2005.05444.x. [DOI] [PubMed] [Google Scholar]
- Cesarman GM, Guevara CA, Hajjar KA. An endothelial cell receptor for plasminogen/tissue plasminogen activator (t-PA). II. Annexin II-mediated enhancement of t-PA-dependent plasminogen activation. J Biol Chem. 1994;269:21198–21203. [PubMed] [Google Scholar]
- Choi KS, Ghuman J, Kassam G, Kang HM, Fitzpatrick SL, et al. Annexin II tetramer inhibits plasmin-dependent fibrinolysis. Biochemistry. 1998;37:648–655. doi: 10.1021/bi971005k. [DOI] [PubMed] [Google Scholar]
- Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. 1998;91:3527–3561. [PubMed] [Google Scholar]
- Cooper JA, King CS. Dephosphorylation or antibody binding to the carboxy terminus stimulates pp60c-src. Mol Cell Biol. 1986;6:4467–4477. doi: 10.1128/mcb.6.12.4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Burke T, Plow EF. Histone H2B as a functionally important plasminogen receptor on macrophages. Blood. 2007;110:3763–3772. doi: 10.1182/blood-2007-03-079392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deora AB, Kreitzer G, Jacovina AT, Hajjar KA. An annexin 2 phosphorylation switch mediates p11-dependent translocation of annexin 2 to the cell surface. J Biol Chem. 2004;279:43411–43418. doi: 10.1074/jbc.M408078200. [DOI] [PubMed] [Google Scholar]
- Diamond SL, Eskin SG, McIntire LV. Fluid flow stimulates tissue plasminogen activator secretion by cultured human endothelial cells. Science. 1989;243:1483–1485. doi: 10.1126/science.2467379. [DOI] [PubMed] [Google Scholar]
- Dichek D, Quertermous T. Thrombin regulation of mRNA levels of tissue plasminogen activator and plasminogen activator inhibitor-1 in cultured human umbilical vein endothelial cells. Blood. 1989;74:222–228. [PubMed] [Google Scholar]
- Durr E, Yu J, Krasinska KM, Carver LA, Yates JR, et al. Direct proteomic mapping of the lung microvascular endothelial cell surface in vivo and in cell culture. Nat Biotechnol. 2004;22:985–992. doi: 10.1038/nbt993. [DOI] [PubMed] [Google Scholar]
- Falcone DJ, Borth W, Khan KM, Hajjar KA. Plasminogen-mediated matrix invasion and degradation by macrophages is dependent on surface expression of annexin II. Blood. 2001;97:777–784. doi: 10.1182/blood.v97.3.777. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick SL, Kassam G, Choi KS, Kang HM, Fogg DK, et al. Regulation of plasmin activity by annexin II tetramer. Biochemistry. 2000;39:1021–1028. doi: 10.1021/bi991411z. [DOI] [PubMed] [Google Scholar]
- Foulkes T, Nassar MA, Lane T, Matthews EA, Baker MD, et al. Deletion of annexin 2 light chain p11 in nociceptors causes deficits in somatosensory coding and pain behavior. J Neurosci. 2006;26:10499–10507. doi: 10.1523/JNEUROSCI.1997-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- Garlanda C, Dejana E. Heterogeneity of endothelial cells. Specific markers. Arterioscler Thromb Vasc Biol. 1997;17:1193–1202. doi: 10.1161/01.atv.17.7.1193. [DOI] [PubMed] [Google Scholar]
- Gerke V, Creutz CE, Moss SE. Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol. 2005;6:449–461. doi: 10.1038/nrm1661. [DOI] [PubMed] [Google Scholar]
- Gimbrone MA. Endothelial dysfunction and the pathogenesis of atheroscerosis. In: Gotto A, editor. Atherosclerosis-V, Proceedings of the Vth International Symposium on Atherosclerosis. Springer-Verlag; New York: 1980. pp. 415–425. [Google Scholar]
- Gimbrone MA. Vascular Endothelium in health and disease. In: Habor E, editor. Molecular Cardiovascular Medicine. Scientific American Medicine; New York: 1995. pp. 67B–70B. [Google Scholar]
- Gimbrone MA. Functions and dysfunctions of vascular endothelium: nature's container for blood. In: De Caterina R, Libby P, editors. Endothelial dysfunctions and vascular disease. Blackwell Futura; Malden, MA: 2007. pp. xi–xii. [Google Scholar]
- Gimbrone MA, Kume N, Cybulsky MI. Vascular endothelial dysfunction and the pathogenesis of atherosclerosis. In: Weber PC, Leaf A, editors. Atherosclerosis Reviews. Reaven Press; New York: 1993. [Google Scholar]
- Glenney JR, Jr., Tack BF. Amino-terminal sequence of p36 and associated p10: identification of the site of tyrosine phosphorylation and homology with S-100. Proc Natl Acad Sci U S A. 1985;82:7884–7888. doi: 10.1073/pnas.82.23.7884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajjar KA. The endothelial cell tissue plasminogen activator receptor. Specific interaction with plasminogen. J Biol Chem. 1991;266:21962–21970. [PubMed] [Google Scholar]
- Hajjar KA. Homocysteine-induced modulation of tissue plasminogen activator binding to its endothelial cell membrane receptor. J Clin Invest. 1993;91:2873–2879. doi: 10.1172/JCI116532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajjar KA. The Molecular Basis of Fibrinolysis. In: Nathan DG, Orkin SH, Ginsberg D, Look AT, Fisher DE, Lux SE, editors. Hematology of Infancy and Childhood. 7th Ed. Saunders Elsevier; Philadelphia: 2009. pp. 1425–1447. [Google Scholar]
- Hajjar KA. Vascular Biology. In: Young NS, Gerson SL, High KA, editors. Clinical hematology. Mosby/Elsevier; Philadelphia: 2006. pp. 123–133. [Google Scholar]
- Hajjar KA, Esmon NL, Marcus AJ, Muller AJ. Vascular Function in Hemostasis. In: Beutler E, Williams WJ, editors. Williams hematology. McGraw-Hill, Medical Publishing Division; New York: 2001. pp. 1451–1471. [Google Scholar]
- Hajjar KA, Francis CW. Fibrinolysis and Thrombolysis. In: Lichtman MA, Williams WJ, editors. Williams hematology. McGraw-Hill; Medical Pub. Division; New York: 2006. pp. 2089–2115. [Google Scholar]
- Hajjar KA, Gavish D, Breslow JL, Nachman RL. Lipoprotein(a) modulation of endothelial cell surface fibrinolysis and its potential role in atherosclerosis. Nature. 1989;339:303–305. doi: 10.1038/339303a0. [DOI] [PubMed] [Google Scholar]
- Hajjar KA, Guevara CA, Lev E, Dowling K, Chacko J. Interaction of the fibrinolytic receptor, annexin II, with the endothelial cell surface. Essential role of endonexin repeat 2. J Biol Chem. 1996;271:21652–21659. doi: 10.1074/jbc.271.35.21652. [DOI] [PubMed] [Google Scholar]
- Hajjar KA, Hamel NM. Identification and characterization of human endothelial cell membrane binding sites for tissue plasminogen activator and urokinase. J Biol Chem. 1990;265:2908–2916. [PubMed] [Google Scholar]
- Hajjar KA, Hamel NM, Harpel PC, Nachman RL. Binding of tissue plasminogen activator to cultured human endothelial cells. J Clin Invest. 1987;80:1712–1719. doi: 10.1172/JCI113262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajjar KA, Harpel PC, Jaffe EA, Nachman RL. Binding of plasminogen to cultured human endothelial cells. J Biol Chem. 1986;261:11656–11662. [PubMed] [Google Scholar]
- Hajjar KA, Jacovina AT. Modulation of annexin II by homocysteine: implications for atherothrombosis. J Investig Med. 1998;46:364–369. [PubMed] [Google Scholar]
- Hajjar KA, Jacovina AT, Chacko J. An endothelial cell receptor for plasminogen/tissue plasminogen activator. I. Identity with annexin II. J Biol Chem. 1994;269:21191–21197. [PubMed] [Google Scholar]
- Hajjar KA, Mauri L, Jacovina AT, Zhong F, Mirza UA, et al. Tissue plasminogen activator binding to the annexin II tail domain. Direct modulation by homocysteine. J Biol Chem. 1998;273:9987–9993. doi: 10.1074/jbc.273.16.9987. [DOI] [PubMed] [Google Scholar]
- He KL, Deora AB, Xiong H, Ling Q, Weksler BB, et al. Endothelial cell annexin A2 regulates polyubiquitination and degradation of its binding partner S100A10/p11. J Biol Chem. 2008;283:19192–19200. doi: 10.1074/jbc.M800100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii H, Yoshida M, Hiraoka M, Hajjar KA, Tanaka A, et al. Recombinant annexin II modulates impaired fibrinolytic activity in vitro and in rat carotid artery. Circ Res. 2001;89:1240–1245. doi: 10.1161/hh2401.101066. [DOI] [PubMed] [Google Scholar]
- Jacovina AT, Zhong F, Khazanova E, Lev E, Deora AB, et al. Neuritogenesis and the nerve growth factor-induced differentiation of PC-12 cells requires annexin II-mediated plasmin generation. J Biol Chem. 2001;276:49350–49358. doi: 10.1074/jbc.M106289200. [DOI] [PubMed] [Google Scholar]
- Kanalas JJ, Makker SP. Identification of the rat Heymann nephritis autoantigen (GP330) as a receptor site for plasminogen. J Biol Chem. 1991;266:10825–10829. [PubMed] [Google Scholar]
- Kassam G, Choi KS, Ghuman J, Kang HM, Fitzpatrick SL, et al. The role of annexin II tetramer in the activation of plasminogen. J Biol Chem. 1998;273:4790–4799. doi: 10.1074/jbc.273.8.4790. [DOI] [PubMed] [Google Scholar]
- Kim J, Hajjar KA. Annexin II: a plasminogen-plasminogen activator co-receptor. Front Biosci. 2002;7:d341–348. doi: 10.2741/kim. [DOI] [PubMed] [Google Scholar]
- Kooistra T, van den Berg J, Tons A, Platenburg G, Rijken DC, et al. Butyrate stimulates tissue-type plasminogen-activator synthesis in cultured human endothelial cells. Biochem J. 1987;247:605–612. doi: 10.1042/bj2470605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan S, Deora AB, Annes JP, Osoria J, Rifkin DB, et al. Annexin II-mediated plasmin generation activates TGF-beta3 during epithelial-mesenchymal transformation in the developing avian heart. Dev Biol. 2004;265:140–154. doi: 10.1016/j.ydbio.2003.08.026. [DOI] [PubMed] [Google Scholar]
- Levin EG, Marotti KR, Santell L. Protein kinase C and the stimulation of tissue plasminogen activator release from human endothelial cells. Dependence on the elevation of messenger RNA. J Biol Chem. 1989;264:16030–16036. [PubMed] [Google Scholar]
- Ling Q, Jacovina AT, Deora A, Febbraio M, Simantov R, et al. Annexin II regulates fibrin homeostasis and neoangiogenesis in vivo. J Clin Invest. 2004;113:38–48. doi: 10.1172/JCI200419684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JW, Shen JJ, Tanzillo-Swarts A, Bhatia B, Maldonado CM, et al. Annexin II expression is reduced or lost in prostate cancer cells and its re-expression inhibits prostate cancer cell migration. Oncogene. 2003;22:1475–1485. doi: 10.1038/sj.onc.1206196. [DOI] [PubMed] [Google Scholar]
- MacLeod TJ, Kwon M, Filipenko NR, Waisman DM. Phospholipid-associated annexin A2-S100A10 heterotetramer and its subunits: characterization of the interaction with tissue plasminogen activator, plasminogen, and plasmin. J Biol Chem. 2003;278:25577–25584. doi: 10.1074/jbc.M301017200. [DOI] [PubMed] [Google Scholar]
- Menell JS, Cesarman GM, Jacovina AT, McLaughlin MA, Lev EA, et al. Annexin II and bleeding in acute promyelocytic leukemia. N Engl J Med. 1999;340:994–1004. doi: 10.1056/NEJM199904013401303. [DOI] [PubMed] [Google Scholar]
- Miles LA, Hawley SB, Baik N, Andronicos NM, Castellino FJ, Parmer RJ. Plasminogen receptors: the sine qua non of cell surface plasminogen activation. Front Biosci. 2005;10:1754–1762. doi: 10.2741/1658. [DOI] [PubMed] [Google Scholar]
- Miles LA, Dahlberg CM, Plescia J, Felez J, Kato K, et al. Role of cell-surface lysines in plasminogen binding to cells: identification of alpha-enolase as a candidate plasminogen receptor. Biochemistry. 1991;30:1682–1691. doi: 10.1021/bi00220a034. [DOI] [PubMed] [Google Scholar]
- Miles LA, Ginsberg MH, White JG, Plow EF. Plasminogen interacts with human platelets through two distinct mechanisms. J Clin Invest. 1986;77:2001–2009. doi: 10.1172/JCI112529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montaville P, Neumann JM, Russo-Marie F, Ochsenbein F, Sanson A. A new consensus sequence for phosphatidylserine recognition by annexins. J Biol Chem. 2002;277:24684–24693. doi: 10.1074/jbc.M109595200. [DOI] [PubMed] [Google Scholar]
- Oh P, Li Y, Yu J, Durr E, Krasinska KM, et al. Subtractive proteomic mapping of the endothelial surface in lung and solid tumours for tissue-specific therapy. Nature. 2004;429:629–635. doi: 10.1038/nature02580. [DOI] [PubMed] [Google Scholar]
- Okuse K, Malik-Hall M, Baker MD, Poon WY, Kong H, et al. Annexin II light chain regulates sensory neuron-specific sodium channel expression. Nature. 2002;417:653–656. doi: 10.1038/nature00781. [DOI] [PubMed] [Google Scholar]
- Parkkinen J, Rauvala H. Interactions of plasminogen and tissue plasminogen activator (t-PA) with amphoterin. Enhancement of t-PA-catalyzed plasminogen activation by amphoterin. J Biol Chem. 1991;266:16730–16735. [PubMed] [Google Scholar]
- Pepper MS, Sappino AP, Stocklin R, Montesano R, Orci L, et al. Upregulation of urokinase receptor expression on migrating endothelial cells. J Cell Biol. 1993;122:673–684. doi: 10.1083/jcb.122.3.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson EA, Sutherland MR, Nesheim ME, Pryzdial EL. Thrombin induces endothelial cell-surface exposure of the plasminogen receptor annexin 2. J Cell Sci. 2003;116:2399–2408. doi: 10.1242/jcs.00434. [DOI] [PubMed] [Google Scholar]
- Ratnoff OD, Forbes CD. Disorders of hemostasis. Grune & Stratton, Orlando. 1984:8. [Google Scholar]
- Rowan WH, 3rd, Sun P, Liu L. Nitration of annexin II tetramer. Biochemistry. 2002;41:1409–1420. doi: 10.1021/bi0119767. [DOI] [PubMed] [Google Scholar]
- Sharma MC, Sharma M. The role of annexin II in angiogenesis and tumor progression: a potential therapeutic target. Curr Pharm Des. 2007;13:3568–3575. doi: 10.2174/138161207782794167. [DOI] [PubMed] [Google Scholar]
- Sullivan DM, Wehr NB, Fergusson MM, Levine RL, Finkel T. Identification of oxidant-sensitive proteins: TNF-alpha induces protein glutathiolation. Biochemistry. 2000;39:11121–11128. doi: 10.1021/bi0007674. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Ishii H, Hiraoka M, Miyasaka N, Kuroiwa T, et al. Efficacy of recombinant annexin 2 for fibrinolytic therapy in a rat embolic stroke model: a magnetic resonance imaging study. Brain Res. 2007;1165:135–143. doi: 10.1016/j.brainres.2007.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel C, Osborn M, Gerke V. The tight association of the tyrosine kinase substrate annexin II with the submembranous cytoskeleton depends on intact p11- and Ca(2+)-binding sites. J Cell Sci. 1992;103(Pt 3):733–742. doi: 10.1242/jcs.103.3.733. [DOI] [PubMed] [Google Scholar]
- Todd AS. Localization of Fibrinolytic Activity in Tissues. Br Med Bull. 1964;20:210–212. doi: 10.1093/oxfordjournals.bmb.a070333. [DOI] [PubMed] [Google Scholar]
- Tsai SJ. The P11, tPA/plasminogen system and brain-derived neurotrophic factor: Implications for the pathogenesis of major depression and the therapeutic mechanism of antidepressants. Med Hypotheses. 2007;68:180–183. doi: 10.1016/j.mehy.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Wojta J, Hoover RL, Daniel TO. Vascular origin determines plasminogen activator expression in human endothelial cells. Renal endothelial cells produce large amounts of single chain urokinase type plasminogen activator. J Biol Chem. 1989;264:2846–2852. [PubMed] [Google Scholar]
- Yamamoto K, Loskutoff DJ. Fibrin deposition in tissues from endotoxin-treated mice correlates with decreases in the expression of urokinase-type but not tissue-type plasminogen activator. J Clin Invest. 1996;97:2440–2451. doi: 10.1172/JCI118691. [DOI] [PMC free article] [PubMed] [Google Scholar]