

Synopsis
The effects of switching the chirality of a single layer of amino acids in a three stranded coiled coil has been investigated. X-ray crystallography reveals that this modification is well tolerated and does not alter the designed structure. In contrast, spectroscopic studies of cadmium binding to both the L- and D- enantiomers of the penicillamine, provide evidence that this switch dramatically alters the metal binding capability, the resulting coordination environment and the position of binding.
Keywords: de novo peptides, crystallography, protein design, D-amino acids, cadmium
De novo metallopeptide design contributes significantly to the understanding of protein folding, protein-protein interactions and metal ion sites in biology.[1,2] Within this rubric, work in our research group has been focused on the biochemistry of Cd(II) thiolate sites. A class of peptides based on the parent peptide TRI, Ac-G(LKALEEK)4G-NH2, was designed to assemble in aqueous solution into amphiphilic α-helices. These peptides aggregate to form three stranded coiled coils above pH 5.5.[3] Substitutions can be made to the interior of these coiled coils to generate metal binding sites, potentially with adjacent cavities.[4–7] One benefit of de novo design is that we are no longer limited to the 20 naturally coded amino acids. We demonstrated using two different strategies, that one can achieve a coordinatively unsaturated trigonal planar CdS3 site, by incorporating non-protein amino acids into the peptide sequence. The first approach was to substitute the coordinating L-cysteine (L-Cys) with the bulkier non-protein analogue L-penicillamine (L-Pen), in which the β-methylene protons have been replaced with larger methyl groups.[8] Our second approach was to modify the chirality of the second coordination sphere ligands directly above the metal binding plane (L-Leu → D-Leu), reorienting the Leu side chain towards the C-terminus and the metal binding site. Presumably, this modification sterically inhibits a fourth ligand from binding to the Cd(II).[9,10]
Although these were significant accomplishments, we soon realized that we neither understood how L-Pen was capable of enforcing a CdS3 coordination number nor how introducing a single layer of D-amino acids in the interior of a coiled coil composed of L-amino acids would affect the structure. We also realized that we could now assess how metal binding would be perturbed by combining increased steric bulk and alternate chirality within the same amino acid. One might expect that simple conversion from an L- to D-ligand would lead to the same coordination geometry with inverted stereochemical configuration; however, since these are diastereopeptides, this stereochemical modification is occurring within a sea of L-amino acids. These issues could be best addressed by performing comparative spectroscopic and structural studies on three-stranded coiled coils containing either L-Pen or D-Pen (Figure S1). We chose to perform experiments on the related peptide Coil Ser (CS),[11] Ac-E WEALEKK LAALESK LQALEKK LEALEHG-NH2, with L- and D-Pen substituted at the 16 position (CSL16L-Pen and CSL16D-Pen), as Coil Ser behaves similarly to the TRI family with which the majority of work was performed,[12] yet has proven to be more readily crystallizable.[13,14]
First, we explored the interactions of these peptides in aqueous solution with Cd(II). 113Cd NMR spectroscopy is a powerful tool for determining Cd(II) coordination environments.[15] The 113Cd NMR spectrum for Cd(II) bound to the L-amino acid analogue of Pen, CSL16L-Pen, shows a single resonance at 683 ppm. This is almost identical to that reported previously for the TRI analogue (684 ppm) confirming that Cd(II) is bound as a trigonal planar three-coordinate species.[8] The D-Pen analogue results in a weaker intensity resonance at 557 ppm which is far from the range associated with CdS3 (680–700 ppm) and more consistent with [15] an S2O2 (or possibly S3O) type first coordination sphere environment (Figure S2).
The binding of Cd(II) to these constructs can also be monitored by UV-visible spectroscopy, and once again was found to be different for the L- and D-Pen analogues (Figure S3). The characteristic LMCT band for Cd(II) bound to L-Pen as Cd(CSL16L-Pen)3− is similar (λmax 227 nm, ε227 36 000 M−1cm−1) to that reported for Cd(TRIL16L-Pen)3−.[16] In contrast, an absorption band with λmax around 210 nm appears for solutions containing Cd(II) in the presence of CSL16D-Pen at pH 9.6, a pH at which we generally expect Cd(II) to be fully bound.
To determine the pH dependence associated with Cd(II) binding we monitored the absorbance as a function of pH. The resulting pH titration curves (Figure S4) reveal that Cd(II) binds to CSL16L-Pen with a high pKa2 (16.3) which is consistent with the behavior of Cd(II) binding to a three-coordinate site.[15,16] However, no significant increase in absorbance is observed for CSL16D-Pen until the pH is >9, at which point unfolding of the apo peptide contributes to the signal (Figure S4). The behavior is not consistent with Cd bound to three thiolates (either CdS3 or CdS3O). However, it would be consistent with a more sulfur poor coordination environment such as CdS2O2 (or CdSO3).
Although the first coordination sphere of the Cd(II) ion is not chiral when bound in the interior of these coiled coils, the peptide environment conveys chirality onto the metal resulting in distinct CD (circular dichroism) difference spectra. Similar transitions, both with negative intensity at 257 nm, are present in the CD difference spectra when Cd(II) is bound to either CSL16L-Pen or TRIL16L-Pen.[17] Again, no transition is observed in analogous experiments performed with CSL16D-Pen (Figure S5).
The combination of 113Cd NMR, UV-vis and CD spectroscopies supports the conclusion that Cd(II) binds as a trigonal three-coordinate species to L-Pen substituted Coil Ser. In contrast, Cd(II) binds to D-Pen radically differently, with only a small fraction of Cd(II) binding and then doing so with a mixed first coordination sphere which is likely four coordinate CdS2O2 (where O are exogenous water molecules).
Clearly, metal binding is very different upon inverting the chirality of a layer of amino acid ligands in the interior of a three-stranded coiled coil. Before interpreting these data fully it is important to understand how chiral inversion changes the peptide construct in the absence of metal ions. CD spectra demonstrate that the construct with a layer of L-Pen results in a well folded coiled coil (negative maxima at 208 and 222 nm; >90% folded), while replacing this ligand with D-Pen the ellipticity at 222 nm reduces in intensity, although still indicative of a right-handed α-helix, suggesting that CSL16D-Pen is less well folded (ca. 50%) than the L-Pen analogue (Figure S6A). Consistent with this conclusion, CSL16D-Pen is less stable with respect to denaturant (guanidine hydrochloride) than CSL16L-Pen, with relative half points at ca. 0.4 and 1.5 M denaturant, respectively (Figure S6B). It has been reported that introduction of a single D-Ala onto the solvent exposed face of Coil Ser, CSL14D-Ala, destabilizes the helix in a two-stranded coiled coil by 0.95 kcal/mol.[18,19] Shorter HPLC retention times (related to the α-helical content of the peptide) between the L- and D-amino acid containing peptides (0–10 min) have been reported to correlate with the destabilizing effect of the substitution.[20–22] Consistent with this, we find that CSL16D-Pen (30 min) elutes 3 min earlier than CSL16L-Pen (33 min). The helix destabilizing propensity of D-amino acids has been shown to be highly dependent on the size of the side chain, with β-branched amino acids (of which D-Pen would belong) being the most effective.[20,21] The substitution of D-Val in the center of a 19 amino acid α-helix results in a significantly less α-helical structure (50%) compared to the L-Val analogue (>90%),[23] while D-Leu (no β-branches) are only medium helix destabilizers, which is consistent with our previous observations.[9]
Structures of D-residues in predominantly L-amino acid sequences are rare, especially in longer polypeptides of 20 residues or more.[24] Furthermore, those containing the LDL type sequence, offer the potential to evaluate conformations that would be accessible to a D-residue within a natural protein environment. Although heteromeric coiled coils consisting of bundles containing both all L- and all D-helices have been reported,[25] to the best of our knowledge, no reports exist on structures of L-amino acid coiled coils containing a single layer of D-amino acids. Despite the differences in stability, both CSL16L-Pen and CSL16D-Pen, crystallized in the C2 space group and have been solved to 1.86 and 1.71 Å resolution, respectively. Both structures are well folded three-stranded coiled coils with the α-helices oriented parallel to one another (Figure 1).
Figure 1.
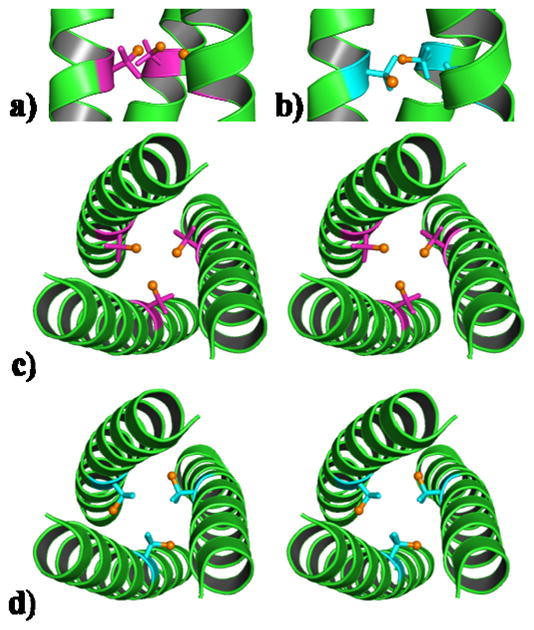
Ribbon diagram of the CSL16X-Pen trimer, illustrating the orientation of the X-Pen side chain. Shown are the main chain atoms represented as helical ribbons (green) and the L- and D-Pen side chain in stick form (L-Pen pink; D-Pen blue) with the thiol group shown as a sphere (orange) in the interior of the parallel three-stranded coiled coil. A side on view shows a) the L-Pen and b) D-Pen side chains oriented towards the N- and C-terminus, respectively; A stereo top down view from the N-terminus of the coiled coil shown for c) L-Pen (major rotamer, 95%) and d) D-Pen.
L -Pen (β,β-dimethyl-cysteine), often used in de novo peptide design,[7,8,16] is considered to be a bulkier analogue of L-Cys. To the best of our knowledge, L -Pen has not been characterized crystallographically in a protein environment. Due to our success in achieving trigonal CdS3 when bound to L-Pen[8,16] we realized it was crucial to determine precisely what this non-protein amino acid looks like within the interior of a coiled coil. The L-Pen residue was built starting from a model structure containing L-Cys. After one round of refinement, density for the methyl groups of L-Pen was clearly present in the Fo−Fc map (Figure S7A). The L-Pen ligand could then be built using this density, by placing the methyl groups at the positions of the L-Cys β-methylene protons. Alternatively, one can envision L-Pen as the thiol containing analogue of L-valine (L-Val) in which the single β-methylene proton is replaced by a thiol group (β-mercapto-valine). On using a rotamer of L-Val in the model we observe density in the Fo−Fc map consistent with a thiol group (Figure S7B). The resulting side chain of L-Pen is oriented towards the N-terminus in the interior of the coiled coil (Figure 1A), in a similar manner to L-Cys. The thiol group of the major rotamer (95%), is positioned towards the interior of the coiled coil (S-S distances of 3.7 Å) as if pre-organized for metal binding (Figure 1C). Intriguingly, the methyl groups can be divided into two distinct classes: those situated above the plane (toward the N-terminus) appear to be oriented away from the helical axis towards the helical interfaces, whereas those positioned below the thiol containing plane point towards the interior of the coiled coil, blocking this face. This structure suggests that the role of the methyl groups may be to improve packing and prevent access to the site, so as to achieve a coordinatively unsaturated CdS3 species. The lower abundance conformation (5%) rotates one of the thiol side arms so that the two methyl groups point towards the interior of the coiled coil and the sulfur towards the helical interface (Figure S8).
The D-Pen side chain structure was determined as described for L-Pen, using a model based on D-Cys (or D-Val). Introduction of a single D-amino acid into an all L-amino acid coiled coil, does not lead to a major perturbation to the α-helical backbone, allowing the two structures to overlay very well (Figure S9). This observation is important as D-amino acids are commonly used to induce turns or terminate designed α-helices[26–29] All non-glycine residues (including D-Pen) fall within the preferred region of the Ramachandran plot for α-helices (Figure S10).[30] This observation is consistent with reported X-ray structures of predominantly L-amino acid α-helices which have been shown to tolerate up to 3 D-amino acids in a 19 amino acid sequence.[31,32] Any energy penalty associated with misorientation of the side chain in a left handed α-helix must be offset by the favorable total hydrophobic packing interactions and the intramolecular hydrogen bonds formed along the α-helical backbone. The (CSL16D-Pen)3 structure illustrates that switching the chirality of an internal amino acid and reorientating the D-Pen side chain towards the C-terminus (Figure 1B) does not prevent the three stranded coiled coil from assembling. The addition of at least one (sterically bulky) D-amino acid is well tolerated in such constructs and is likely to be tolerated in analogous designed coiled coils.
The thiol group in D-Pen is oriented toward the C-terminus, away from the interior and directed at the helical interface (S-S distances of 6.5 Å), whereas both of the methyl groups are now positioned within the hydrophobic interior of the coiled coil (Figure 1D). Though the major rotamer of L-Pen appears to be more closely related to β,β-dimethyl-cysteine, the D-Pen residue behaves more like the thiol containing analogue of D-Val. In contrast to that of L-Pen (Figure 1C), this site is not pre-organized for metal binding and is incapable of forming CdS3 or CdS3O forms within the coiled coil. However, there is evidence that a small amount of Cd(II) is capable of binding, most likely as CdS2O2. This structure may be realized by orientating the thiol groups of two D-Pen ligands towards the same helical interface at which point Cd(II) could bind to the two thiol ligands in addition to two molecules of solvent (water). The Pen side chain resembles a propeller (three groups on β-carbon) with bulky blades, which may not readily rotate. In the case of D-Pen, which does not provide a preorganized metal binding site, the energy penalty to reposition the thiol groups so as to be favorable for metal binding, may be too high. Figure 2 illustrates the two probable metal binding modes found in L- and D-Pen.
Figure 2.

ChemDraw representation of A) the established CdS3 binding to L-Pen in the interior of a three stranded coiled coil. Two possible modes are illustrated for Cd binding to D-Pen at the helical interface, as either B) CdSO3 or C) CdS2O2. The latter (CdS2O2) would require reorientation of one D-Pen residue for Cd binding.
The data in this work provides new strategies for protein design by recognizing the relationship between L- and D-Pen as analogues of Cys and Val. Previously we showed that one can control Cd(II) coordination geometry by modifying the chirality of a second coordination sphere ligand.[9] Here we provide the first report that changing the chirality of the coordinating ligand can not only drastically alter the coordination environment of the metal within a protein, but it may change the physical location of the metal ion within the construct. X-ray crystallography suggests that metal binding differences may arise because D-Pen may behave like a pseudo D-Val, whereas L-Pen behaves more like L-Cys (with a preorganized metal binding site). We have shown that the incorporation of D-amino acids into predominantly L-amino acid coiled coils is well tolerated, allowing full application of D-amino in the field of protein design.
Experimental Procedures
Peptide Synthesis and Purification: Peptides were synthesized on an Applied Biosystem 433A peptide synthesizer using standard protocols,[33] and characterized by electrospray mass spectrometry. Peptides were purified by preparative reversed-phase C18 HPLC using a flow rate of 10 mL/min and a linear gradient of 0.1% trifluoroacetic acid (TFA) in water to 0.1% TFA in 9:1 CH3N:H2O over 50 min. Stock solutions were prepared in degassed doubly distilled water and their concentrations determined using a previously published assay.[34]
113Cd NMR Spectroscopy: Spectra were collected and data processed as reported previously.[9]
Circular Dichroism and Ultraviolet-Visible Spectroscopy: CD and UV-Vis spectra were recorded in quartz cuvettes at 298 K on an AVIV 62DS and Cary 100 Bio UV-vis spectrometer, respectively. The observed CD ellipticity in millidegrees has been converted to molar ellipticity, [Θ], and is reported in units of deg dmol−1 cm2. Difference spectra were obtained by subtracting the background spectrum of peptide in the absence of metal. pH Titrations were performed as reported previously, and the experimental data for CSL16D-Pen was fit to a model for the simultaneous release of two protons on binding Cd(II) to three thiolates.[9,35] Guanidine hydrochloride CD titrations were performed at pH 8.5 as described previously.[3]
Crystallization, data collection and refinement: Crystals of CSL16L-Pen were grown by vapour diffusion at 20°C in a sitting drop with equal volumes of peptide (20 mg/ml CSL16L-Pen, 2.0 mM CdCl2, 20 mM Zn(OAc)2, 5 mM Tris buffer pH 8.5) and precipitant (100 mM cacodylate pH 6.5, 200 mM MgCl2, 50% PEG 200). Crystals of CSL16D-Pen were grown by vapor diffusion at 20°C in a hanging drop with equal volumes of peptide (15.8 mg/ml CSL16D-Pen, 8.4 mM CdCl2, 8.4 mM Zn(OAc)2, 5 mM Tris buffer pH 8.5) and precipitant (100 mM MES pH 6.0, 30% PEG-200, 5% PEG-3000). Crystals were frozen in their mother liquor for data collection.
Data were collected at the Advanced Photon Source in the Argonne National Laboratory on the LS-CAT Beamline 21-ID equipped with a Mar 225 CCD (Mar USA, Evanston, IL). Three hundred sixty frames of data were collected at −180 °C with a 1° rotation and 2-s exposure. Data were processed and scaled with the program HKL-2000.[36] The structures were solved by molecular replacement with using Phaser in the CCP4 suite of programs,[37,38] using as a model As(CSL9C)3 (Protein Data Bank ID 2jgo)[14] with the metal ions removed, but the side chains included. These models were refined with restrained refinement in Refmac 5 in the CCP4 suite of programs[39] and built in Coot[40] using the 2F0 − Fc and F0 − Fc electron density maps generated in Refmac 5. The structures of CSL16L-Pen and CSL16D-Pen were refined to 1.86 Å (Rworking = 19.9% and Rfree = 25.6%) and 1.71 Å (Rworking = 18.1% and Rfree = 22.5%), respectively. Data collection and refinement statistics are given in Table S1. The atomic coordinates and structure factors for CSL16L-Pen and CSL16D-Pen have been deposited in the Protein Data Bank with the ID codes 3H5F and 3H5G, respectively. Figures were generated in Pymol.[41]
Acknowledgments
V.L.P. thanks the National Institute of Health for support of this research (ES012236) and J.A.S. thanks the University of Michigan Center for Structural Biology.
Contributor Information
Dr. Anna F. A. Peacock, Department of Chemistry, University of Michigan, Ann Arbor, MI 48109 (USA)
Prof. Jeanne A. Stuckey, Life Sciences Institute, University and Michigan, Ann Arbor, MI 48109 (USA)
Prof. Vincent L. Pecoraro, Department of Chemistry, University of Michigan, Ann Arbor, MI 48109 (USA).
References
- 1.DeGrado WF, Summa CM, Pavone V, Nastri F, Lombardi A. Annu Rev Biochem. 1999;68:779. doi: 10.1146/annurev.biochem.68.1.779. [DOI] [PubMed] [Google Scholar]
- 2.Ghosh D, Pecoraro VL. Inorg Chem. 2004;43:7902. doi: 10.1021/ic048939z. [DOI] [PubMed] [Google Scholar]
- 3.Dieckmann GR, McRorie DK, Lear JD, Sharp KA, DeGrado WF, Pecoraro VL. J Mol Biol. 1998;280:897. doi: 10.1006/jmbi.1998.1891. [DOI] [PubMed] [Google Scholar]
- 4.Yadav MK, Redman JE, Leman LJ, Alvarez-Gutiérrez JM, Zhang Y, Stout CD, Ghadiri MR. Biochemistry. 2005;44:9723. doi: 10.1021/bi050742a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee KH, Matzapetakis M, Mitra S, Marsh ENG, Pecoraro VL. J Am Chem Soc. 2004;126:9178. doi: 10.1021/ja048839s. [DOI] [PubMed] [Google Scholar]
- 6.Dieckmann GR, McRorie DK, Tierney DL, Utschig LM, Singer CP, O’Halloran TV, Penner-Hahn JE, DeGrado WF, Pecoraro VL. J Am Chem Soc. 1997;119:6195. [Google Scholar]
- 7.Petros AK, Shaner SE, Costello AL, Tierney DL, Gibney BR. Inorg Chem. 2004;43:4793. doi: 10.1021/ic0497679. [DOI] [PubMed] [Google Scholar]
- 8.Lee K-H, Cabello C, Hemmingsen L, Marsh ENG, Pecoraro VL. Angew Chem. 2006;118:2930. doi: 10.1002/anie.200504548. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2006;45:2864. [Google Scholar]
- 9.Peacock AFA, Hemmingsen L, Pecoraro VL. Proc Natl Acad Sci, USA. 2008;105:16566. doi: 10.1073/pnas.0806792105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peacock AFA, Iranzo O, Pecoraro VL. J Chem Soc, Dalton Trans. 2009;13:2271. doi: 10.1039/b818306f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Neil KT, DeGrado WF. Science. 1990;250:646. doi: 10.1126/science.2237415. [DOI] [PubMed] [Google Scholar]
- 12.Iranzo O, Ghosh D, Pecoraro VL. Inorg Chem. 2006;45:9959. doi: 10.1021/ic061183e. [DOI] [PubMed] [Google Scholar]
- 13.Lovejoy B, Choe S, Cascio D, McRorie DK, DeGrado WF, Eisenberg D. Science. 1993;259:1288. doi: 10.1126/science.8446897. [DOI] [PubMed] [Google Scholar]
- 14.Touw DS, Nordman CE, Stuckey JE, Pecoraro VL. Proc Natl Acad Sci, USA. 2007;104:11969. doi: 10.1073/pnas.0701979104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iranzo O, Stachura M, Jakusch T, Lee KH, Hemmingsen L, Pecoraro VL. Chem Eur J. 2007;13:9178. [Google Scholar]
- 16.Iranzo O, Cabello C, Pecoraro VL. Angew Chem. 2007;119:6808. doi: 10.1002/anie.200701729. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:6688. [Google Scholar]
- 17.Iranzo O, Pecoraro VL. Unpublished data [Google Scholar]
- 18.Fairman R, Anthony-Cahill SJ, DeGrado WF. J Am Chem Soc. 1992;114:5458. [Google Scholar]
- 19.Hermans J, Anderson AG, Yun RH. Biochemistry. 1992;31:5646. doi: 10.1021/bi00139a031. [DOI] [PubMed] [Google Scholar]
- 20.Krause E, Bienert M, Schmieder P, Wenschuh H. J Am Chem Soc. 2000;122:4865. [Google Scholar]
- 21.Chen Y, Mant CT, Hodges RS. J Pept Res. 2002;59:18. doi: 10.1046/j.1397-002x.2001.10994.x. [DOI] [PubMed] [Google Scholar]
- 22.Wendt H, Berger C, Baici A, Thomas RM, Bosshard HR. Biochemistry. 1995;34:4097. doi: 10.1021/bi00012a028. [DOI] [PubMed] [Google Scholar]
- 23.Krause E, Beyermann M, Dathe M, Rothemund S, Bienert M. Anal Chem. 1995;67:252. doi: 10.1021/ac00098a003. [DOI] [PubMed] [Google Scholar]
- 24.Mitchell JBO, Smith J. Proteins. 2003;50:563. doi: 10.1002/prot.10320. [DOI] [PubMed] [Google Scholar]
- 25.Sia SK, Kim PS. Biochemistry. 2001;40:8981. doi: 10.1021/bi010725v. [DOI] [PubMed] [Google Scholar]
- 26.Haack T, González MJ, Sánchez Y, Giralt E. Lett Pept Sci. 1997;4:377. doi: 10.1002/(SICI)1099-1387(199707)3:4%3C299::AID-PSC121%3E3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 27.Aravinda S, Shamala N, Roy RS, Balaram P. Proc Ind Acad Sci. 2003;115:373. [Google Scholar]
- 28.Mahalakshmi R, Balaram P. D-Amino Acids: A New Frontier in Amino Acid and Protein Research. In: Konno R, Fisher GH, Brueckner H, d’Aniello A, Fujii N, Homma H, editors. The use of D-amino acids in peptide design. Nova Science; NY: 2006. pp. 415–430. [Google Scholar]
- 29.Durani S. Acc Chem Res. 2008;41:1301. doi: 10.1021/ar700265t. [DOI] [PubMed] [Google Scholar]
- 30.Ramachandran GN, Sasisekharan V. Adv Protein Chem. 1968;23:283. doi: 10.1016/s0065-3233(08)60402-7. [DOI] [PubMed] [Google Scholar]
- 31.Aravinda S, Shamala N, Desiraju S, Balaram P. J Chem Soc Chem Comm. 2002:2454. doi: 10.1039/b207960g. [DOI] [PubMed] [Google Scholar]
- 32.Karle IL, Gopi HN, Balaram P. Proc Natl Acad Sci, USA. 2003;100:13946. doi: 10.1073/pnas.2336106100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan WC, White PD. In: Fmoc Solid Phase Peptide Synthesis: A Practical Approach. Hames BD, editor. Oxford Univ. Press; New York: 2000. p. 346. [Google Scholar]
- 34.Mantle M, Stewart G, Zayas G, King M. Biochem J. 1990;266:597. [PMC free article] [PubMed] [Google Scholar]
- 35.Matzapetakis M, Ghosh D, Weng TC, Penner-Hahn JE, Pecoraro VL. J Biol Inorg Chem. 2006;11:876. doi: 10.1007/s00775-006-0140-7. [DOI] [PubMed] [Google Scholar]
- 36.Otwinowski Z, Minor W. Enzymol. 1997;276:307. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 37.Potterton E, Briggs P, Turkenburg M, Dodson E. Acta Crystallogr D. 2003;59:1131. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 38.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. J Appl Crystallogr. 2007;40:658. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallogr D. 1997;53:240. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 40.Emsley P, Cowtan K. Acta Crystallogr D. 2004;20:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 41.Delano WL. The PyMol Molecular Graphics System. DeLano Scientific; Palo Alto, California, USA: 2005. http://www.pymol.org. [Google Scholar]