

Abstract
4-hydroxy-2-nonenal (4-HNE) is a lipid peroxidation product formed during oxidative stress that can alter protein function via adduction of nucleophilic amino acid residues. 4-HNE detoxification occurs mainly via GSH conjugation and transporter-mediated efflux. This results in a net loss of cellular GSH and restoration of GSH homeostasis requires de novo GSH biosynthesis. The rate-limiting step in GSH biosynthesis is catalyzed by glutamate cysteine ligase (GCL), a heterodimeric holoenzyme composed of a catalytic (GCLC) and modulatory (GCLM) subunit. The relative levels of the GCL subunits are a major determinant of cellular GSH biosynthetic capacity and 4-HNE induces the expression of both GCL subunits. In this study, we demonstrate that 4-HNE can alter GCL holoenzyme formation and activity via direct post-translational modification of the GCL subunits in vitro. 4-HNE directly modified Cys553 of GCLC and Cys35 of GCLM in vitro, which significantly increased monomeric GCLC enzymatic activity, but reduced GCL holoenzyme activity and formation of the GCL holoenzyme complex. In silico molecular modeling studies also indicate these residues are likely to be functionally relevant. Within a cellular context, this novel post-translational regulation of GCL activity could significantly affect cellular GSH homeostasis and GSH-dependent detoxification during periods of oxidative stress.
Keywords: Oxidative stress, Glutamate cysteine ligase, 4-Hydroxynonenal, Glutathione, Antioxidants
Introduction
Cellular oxidative stress and peroxidation of membrane lipids are thought to play a role in the pathogenesis of a host of human diseases, including Alzheimer’s disease [1], liver disease [2], and chronic obstructive pulmonary disease [3]. 4-hydroxy-2-nonenal (4-HNE) is a major cytotoxic α,β-unsaturated aldehyde produced during the peroxidation of omega-6 polyunsaturated fatty acids (PUFAs) (e.g. arachidonic acid or linoleic acid) [4]. 4-HNE is relatively long-lived and can diffuse from its site of generation to react with various cellular targets, including DNA and protein [4-6]. 4-HNE and other products of lipid peroxidation are highly reactive and capable of covalently modifying nucleophilic amino acid residues of cellular proteins via Michael addition [4, 7] with a reactivity profile of Cys≫His>Lys [8, 9]. 4-HNE can also undergo Schiff base formation with Lys residues [4, 5]. Together these reactions can lead to intra- and/or inter-molecular cross-linking of proteins via Michael addition at one site and subsequent Schiff base formation with a neighboring Lys residue [10-12]. Covalent modification by 4-HNE almost invariably leads to an impairment of protein function [2, 12-18] and 4-HNE-mediated adduction/inactivation of specific cellular proteins can have dramatic effects on cell function [19-21].
The cytotoxic effects of 4-HNE resulting from the functional inactivation of proteins that are vital for cell viability necessitates its rapid and efficient removal from the cell. 4-HNE and other α,β-unsaturated aldehydes are metabolized via spontaneous or GST-mediated conjugation with glutathione (GSH) and export of the GSH-4-HNE conjugate from the cell via transporter-mediated efflux [22]. This results in a net loss of cellular GSH and without replenishment of GSH stores this can rapidly compromise cellular antioxidant capacity leading to increased production of reactive oxygen species (ROS) and disruption of the cellular redox balance [23]. As most cells do not import significant amounts of GSH, restoration of cellular GSH levels requires de novo GSH biosynthesis. GSH is synthesized by two successive ATP-dependent reactions catalyzed by glutamate cysteine ligase (GCL, EC 6.3.2.2) [24] and glutathione synthetase (GS, EC 6.3.2.3) [25]. GCL catalyzes the first and rate-limiting step in GSH biosynthesis [25]. GCL is a heterodimeric holoenzyme complex consisting of a catalytic subunit (GCLC, 73 kDa), which contributes all the enzymatic activity and contains all the substrate and cofactor binding sites of GCL, and a modifier subunit (GCLM, 31 kDa), which modulates GCLC activity and affinity for substrates and inhibitors [25]. While GCLM possesses no enzymatic activity alone, heterodimerization with GCLC and formation of GCL holoenzyme increases the Vmax of GCLC approximately 5-fold, lowers the Km for L-glutamate and ATP, and increases the Ki for GSH-mediated feedback inhibition [26-28]. The relative levels of the GCL subunits generally dictate cellular GSH biosynthetic capacity and both genes are highly regulated at the transcriptional and post-transcriptional levels [29, 30]. It is generally thought that 4-HNE increases GCL activity via induction of GCL subunit expression as a compensatory response to GSH depletion and this is necessary and sufficient to reestablish GSH homeostasis [29, 31-36]. However, there is a temporal discrepancy between 4-HNE-induced GCL expression and restoration of GSH homeostasis. Such findings suggest that post-translational modification and regulation of pre-existing GCL protein may contribute to the rapid increase in cellular GSH biosynthetic capacity and reestablishment of GSH homeostasis in response to 4-HNE exposure [37-40].
Multiple Cys and Lys residues are thought to be important for both the constitutive enzymatic activity of GCLC [41, 42] and the dramatic increase in GCLC activity upon heterodimerization with GCLM [26, 43]. In this regard, GCL holoenzyme formation is redox sensitive and is thought to involve intermolecular disulfide bond formation between Cys residues of the GCL subunits [41, 43]. However, while we have found that oxidative activation of GCL results from increased holoenzyme formation, this does not involve a labile disulfide bridge between GCLC and GCLM [44]. Studies employing sulfhydryl modifying agents suggest that Cys residues within the active site of GCLC are also important for GCLC activity [45]. Chemical modification and mutagenesis studies indicate that Lys residues may also be important in the regulation of GCLC specific activity and substrate binding affinity [42]. Given the propensity of 4-HNE to covalently modify Cys and Lys residues, direct adduction of one or both of the GCL subunits has the potential to rapidly affect GCLC and GCL holoenzyme function and cellular GSH biosynthetic capacity during periods of oxidative stress. GSH plays an important role in cellular antioxidant defense, maintenance of cellular redox state, and in detoxification of xenobiotics via GSH conjugation reactions. Thus, alterations in cellular GSH biosynthetic capacity caused by 4-HNE-mediated adduction of GCL has the potential to not only affect cellular antioxidant capacity and the ability to protect against oxidative damage, but also cellular detoxification capacity and the ability to metabolize cytotoxic and genotoxic xenobiotics. In this study, we demonstrate that both GCLC and GCLM are direct targets for covalent modification by the α,β-unsaturated aldehyde 4-HNE in vitro. While previously identified post-translational modifications of GCLC (e.g. phosphorylation, caspase-mediated cleavage) appear to have relatively modest effects on GCL activity [37, 46, 47], post-translational modification of the GCL subunits by 4-HNE dramatically affects GCL activity. Surprisingly, while 4-HNE-mediated adduction usually inhibits protein function, adduction significantly increased monomeric GCLC enzymatic activity. Although it is generally agreed that transcriptional induction of the GCL subunits leads to a compensatory increase in cellular GSH biosynthetic capacity following exposure to 4-HNE, these findings suggest that direct post-translational modification and activation of pre-existing GCLC may also contribute to the reestablishment of GSH homeostasis following oxidative insult. The ability of 4-HNE to affect cellular GSH homeostasis at multiple levels (conjugation/depletion of GSH, direct inhibition/activation of GSH synthesis, and transcriptional up regulation of the GCL subunits) highlights the potential importance of the GSH antioxidant defense system in protecting cells against α,β-unsaturated aldehyde-induced oxidative damage.
Materials and Methods
Chemicals and Reagents
All chemicals and reagents were obtained from Sigma-Aldrich (St. Louis, MO), unless otherwise noted. Sulfosalicylic acid (SSA) and tris base were obtained from Fisher Scientific (Fair Lawn, NJ). Non-fat powdered milk was obtained from Bio-Rad (Hercules, CA). Sulfo-NHS-Acetate (NHS) was obtained from Pierce Biotechnology (Rockford, IL). Complete Protease Inhibitor Cocktail tablets were obtained from Roche (Indianapolis, IN). α-GCLC and α-GCLM antibodies were generously provided by Dr. Terrance Kavanagh (University of Washington). 4-HNE and 4-HNE antibody were prepared as described previously [8, 48].
Cell Culture and Treatments
A549 cells (ATCC, Manassas, VA) were cultured in DMEM with 10% FBS, 2 mM L-glutamate, and 1% penicillin/streptomycin in a humidified incubator at 37°C with 5% CO2. Cells were seeded at equal densities in 100 mm dishes and grown to a confluency of ~90% before use in experiments. For dose-response and time course experiments, cells were incubated at 37°C in serum-free media with increasing concentrations of 4-HNE or equivalent acetonitrile vehicle control for 30 min, or incubated with 50 μM 4-HNE for up to 60 min. Cells were also pretreated with or without cycloheximide (CHX; 10 μg/ml) in serum-free media for 1 h, followed by the addition of 50 μM 4-HNE or vehicle control for 30 min. At the time of harvest, cells were washed once with warm, serum-containing media and once with ice cold PBS, collected by scraping into 5 ml ice cold PBS, and the resulting pellets stored at -80°C until use. Cellular extracts were prepared by sonicating the pellets in TES/SB buffer (20 mM Tris pH 7.5, 1 mM EDTA, 250 mM sucrose, 1 mM L-serine, 20 mM boric acid) with 1x Complete Protease Inhibitor Cocktail on ice 2 × 10 s, followed by centrifugation at 16,000 × g at 4°C for 10 min, and the resulting supernatant used for immunoblotting and GCL activity assays.
Recombinant Proteins and Treatments
The pET29a-GCLC-6His vector encoding a C-terminal 6His-tagged GCLC fusion protein has been described previously [35, 49]. The vector encoding a 6His-tagged GCLM fusion protein was prepared by subcloning a murine GCLM PCR product in-frame between the Nde I and Xho I sites of pET21a (Clontech, Mountain View, CA) [49]. The fidelity of these constructs was verified by DNA sequencing. BL21(DE3) bacteria (Stratagene, San Diego, CA) expressing these plasmids were grown at 37°C to an OD600 of 0.5 and then induced for 5-6 h with 1 mM (GCLC) or 0.25 mM (GCLM) isopropyl-β-D-1-thiogalactopyranoside (IPTG). Bacteria were harvested and lysed either using BugBuster® protein extraction reagent (GCLC) (Novagen, Madison, WI) or by sonication in TALON® Metal Affinity Resin wash buffer (GCLM) (Clontech). Lysis buffers contained 1X Complete EDTA-free Protease Inhibitor Cocktail, 25 U/mL benzonase nuclease (Novagen), and 0.1 μL/mL lysozyme (Novagen). Recombinant proteins were recovered from the clarified lysate using TALON® Metal Affinity Resin (Clontech). The immobilized fusion proteins were washed extensively and eluted in a PBS solution containing 150 mM imidazole. Protein concentrations were determined by Bradford assay and purity assessed via Coomassie-stained polyacrylamide gel. Protein aliquots were stored with 10% glycerol at -80°C. Just prior to use, protein aliquots were thawed on ice and the glycerol and imidazole removed using PD-10 protein desalting spin columns (Pierce Biotechnology, Rockford, IL). Recombinant GCLC (0.33 μg/ul, ~4.4 μM) and GCLM (0.15 μg/ul, ~4.4 μM) were treated either individually or after formation of GCL holoenzyme with various molar ratios of 4-HNE, N-ethylmaleimide (NEM), NHS, or equivalent volume of vehicle (acetonitrile) in 50 mM sodium phosphate (pH 7.4) for 30 min at 37°C. Molar ratios were calculated per mole of protein. However, murine GCLC and GCLM contain 14 and 6 cysteine residues, and 36 and 16 lysine residues, respectively. Thus, a 100X molar excess of NEM or NHS would correspond to a 7.1X and 16.7X molar excess per cysteine residue, or a 2.5X and 6.7X molar excess per lysine residue, respectively. In the case of GCL holoenzyme (20 cysteines and 52 lysines), 100X molar excess would correlate with 10X NEM per cysteine residue and 3.8X NHS per lysine residue. Formation of holoenzyme was achieved by incubating GCLC and GCLM together at a molar ratio of 1:1 for 10 min at 37°C either prior to or after 4-HNE/NEM/NHS treatment. Free 4-HNE, NEM, or NHS was removed using PD-10 desalting columns prior to all subsequent manipulations and analyses.
PAGE and Immunoblot Analysis
Soluble cellular protein (20 μg) or recombinant GCLC and/or GCLM (150 ng) were resolved on polyacrylamide gels under either non-denaturing, non-reducing (Native), or denaturing, reducing (SDS) conditions. Native samples were mixed with ice-cold protein loading buffer without 2-mercaptoethanol and were not boiled. Samples were loaded on 8% polyacrylamide gels without stacking gel or SDS and run at 70 V at 4°C for 3 h. Denatured samples were prepared by heating at 95°C for 5 min in the presence of 2-mercaptoethanol (0.7 M). Samples were loaded on 10% polyacrylamide gels with 5% stacking gels both containing 0.1% SDS and run at 100 V at room temperature for 90 min. Gels were transferred overnight at 4°C to HyBond-C Extra membranes (Amersham Biosciences, Piscataway, NJ). Membranes were blocked in TBS-T containing 5% powdered milk for 1 h, probed with the indicated antibodies, and immune complexes detected by Western Lighting chemoluminescent reagent (PerkinElmer, Boston, MA).
GSH and GCL Activity Assays
GCL activity and GSH content (GSH + GSSG) of cellular extracts or recombinant proteins was determined as described previously [50]. For GCL activity measurements, cellular extracts (150 μg) or recombinant proteins (1 μg) were incubated at 37°C in microtiter plates with L-glutamate (10 mM), L-cysteine (2 mM), and ATP (5 mM) for 30 min or 10 min, respectively. Substrate kinetic determinations were performed by incubating recombinant proteins (1 μg) with varying concentrations of L-glutamate (0-10 mM), ATP (0-5 mM), or L-cysteine (2 mM) for 10 min at 37°C. Reactions were halted and protein precipitated by the addition of 200 mM SSA. The plates were centrifuged at 1500 rpm for 10 min at 4°C and four aliquots of each well were derivatized with naphthalene-2,3-dicarboxyaldehyde (NDA) in the dark for 30 min at room temperature. NDA fluoresces upon reaction with glutathione (GSH) or γ-glutamylcysteine (γ-GC) and fluorescence (472 nm excitation, 528 nm emission) was measured on a SpectraMax Gemini EM plate reader (Molecular Devices, Sunnyvale, CA) and GCL activity is expressed as the rate of γ-GC formation (nmol/mg protein/min for cell extracts or nmol/μg protein/min for recombinant proteins).
MALDI-TOF/TOF Analysis of 4-HNE- and NEM-Modified GCLM, GCLC, and GCL Holoenzyme
Recombinant proteins (~4 μM for GCLC, ~8.2 μM for GCLM and ~2.8 μM GCL holoenzyme) in 50 mM NaH2PO4 (pH 7.4) were adducted by incubation with a 25-fold molar excess of 4-HNE for 30 min at 37°C. Sodium borohydride (NaBH4) was then added to a final concentration of 5 mM and the mixture incubated for an additional 30 min at 25°C to stabilize the adducts and quench free aldehyde. Alternatively, proteins were treated with a 10-fold molar excess of NEM for 30 min at 37°C to identify solvent accessible cysteine residues that might serve as functionally relevant targets for 4-HNE-mediated adduction. The samples were heated at 95°C for 5 min in SDS loading buffer and then run in triplicate on a 12% SDS-PAGE gel and stained with Coomassie dye. Gels were destained and bands containing 5 μg protein were excised for digestion separately with trypsin, Glu-C, or chymotrypsin (Promega) as directed by the product inserts. The digested samples were desalted using C18 ZipTips (Millipore), mixed with matrix (10 mg/mL α-cyano-4-hydroxycinnamic acid in 60% acetonitrile/0.1% TFA) at a ratio of 1:1 and spotted in 1 μl aliquots onto a Opti-TOF 384 Well Insert (Applied Biosystems, Foster City, CA). Samples were analyzed on an ABI 4800 Plus MALDI-TOF/TOF instrument (Applied Biosystems) operated under standard conditions for peptides at laser power 2300 with calibration using a ProteoMass Calibration Kit (Sigma). MS/MS collision induced dissociation (CID) was performed at 1 kV and the collision gas was air. MS/MS spectra were collected using laser power of 3300 with 2000 laser shots or until a minimum S/N was obtained. The precursor mass window was 300 relative resolution (fwhm). Data analysis was performed with Mascot (v 2.1.04, www.matrixscience.com) and Applied Biosystems 4000 Series Explorer v 3.5.
GCL Homology Modeling, Residue pKa Analysis, and Rendering
The protein sequences of murine GCLM and GCLC were submitted to the ModWeb protein structure modeling server (http://modbase.compbio.ucsf.edu/ModWeb20-html/modweb.html) [51] to generate homology models of each individual subunit. Based on sequence identity, the templates chosen were human aldehyde reductase (PDB ID: 2ALR; ~20% identity) and S. cerevisiae GCL (PDB ID: 3IG5; ~44% identity) for GCLM and GCLC, respectively. Cys residue pKa calculations were determined using the Vega ZZ program [52] with the PROPKA plugin [53]. Likely subunit interactions were predicted with the Escher NG plugin [54] using a probe radius of 1.0 angstrom, rotation step of 5 degrees, and a max collisions/min charge of 300 and -300, respectively. The resulting structures were rendered in Lightwave 3D 9.6 (NewTek Inc., San Antonio, TX) for figure presentation.
Statistical Analysis
All studies were performed a minimum of three times. Statistical analysis was performed using GraphPad Prism 4.0 (GraphPad Software, La Jolla, CA) and the error bars in each figure represent the standard error of the mean. Results were considered statistically significant if p < 0.05 as determined by one-way ANOVA with Tukey’s multiple comparison test.
Results
Rapid activation of cellular GCL by 4-HNE
4-HNE can increase cellular GCL activity via transcriptional induction and increased protein expression of one or both of the GCL subunits [30, 32, 33]. However, it is not known whether 4-HNE may also regulate GCL activity via direct post-translational modification of the GCL subunits. To assess the kinetics of 4-HNE-induced cellular GCL activity, A549 cells, which express high levels of both GCL subunits [55], were treated with 4-HNE and GCL activity was measured. 4-HNE caused a rapid dose- and time-dependent increase in A549 cell GCL activity (Figure 1A and 1B). Maximal activation of GCL occurred at 30 min in response to 50 μM 4-HNE. These concentrations are relatively high, but are within reported physiological concentrations 4-HNE [56]. A549 cells also have high intracellular GSH content which likely necessitates the use of higher 4-HNE concentrations due to increased metabolism of 4-HNE via spontaneous or GST-mediated GSH conjugation. In any case, these studies provide proof-of-principle that 4-HNE can rapidly increase cellular GCL enzymatic activity. This response appears to be functionally relevant as the reestablishment of GSH homeostasis following a transient 4-HNE-induced reduction in cellular GSH levels (Figure 1B, line) correlated temporally with 4-HNE-induced GCL activity (Figure 1B, bars). Importantly, this acute increase in GCL activity was not accompanied by any detectable change in the expression of either GCLC or GCLM (Figure 1D). Furthermore, pre-treatment with the protein synthesis inhibitor cycloheximide (CHX) had no effect on 4-HNE-induced GCL activity (Figure 1C and 1D), confirming that this response was not due to increased GCL subunit protein expression. These results indicate that this rapid 4-HNE-induced GCL activity occurs by a transcription-independent mechanism that could involve post-translational modification of one or both of the GCL subunits.
Figure 1. 4-HNE rapidly increases cellular GCL activity independent of de novo protein synthesis.
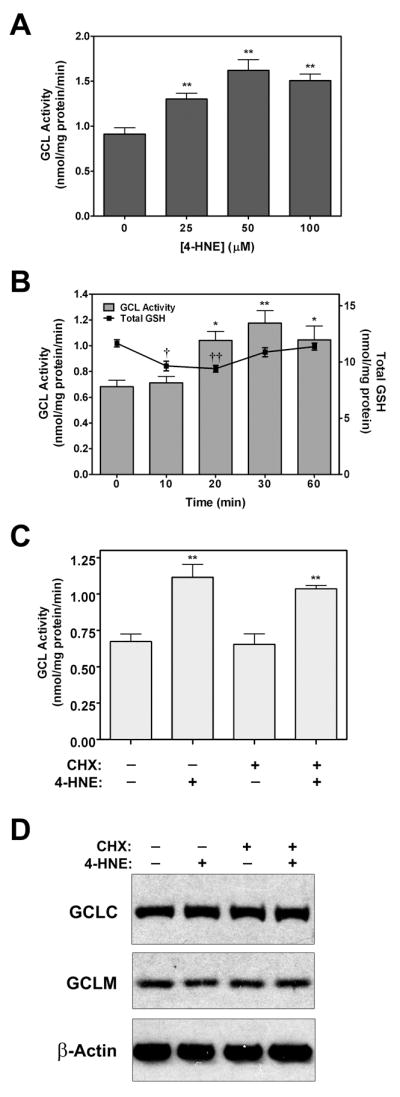
(A) A549 cells were treated for 30 min with the indicated concentrations of 4-HNE. (B) A549 cells were treated with 50 μM 4-HNE for the times indicated. (C and D) A549 cells were pretreated for 1 h in the absence or presence of cycloheximide (CHX; 10 μg/ml) prior to treatment with 4-HNE (50 μM) for 30 min. (A-C) GCL activity and total GSH content (GSH + GSSG) of cellular extracts were assessed by the fluorescence-based NDA assay and a modified Tietze assay, respectively (*†p<0.01, **††p<0.001). (D) GCLC, GCLM, and β-actin protein expression were analyzed by immunoblotting as described in the Materials and Methods section.
In vitro analysis of GCL holoenzyme formation and activity using purified recombinant GCL fusion proteins
Purified recombinant His-tagged GCLC and GCLM fusion proteins [35, 49] were employed to determine whether the GCL subunits were direct targets for 4-HNE-mediated adduction in vitro and to identify the sites of adduction and molecular mechanism(s) mediating 4-HNE-induced GCL activity. To functionally validate this model system, the GCL subunit fusion proteins were mixed at increasing GCLM:GCLC molar ratios and GCL holoenzyme formation was assessed by native PAGE and immunoblotting and GCL activity was analyzed by a fluorescence microtiter plate assay [50]. Increasing amounts of GCLM promoted GCL holoenzyme formation with nearly complete GCLC heterodimerization achieved at a 1:1 molar ratio of GCLM:GCLC (Figure 2A). Importantly, GCL holoenzyme formation correlated with increased GCL activity, with an ~5-fold increase observed upon complete GCL holoenzyme formation (Figure 2B). The specific activities of the recombinant murine monomeric GCLC and GCL holoenzyme and the molar ratios required for quantitative GCL holoenzyme formation are consistent with previously reported values [26]. All subsequent studies examining GCL holoenzyme formation and activity were performed using a 1:1 molar ratio of GCLM:GCLC. These results validate the use of these recombinant His-tagged GCLC and GCLM fusion proteins to examine the functional effects of 4-HNE on GCL holoenzyme formation and enzyme activity in vitro.
Figure 2. In vitro analysis of GCL holoenzyme formation and activity using His-tagged recombinant GCL fusion proteins.
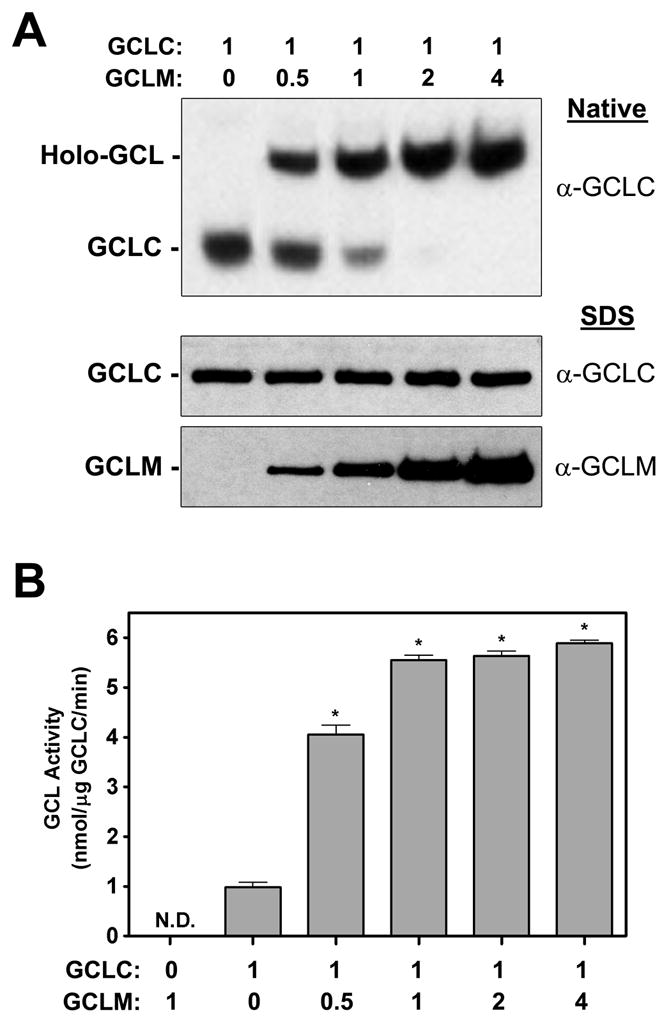
Purified recombinant GCLC-His and GCLM-His fusion proteins were mixed at the indicated molar ratios. (A) GCL holoenzyme formation was analyzed by native PAGE and immunoblotting for GCLC (top panel). The relative amounts of each GCL subunit in the mixture were analyzed by SDS-PAGE and immunoblotting for GCLC or GCLM (bottom panels). (B) GCL activity was measured by the fluorescence-based NDA assay (* p<0.001, N.D. = not detectible).
Direct post-translational modification and regulation of GCL activity by 4-HNE in vitro
The rapid increase in cellular GCL activity in response to 4-HNE treatment suggested that direct post-translational modification of one or both of the GCL subunits may be involved in enzyme activation. To test this hypothesis, recombinant GCLC, GCLM, and GCL holoenzyme were incubated with increasing molar ratios of 4-HNE at 37°C for 30 min and protein modification was assessed by immunoblotting with an antibody that detects 4-HNE adducts [48]. Treatment with 4-HNE resulted in the dose-dependent adduction of both monomeric GCLC and GCLM (Figure 3A and 3B). GCLC and GCLM were also adducted in a similar manner when present in a heterodimeric GCL holoenzyme complex (Figure 3C). Importantly, while 4-HNE-mediated protein adduction almost invariably results in impaired protein function [2, 12-17], 4-HNE-mediated adduction of monomeric GCLC resulted in a dose-dependent increase in GCLC enzymatic activity compared to vehicle control (Figure 3D, open bars). A 25-fold molar excess (25x), which equates to the concentration of 4-HNE (100 μM) that stimulated A549 cell GCL activity in Figure 1, led to an almost 2-fold increase in GCLC activity in vitro. In contrast, treatment of GCL holoenzyme caused a dose-dependent decrease in GCL activity with 25x 4-HNE resulting in an ~15-20% reduction in activity (Figure 3D, closed bars). This 4-HNE-mediated inhibition of GCL holoenzyme activity is associated with a reduction in the levels of heterodimeric GCL holoenzyme complex (Figure 3E). However, this is not due to dissociation of the GCL subunits. Rather, there is an increase in high molecular weight complexes that are immunoreactive towards GCLC, likely resulting from 4-HNE-mediated cross-linking between the GCL holoenzyme complexes. Interestingly, formation of these high molecular weight complexes does not abrogate GCL holoenzyme activity, which is still increased ~4.5-fold over untreated monomeric GCLC after exposure to a 25-fold molar excess of 4-HNE (Figure 3D). It should be noted that these GCL activity measurements were performed in the presence of saturating concentrations of substrates and represent measurements of Vmax. To determine whether 4-HNE-mediated adduction of GCLC might also affect GCLC substrate binding affinity, we examined the substrate kinetics of adducted and non-adducted monomeric GCLC. 4-HNE adduction of GCLC not only increased the Vmax of GCLC, but it also lowered the Km for both L-glutamate and ATP (Figure 4A and 4B; Table 1). Interestingly, 4-HNE-mediated adduction of GCLC partially mimics the functional effects caused by heterodimerization with GCLM and formation of GCL holoenzyme, including enhanced GCLC specific activity and substrate binding affinities [26]. These findings suggest that direct post-translational modification and regulation of GCLC activity likely contributes to the rapid increase in GCL activity observed in 4-HNE-treated A549 cells.
Figure 3. 4-HNE-mediated adduction of the GCL subunits differentially affects GCLC and GCL holoenzyme activity.
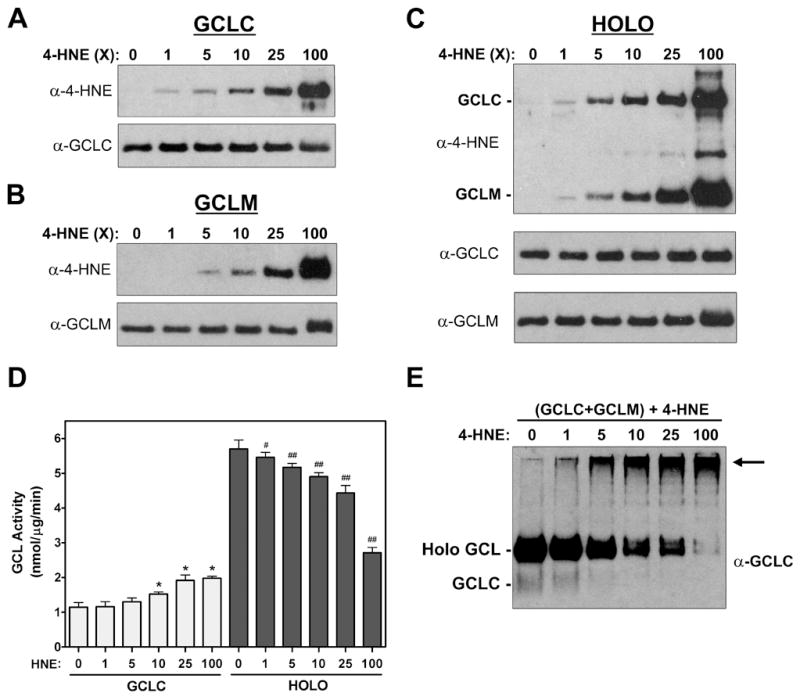
Purified recombinant (A) GCLC, (B) GCLM, or (C) GCL holoenzyme (GCL subunits pre-mixed at a 1:1 molar ratio) were incubated with the indicated molar ratio of 4-HNE for 30 min at 37°C. (A-C) 4-HNE-mediated adduction of the GCL subunits was assessed by SDS-PAGE and immunoblotting with α-4-HNE (top panels). Equivalent loading of the GCL subunits was confirmed by immunoblotting for GCLC or GCLM (bottom panels). (D) Monomeric GCLC (open bars) and GCL holoenzyme (closed bars) were treated in the absence or presence of the indicated molar ratio of 4-HNE for 30 min at 37°C. GCL activity was determined by the fluorescence-based NDA assay (* p<0.001 vs monomeric GCLC control; # p<0.05, ## p<0.001 vs GCL holoenzyme control). (E) GCL holoenzyme was treated as described above and the amount of GCLC present in the monomeric, holoenzyme, and high molecular weight fractions was assessed by native PAGE and immunoblotting for GCLC. The arrow denotes the presence of high molecular weight complexes that contain GCLC.
Figure 4. 4-HNE- and NEM-mediated adduction of monomeric GCLC partially mimics the effects of GCL holoenzyme formation on GCLC enzyme kinetics.
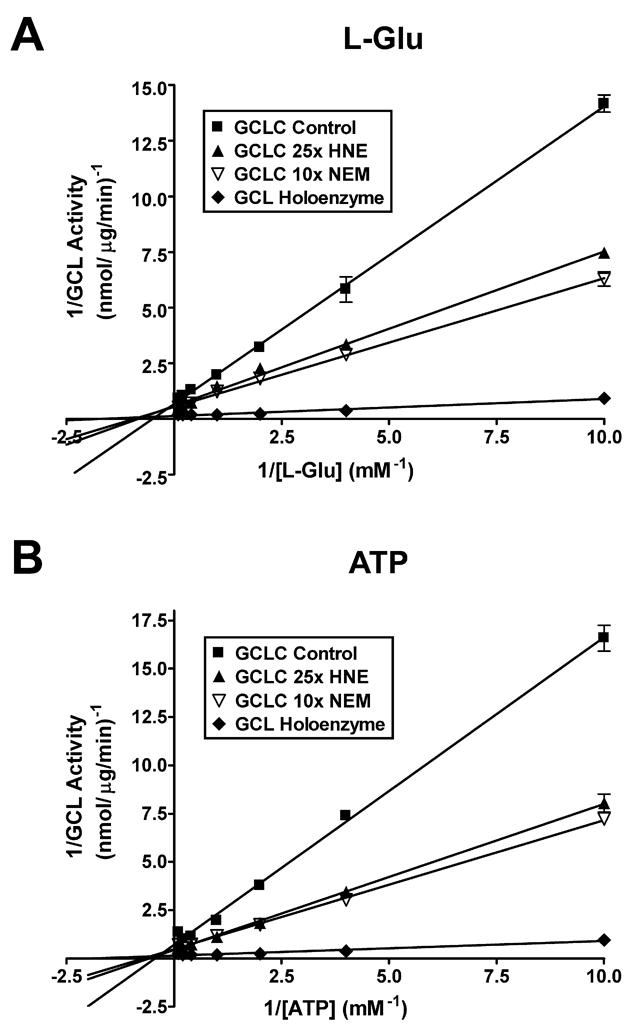
Purified recombinant GCLC was incubated in the absence or presence of a 25-fold molar excess of 4-HNE, a 10-fold molar excess of NEM, or an equivalent molar amount of GCLM for 30 min at 37°C. GCL specific activity was determined under conditions of variable substrate concentrations and Lineweaver-Burke plots generated using GraphPad Prism 4.0 for (A) L-glutamate and (B) ATP.
Table 1. Summary of the effects of 4-HNE and NEM modification on monomeric GCLC enzyme kinetics compared to GCL holoenzyme.
Purified recombinant GCLC was incubated in the absence or presence of a 25-fold molar excess of 4-HNE, a 10-fold molar excess of NEM, or an equivalent molar amount of GCLM for 30 min at 37°C. GCL specific activity (Vmax) was determined under saturating concentrations of substrates while variable substrate concentrations were employed to determine Km values for L-glutamate and ATP.
| GCLC Control | GCLC 25x HNE | GCLC 10x NEM | GCL Holoenzyme | |
|---|---|---|---|---|
| Km L-Glu (mM) | 1.98 +/- 0.22 | 1.18 +/- 0.07** | 1.07 +/- 0.11** | 0.55 +/- 0.02*** |
| Km ATP (mM) | 2.24 +/- 0.16 | 1.59 +/- 0.13* | 1.43 +/- 0.14** | 0.48 +/- 0.01*** |
| Vmax (nmol/μg/min) | 1.12 +/- 0.03 | 1.91 +/- 0.05*** | 2.05 +/- 0.13*** | 5.48 +/- 0.07*** |
All values are reported as the means +/- the standard error for three experiments
p<0.05,
p<0.01
p<0.001 vs GCLC control
4-HNE-mediated adduction of the GCL subunits prevents GCL holoenzyme formation
While 4-HNE did not cause the dissociation of pre-formed GCL holoenzyme into its monomeric subunits, it was of interest to determine whether prior adduction of the GCL subunits might affect their ability to heterodimerize and form functional GCL holoenzyme. Monomeric GCLC and GCLM were treated separately with increasing molar ratios of 4-HNE at 37°C for 30 min prior to mixing at a 1:1 molar ratio and GCL holoenzyme formation and activity were examined. In contrast to the cross-linking effects of 4-HNE on pre-formed GCL holoenzyme, adduction of both GCL subunits with increasing concentrations of 4-HNE prior to mixing prevented GCL holoenzyme formation, with complete inhibition of GCL subunit heterodimerization observed at 4-HNE molar ratios above 25x (Figure 5A). 4-HNE-mediated inhibition of GCL holoenzyme formation resulted in a proportionally lower GCL activity upon mixing of the subunits (Figure 5B). However, GCL activity was still ~2-fold greater than untreated GCLC at 4-HNE concentrations that abrogated GCL holoenzyme formation. This is likely due to 4-HNE-mediated adduction and activation of monomeric GCLC, which resulted in a similar level of GCL activity (see Figure 3D). Interestingly, prior adduction of either GCLC or GCLM alone was sufficient to impair or prevent, respectively, GCL holoenzyme complex formation (Figure 5C, lanes 4 and 5) and significantly reduce GCL activity (Figure 5D) upon mixing of the GCL subunits. These findings indicate that 4-HNE modification simultaneously activates monomeric GCLC activity and prevents its ability to heterodimerize with GCLM and form functional GCL holoenzyme.
Figure 5. 4-HNE-mediated adduction of the individual GCL subunits prevents heterodimerization and GCL holoenzyme formation.
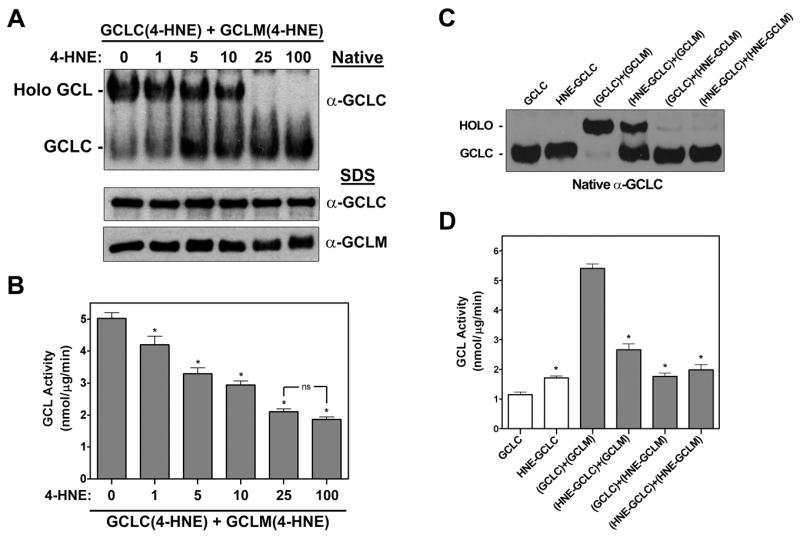
Purified recombinant GCLC and GCLM were incubated separately with (A and B) the indicated molar ratio of 4-HNE or (C and D) 100X molar excess of 4-HNE for 30 min at 37°C prior to mixing at a 1:1 molar ratio to form GCL holoenzyme. (A and C) GCL holoenzyme formation was assessed by native PAGE and immunoblotting for GCLC (top panel). The relative levels of the individual subunits were assessed by SDS-PAGE and immunoblotting for GCLC or GCLM (bottom panels). (B and D) GCL activity was determined by the fluorescence-based NDA assay (* p<0.001, ns = not significant).
Identification of 4-HNE-adducted residues of the GCL subunits by mass spectrometry
To identify the specific residue(s) of the GCL subunits that were adducted by 4-HNE, proteolytic digests of untreated and 4-HNE-adducted GCLM, GCLC, and GCL holoenzyme were analyzed by MALDI-TOF/TOF. MS analysis of 4-HNE-adducted monomeric GCLM identified one 4-HNE-modified Cys residue on the peptide fragment CSILNYLK that corresponded to Cys35 of GCLM (Figure 6A). Likewise, MS analysis of 4-HNE-adducted monomeric GCLC identified a single modified Cys residue on the peptide fragment CPSTHSEELR that corresponded to Cys553 of GCLC (Figure 6B). No other 4-HNE-modified residues were detected by MS analysis even though coverage was >95% for both GCLC and GCLM (data not shown). Interestingly, Cys35 of GCLM was modified upon treatment of either monomeric GCLM or pre-formed GCL holoenzyme, while modification of GCLC Cys553 was only detected upon treatment of monomeric GCLC (data not shown). These findings suggest that heterodimerization with GCLM and formation of GCL holoenzyme either directly masks Cys553 of GCLC or induces a conformational change that indirectly masks Cys553 of GCLC making it inaccessible to adduction by 4-HNE.
Figure 6. Identification of GCLM(Cys35) and GCLC(Cys553) as sites for 4-HNE-mediated adduction in vitro.
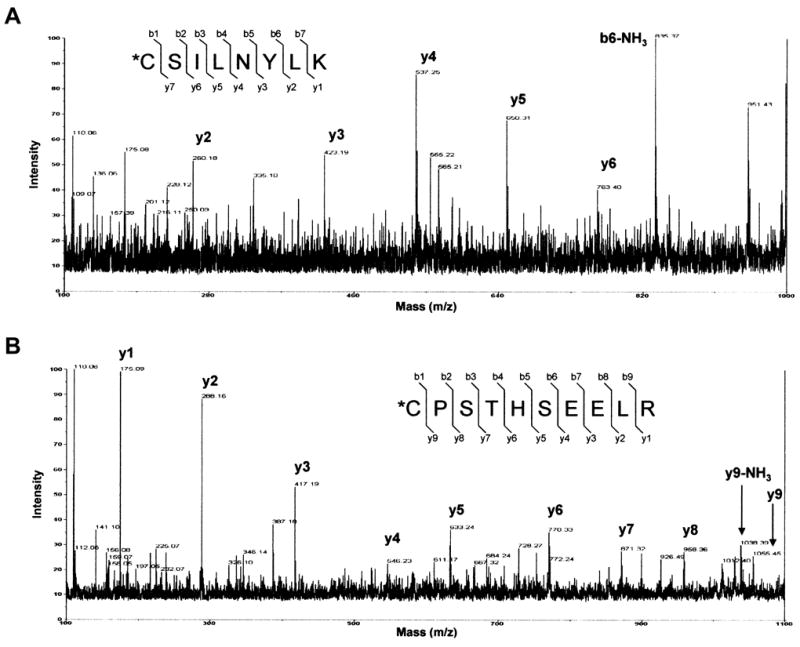
Monomeric GCLC and GCLM were incubated with a 25X molar excess of 4-HNE for 30 min at 37°C. Samples were reduced with NaBH4, resolved by SDS-PAGE, and digested with trypsin, Glu-C, or chymotrypsin and peptide fragments were analyzed by MALDI-TOF/TOF mass spectrometry. (A) MS/MS spectra of the [M+H]+ ion at m/z = 1316.4 from the 4-HNE-modified GCLM peptide containing Cys-35, *CPSTHSEELR. (B) MS/MS spectra of the [M+H]+ ion at m/z = 1111.5 from the 4-HNE-modified GCLC peptide containing Cys-553, *CSILNYLK. The asterisk indicates the site of adduction. (b/y fragmentation predicted using ProteinProspector, UCSF).
Differential regulation of GCLC and GCL holoenzyme activity by selective modification of cysteine and lysine residues
Identification of Cys553 of GCLC as a target for 4-HNE-mediated adduction was of great interest as this residue has been implicated in GCLC enzyme activation upon GCL holoenzyme formation [41]. To assess the potential involvement of Cys modification in GCLC activation, we examined the functional effects of the Cys-modifying agent, N-ethylmaleimide (NEM). Interestingly, treatment of monomeric GCLC with NEM resulted in a dose-dependent increase in GCLC enzymatic activity (Figure 7A, left) that was similar to that observed for 4-HNE (see Figure 3D). However, NEM was a more effective activator of monomeric GCLC than 4-HNE, with maximal GCLC activation occurring upon treatment with a 10-fold molar excess of NEM. In contrast to 4-HNE, NEM had a biphasic effect on GCLC activity, with a 100-fold molar excess nearly abolishing the enzymatic activity of the monomeric subunit. These differential functional effects are likely attributable to the ability of NEM to modify a number of additional solvent accessible Cys residues within GCLC. In this regard, MS analysis of NEM-treated GCLC identified 12 modified residues, including Cys553, which was also adducted by 4-HNE (Table 2). NEM caused a dose-dependent reduction in GCL holoenzyme activity similar to that of 4-HNE (Figure 7A, right). However, NEM was a much more effective and potent inhibitor of GCL enzymatic activity compared to 4-HNE. While MS analysis of the 4-HNE-treated GCL subunits only identified a single adducted Cys residue on GCLC and GCLM, Lys residues are also highly susceptible to 4-HNE-mediated adduction yet are notoriously unstable and difficult to detect [5, 18, 57]. To indirectly examine whether Lys adduction could mediate some of the functional effects of 4-HNE we examined the effects of the Lys-specific modifying agent Sulfo-NHS-Acetate (NHS) on GCL activity. Selective covalent modification of Lys residues with NHS led to the dose-dependent inhibition of GCL holoenzyme activity (Figure 7B, right) with a nearly identical efficacy and potency to that of 4-HNE (see Figure 3D, right). In contrast, modification of Lys residues with NHS was not capable of recapitulating the stimulatory effects of 4-HNE on monomeric GCLC activity (Figure 7B, left). While Lys-specific modification inhibited GCL holoenzyme activity by ~50% (Figure 7B and 8B, black bars), this was not due to dissociation of the GCL heterodimeric complex. The NHS-modified GCL holoenzyme complex remained intact even though it migrated aberrantly on native gels (Figure 8A, lanes 7 and 8). In contrast, prior modification of one or both GCL subunits was sufficient to impair formation of the GCL holoenzyme complex (Figure 8A, lanes 4-6) and significantly reduce GCL activity (Figure 8B, grey bars) upon mixing of the GCL subunits. Taken together these results suggest that Cys and Lys residues play a critical role in regulating GCL holoenzyme formation and activity and suggest that 4-HNE-mediated alterations in GCL activity may be due, at least in part, to modification of Cys and Lys residues on one or both of the GCL subunits.
Figure 7. Cys- and Lys-specific modifications differentially affect GCLC and GCL holoenzyme activity.
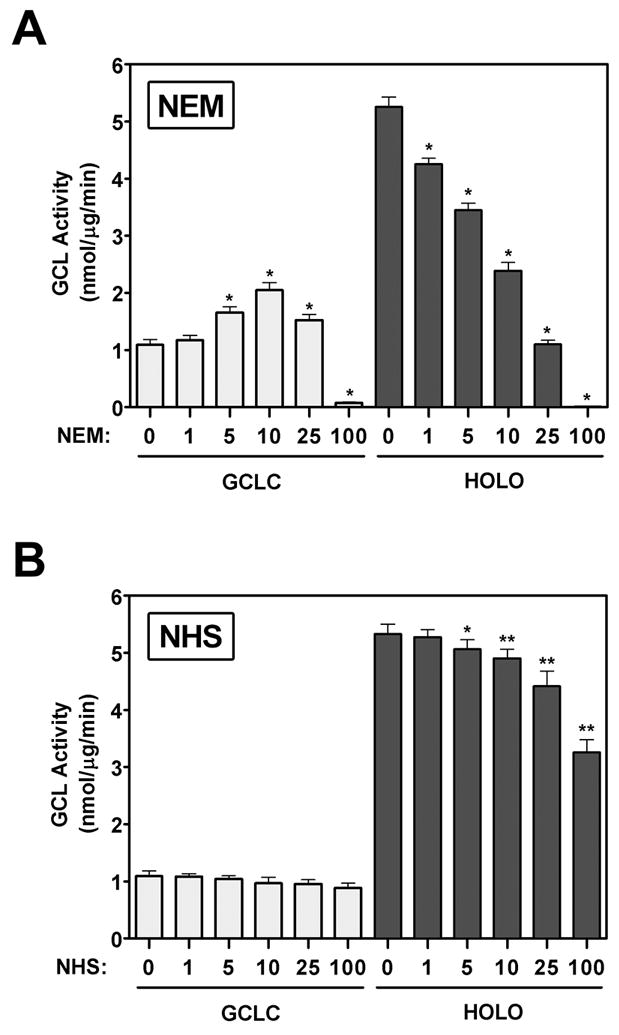
Purified recombinant monomeric GCLC (open bars) or GCL holoenzyme (closed bars) were incubated in the absence or presence of the indicated molar excess of (A) NEM or (B) NHS for 30 min at 37°C. GCL activity was determined by the fluorescence-based NDA assay (* p<0.001 vs. respective NEM controls; *p<0.01, ** p<0.001 vs. respective NHS controls).
Table 2. Summary of GCL subunit residues modified by 4-HNE and NEM.
Recombinant monomeric GCLC or GCLM were treated with a 25-fold molar excess of 4-HNE or a 10-fold molar excess of NEM for 30 min at 37°C. Modified residues were detected by MS analysis as described in the Materials and Methods.
| 4-HNE | NEM | ||
|---|---|---|---|
| GCLM | Cys 35 | Cys 35 | |
| GCLC | Cys 553 | Cys 142 | Cys 152 |
| Cys 248 | Cys 249 | ||
| Cys 257 | Cys 295 | ||
| Cys 329 | Cys 501 | ||
| Cys 553 | Cys 605 | ||
| Cys 613 | Cys 615 | ||
Figure 8. Lys-specific modification of the GCL subunits attenuates GCL holoenzyme complex formation and activity.
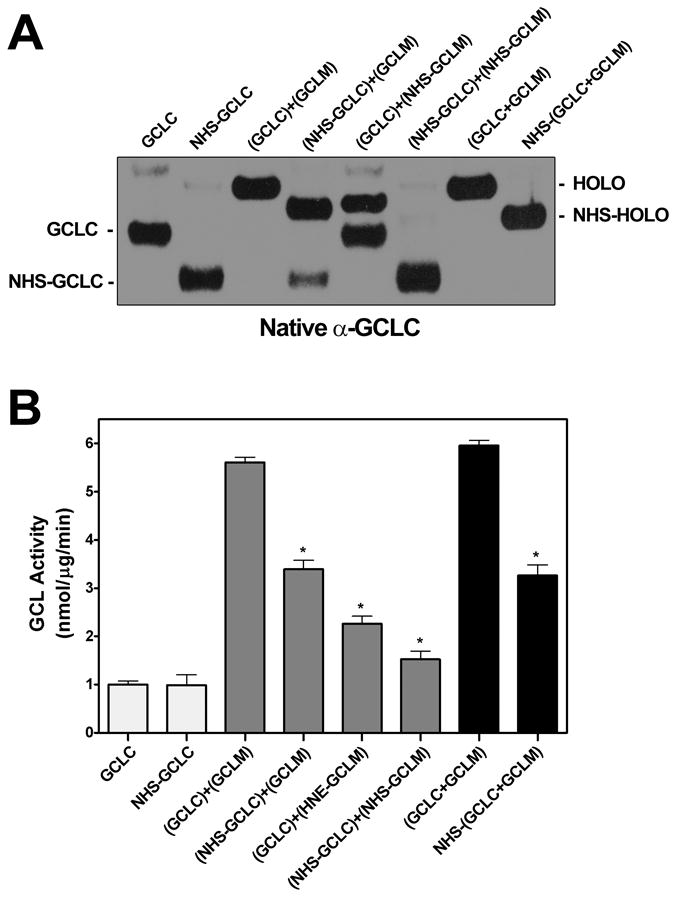
Purified recombinant GCLC and/or GCLM were incubated separately with 100X molar excess of 4-HNE for 30 min at 37°C prior to mixing at a 1:1 molar ratio to form GCL holoenzyme as indicated. The GCL subunits were mixed prior to treatment in lanes 7 and 8. (A) GCL holoenzyme formation was assessed by native PAGE and immunoblotting for GCLC. NHS-modified GCLC migrated aberrantly upon native PAGE both as a monomer (NHS-GCLC) and a GCL holoenzyme complex (NHS-HOLO) as indicated. (B) GCL activity was determined by the fluorescence-based NDA assay (* p<0.001 vs. GCL holoenzyme activity controls).
Homology modeling of monomeric GCL subunits and protein-protein interaction modeling of GCL holoenzyme
As the crystal structure of human and rodent GCLC and GCLM have not been elucidated, homology modeling of murine GCLM and GCLC was employed to examine the structural characteristics of the subunits and how 4-HNE-mediated adduction may affect GCLC and GCL holoenzyme formation and/or activity. Homology models of murine GCLM and GCLC were constructed by sequence submission to the ModWeb homology modeling server (Figure 9A & 9B) [51]. The structure of the yeast homolog of GCL was recently determined by Biterova, et al [58]. This structure was used to produce a homology model of murine GCLC from a template with a high degree of similarity in amino acid sequence (~44% identity, 63% similarity) (Figure 9B). The C-terminus of GCLC has been postulated to contain a GCLM-interaction domain involved in both GCL holoenzyme formation and enzyme activation [28, 37, 41]. While Cys553 of GCLC is located at a C-terminal site distant from the N-terminal active site of GCLC, it has been shown to be important for heterodimerization-induced activation of GCLC [27, 28]. As 4-HNE adduction of GCLC has similar functional effects on GCLC enzyme kinetics (increased Vmax and substrate binding affinities), it is intriguing to speculate that 4-HNE-mediated adduction of Cys553 of GCLC and GCL holoenzyme formation evoke a similar conformational change at the active site of GCLC. Consistent with this model, Cys553 of GCLC was not adducted by 4-HNE when present as GCL holoenzyme, suggesting that the region surrounding Cys553 likely resides within the interaction interface between GCLC and GCLM. Possible interactions between the GCLC and GCLM homology models were examined using Escher NG, which predicts likely protein-protein interactions based on shape complementarity and steric interactions [54]. Many of the resulting interactions involved the region of GCLC containing Cys553 and a representative structure is given in Figure 9C. Similar to Cys553 of GCLC, Cys193 and Cys194 of murine GCLM are homologues of conserved residues Cys213 and Cys214 within Drosophila GCLM that have been implicated in enhanced GCL enzyme activation upon holoenzyme formation, but have no effect on heterodimerization per se [59]. Interestingly, several of the predicted interaction models between GCLC and GCLM involved direct interaction between Cys553 and Cys193/Cys194. In contrast, while Cys35 of GCLM is conserved in many species, the functional relevance of 4-HNE-mediated modification of Cys35 is not clear as there is no known structural or functional role associated with this residue. While homology modeling of murine GCLM indicates that Cys35 of GCLM is not solvent accessible, modification of Cys35 is readily detected upon treatment with either 4-HNE or NEM (Table 2). Furthermore, adduction of GCLM alone with 4-HNE or NEM is sufficient to abolish GCL holoenzyme formation when mixed with unmodified GCLC (data not shown). Such findings suggest that modification of Cys35 of GCLM is likely functionally relevant, but does not preclude the potential involvement of undetected Lys modification(s) in mediating the effects of 4-HNE.
Figure 9. Homology model of murine GCLM, GCLC, and GCL holoenzyme.
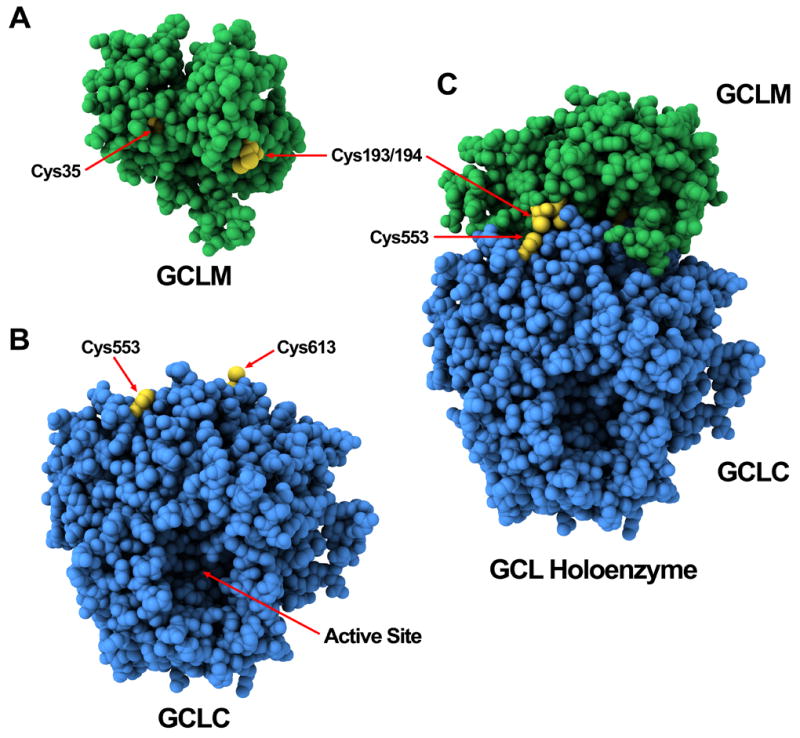
The murine GCLM and GCLC protein sequences were submitted to the ModWeb protein modeling server and the resulting protein structures of (A) GCLM and (B) GCLC were rendered in Lightwave 3D 9.6. Cys residues of interest are colored in yellow and indicated with arrows. The remaining residues of GCLM are colored in green and those of GCLC in blue. (C) Prediction of protein-protein interaction between the GCLM and GCLC homology models was calculated with Escher NG and a representative solution is depicted.
Discussion
It is well established that exposure to inducers of oxidative stress, including 4-HNE, lead to a reduction in cellular GSH levels and a compensatory increase in the transcription and translation of one or both subunits of GCL [30, 32, 60, 61]. This adaptive response is thought to result in increased GCL enzymatic activity and cellular GSH biosynthetic capacity, which correlate with the relative levels of the GCL subunits [37]. However, there is a temporal disconnect between the reestablishment of GSH homeostasis and induction of the GCL subunits. GSH levels are rapidly restored, while upregulation of the GCL subunits is not observed for several hours following 4-HNE treatment [34-36, 62]. Such findings have led us to hypothesize that post-translational control mechanisms may contribute to the rapid activation of GCL following 4-HNE exposure. Consistent with this model cellular GCL activity in A549 cells is rapidly increased (<30 min) following 4-HNE treatment. Furthermore, there was no detectable increase in GCL subunit protein expression within this time frame and 4-HNE-induced GCL activity was insensitive to the protein synthesis inhibitor CHX. These findings preclude the involvement of de novo GCL subunit protein expression in mediating this adaptive increase in GCL activity. While the relative levels of the GCL subunits are a major determinant of cellular GSH biosynthetic capacity, cellular GCL activity can also be regulated by post-translational modification of one or both of the GCL subunits [38-40]. In this regard, we have recently demonstrated that certain forms of oxidative stress (H2O2 and phorone) can post-translationally activate GCL by causing a shift of GCLC from its low activity monomeric form to a high activity holoenzyme form [44]. However, 4-HNE-induced GCL activity in A549 cells was not associated with a change in the dynamic equilibrium between the monomeric and holoenzyme forms of GCLC (data not shown). 4-HNE is known to form covalent adducts with nucleophilic residues on cellular proteins with a reactivity profile of Cys≫His>Lys [8, 9] and a wide variety of cellular proteins and enzymes are known to be susceptible to 4-HNE adduction, including tubulin [12], protein disulfide isomerase [13], peroxiredoxin 6 [18], glutathione-S-transferase [15], and glutathione reductase [14]. 4-HNE-mediated adduction of these and other proteins almost invariably leads to the suppression or complete inactivation of protein function [2, 12-17]. A salient aspect of the current study is the novel finding that 4-HNE-mediated post-translational modification of GCLC stimulates its enzymatic activity. Within a cellular context, this rapid post-translational regulation of GCL activity could significantly affect cellular GSH homeostasis and antioxidant capacity, and GSH-dependent detoxification during periods of oxidative stress.
Cys residues are particularly susceptible to adduction by α,β-unsaturated aldehydes [4, 5, 7], and Cys modification by 4-HNE is a common mechanism of protein inactivation [10, 13, 63]. Multiple Cys residues of the GCL subunits play important structural and functional roles in regulating GCL holoenzyme formation and activity [28, 37, 41, 45]. Importantly, GCL holoenzyme formation is thought to involve intermolecular sulfhydryl bond formation between Cys residues of the GCL subunits [43, 64]. MS analysis identified Cys553 of GCLC as a site of 4-HNE adduction. Modification at this site is likely functionally relevant as Cys553 of GCLC has been implicated in GCLC enzyme activation upon holoenzyme formation [41]. While mutation of GCLC Cys553 has no effect on monomeric GCLC activity, it significantly reduces GCL holoenzyme activity yet does not prevent GCL holoenzyme formation per se [41]. Thus, Cys553 does not necessarily participate in the heterodimerization with GCLM, but is required for heterodimerization-induced enhancement of GCLC enzymatic activity. Interestingly, this site was not detected in 4-HNE-treated GCL holoenzyme, suggesting that 4-HNE accessibility to this residue is obstructed by the formation of the heterodimer. Protein interaction modeling suggests the C-terminus of GCLC is a likely area of interaction with GCLM based on shape complementarity and steric interactions. The importance of Cys553 and other Cys residues in regulating GCLC enzyme activity was further highlighted by studies employing the Cys-modifying agent NEM. While Cys553 was the only 4-HNE adduct detected on GCLC, NEM treatment modified a number of Cys residues, including Cys553 and Cys613 within the C-terminus of GCLC, indicating that both residues are solvent accessible and could participate in sulfhydryl bond formation with GCLM. These findings are consistent with homology modeling of GCLC which suggests that Cys613 could be involved in heterodimerization with GCLM. Interestingly, yeast do not express a GCLM subunit and the yeast homolog of GCLC, which lacks Cys553 and Cys613, functions as a monomer [58]. Not only did both 4-HNE and NEM modify Cys553 of GCLC, but treatment with low concentrations of NEM activated monomeric GCLC in a manner identical to that of 4-HNE. Furthermore, both 4-HNE and NEM altered GCLC enzyme kinetics in a manner similar to that observed following heterodimerization with GCLM and formation of GCL holoenzyme, including increased Vmax and lowered Km for both L-glutamate and ATP. Based on these findings, it is intriguing to speculate that 4-HNE- or NEM-mediated covalent modification of specific Cys residues within GCLC may be sufficient to induce a partial enhancement of enzyme activity by simulating the conformational changes elicited by GCL holoenzyme formation. However, 4-HNE and NEM increased GCLC activity much less than GCL holoenzyme formation, indicating that Cys modification does not fully recapitulate the functional effects associated with heterodimerization and that additional molecular events contribute to GCLC activation upon formation of the GCL holoenzyme complex.
Unlike 4-HNE, NEM treatment caused a biphasic dose-dependent effect on monomeric GCLC activity. While low doses of NEM significantly increased GCLC activity, higher concentrations of NEM nearly abolished both GCLC and GCL holoenzyme activity. These differential functional effects may be due to the ability of NEM to adduct Cys248 and Cys249 of GCLC. These Cys residues are thought to reside within the active site of GCLC [28], which is in agreement with the location of these residues in our in silico homology modeling studies (data not shown). Mutation of Cys248 and Cys249 attenuates, but does not abrogate GCLC activity [28]. While Cys248/Cys249 may not be essential for the enzymatic activity of monomeric GCLC or GCL holoenzyme, covalent modification of one or both of these residues would introduce functional groups into the active site that could affect substrate binding affinity and/or catalytic activity. Indeed, modification of these Cys residues likely provides the molecular basis for NEM-mediated inhibition of GCLC and GCL holoenzyme activity observed at higher concentrations of NEM.
4-HNE and NEM clearly have differential dose-dependent effects on GCLC and GCL holoenzyme activity. The selective adduction of Cys553 by 4-HNE may be derivative of its reactivity towards various Cys residues within GCLC, which is influenced by the ionization state of the thiol, with the thiolate anion form the more reactive of the two [7]. Cys553 has a predicted pKa of 7.59, compared to 14.51 and 10.98 for Cys248 and Cys249, respectively (Table 3), indicating that Cys553 is more likely to be in the thiolate form, and hence more reactive towards 4-HNE than either of the active site Cys residues at physiological pH. In fact, the predicted pKa’s of each Cys residue varies considerably, suggesting that there is a range of reactivities towards the individual Cys residues. In the case of NEM, we were unable to distinguish the relative quantities of each adduct by MS analysis and our results represent a population of all the possible NEM-mediated modifications. However, when taken together with the analysis of GCL activity and the predicted pKa values, it is likely that the relative level of adduction at any given NEM concentration varies by residue. For example, NEM-induced GCL activity at lower doses of NEM may result from extensive modification of Cys553, while the inhibition of GCL activity observed at higher NEM doses may be due to increased adduction of the active site Cys residues.
Table 3. Predicted pKa of selected Cys residues adducted by 4-HNE and/or NEM.
| Residue | pKa |
|---|---|
| Cys 248 | 14.51 |
| Cys 249 | 10.98 |
| Cys 553 | 7.59 |
| Cys 613 | 9.07 |
No Lys adducts were detected on either GCL subunit when analyzed by MS. However, 4-HNE adduction of Lys residues tends to be reversible and difficult to detect by MS [5, 18]. Indeed, several lines of evidence indicate that 4-HNE-mediated modification of Lys residues does occur and may be functionally relevant. First, there is significant 4-HNE-induced cross-linking between the GCL subunits (Figure 3C and 3E), which likely results from Michael addition and subsequent Schiff base formation at a neighboring Lys residue [5, 7, 18, 19]. Pretreatment with the Lys-specific modifier Sulfo-NHS-Acetate (NHS) also substantially reduced 4-HNE-mediated adduction of both monomeric GCL subunits and GCL holoenzyme as detected by immunoblotting (data not shown). These residue-masking studies provide additional evidence that 4-HNE likely modifies one or more Lys residue(s) on both GCLC and GCLM. Studies are underway to identify the specific residues adducted. NHS was also employed to assess the functional effect(s) of any potential adduction of Lys residues. NHS had no effect on monomeric GCLC activity, suggesting that 4-HNE-induced activation of monomeric GCLC is likely mediated by adduction of Cys residues alone. In contrast, NHS inhibited GCL holoenzyme activity with a potency and efficacy almost identical to that observed for 4-HNE, suggesting that 4-HNE-induced inhibition of GCL holoenzyme activity may involve adduction of Lys residues. Interestingly, while NHS treatment did not result in the dissociation of pre-formed GCL holoenzyme, NHS pretreatment of either GCL subunit prior to mixing impaired heterodimerization and formation of the high activity GCL holoenzyme complex. These results support the importance of Lys residues of both GCLC and GCLM in regulating heterodimerization and GCL holoenzyme activity. Although we have identified only Cys adducts by MS and our data appear to indicate that the activating effects of 4-HNE and NEM on GCLC are mediated by Cys modification, it is likely that Cys adducts do not represent the only post-translational modifications of 4-HNE on the GCL subunits and Lys modification likely plays an important role in the impairment of GCL holoenzyme formation and activity.
In most cell types, GCLM expression is limiting in the formation of the GCL holoenzyme complex and total cellular GCL activity is the sum of the activities of both monomeric GCLC and GCL holoenzyme [26, 35, 37, 65]. Under in vitro conditions we observed both an increase in monomeric GCLC activity and an inhibition of holoenzyme activity upon 4-HNE adduction. Under conditions of excess monomeric GCLC, as would be expected in most tissues and cell types, 4-HNE modification of the GCL subunits would likely result in a net increase in cellular GCL activity, as activation of GCLC occurred at 4-HNE concentrations that had little effect on GCL holoenzyme activity. A549 cells have an excess of GCLC compared to GCLM, suggesting that adduction and activation of monomeric GCLC is likely responsible for the rapid increase in cellular GCL activity in response to 4-HNE treatment. In contrast, 4-HNE exposure may lead to a decrease in cellular GCL activity and impaired GSH biosynthetic capacity in tissues that express more equivalent molar ratios of GCLC and GCLM, such as heart [35] or kidney [26]. 4-HNE-mediated depletion of intracellular GSH due to spontaneous and/or GST-mediated conjugation of 4-HNE could also contribute to increased cellular GCL activity via reduced GSH feedback inhibition of GCLC [25]. However, A549 cells have very high GSH levels and the relatively small reduction in GSH levels caused by 4-HNE would only increase cellular GCL activity by 10-20% based on in vitro studies utilizing purified recombinant proteins [27]. 4-HNE may cause a more dramatic depletion of GSH in cells/tissues with low initial levels of GSH. Under such conditions, it would be expected that reduced GSH feedback inhibition of GCLC may play a much more important role in regulating GCL activity in response to 4-HNE. Ultimately, the functional effects of 4-HNE on cellular GCL activity and GSH biosynthetic capacity are likely complex, depending on 4-HNE concentration and the relative levels GSH and the GCL subunits.
Increased GSH biosynthesis resulting from transcriptional induction and increased expression of the GCL subunits is thought to play a major role in reestablishing GSH homeostasis following oxidative insult [31-36]. However, there is little experimental evidence to demonstrate that this transcriptional response is necessary, sufficient, or constitutes the sole regulatory mechanism involved in the reestablishment of cellular GSH homeostasis. Our findings suggest that direct post-translational regulation of GCL activity by the lipid peroxidation product 4-HNE may contribute to this compensatory cytoprotective response. Our demonstration that 4-HNE can directly adduct the GCL subunits and alter GCL enzymatic activity represents a novel mechanism of post-translational modification and regulation of GCL enzymatic activity. Within a cellular context, this direct activation of GCL could serve a cytoprotective function by rapidly increasing cellular GSH biosynthetic capacity and dramatically affecting cellular antioxidant capacity and GSH-dependent detoxification during periods of acute and chronic oxidative stress.
Acknowledgments
This work was supported by NIH grants CA75316, CA90473, and ES07033 (CCF), R37AA09300 (DRP), F31AA016710 (JRR). The authors thank Drs. Terrance J. Kavanagh and Ben J. Stewart for numerous valuable discussions and Dr. Kavanagh for providing the GCL antibodies and other reagents used in these studies. The authors also thank Dr. Philip Reigan and the University of Colorado Denver, School of Pharmacy, Computational Chemistry and Biology Core Facility for assistance with homology and interaction modeling and the UCD SOP Mass Spectrometry Facility and Mr. Joe Gomez for assistance with the analysis of samples.
List of Abbreviations
- 4-HNE
4-hydroxy-2-nonenal
- GCL
glutamate-cysteine ligase
- GCLC
catalytic subunit of glutamate-cysteine ligase
- GCLM
modifier subunit of glutamate-cysteine ligase
- GST
glutathione S-transferase
- GSH
glutathione
- Cys
L-cysteine
- His
L-histidine
- Lys
L-lysine
- NEM
N-ethylmaleimide
- NHS
Sulfo-NHS-Acetate
- CHX
cycloheximide
- NDA
naphthalene-2,3-dicarboxaldehyde
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ando Y, Brannstrom T, Uchida K, Nyhlin N, Nasman B, Suhr O, Yamashita T, Olsson T, El Salhy M, Uchino M, Ando M. Histochemical detection of 4-hydroxynonenal protein in Alzheimer amyloid. J Neurol Sci. 1998;156:172–176. doi: 10.1016/s0022-510x(98)00042-2. [DOI] [PubMed] [Google Scholar]
- 2.Carbone DL, Doorn JA, Kiebler Z, Ickes BR, Petersen DR. Modification of heat shock protein 90 by 4-hydroxynonenal in a rat model of chronic alcoholic liver disease. J Pharmacol Exp Ther. 2005;315:8–15. doi: 10.1124/jpet.105.088088. [DOI] [PubMed] [Google Scholar]
- 3.Rahman I, van Schadewijk AA, Crowther AJ, Hiemstra PS, Stolk J, MacNee W, De Boer WI. 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2002;166:490–495. doi: 10.1164/rccm.2110101. [DOI] [PubMed] [Google Scholar]
- 4.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 5.Sayre LM, Lin D, Yuan Q, Zhu X, Tang X. Protein adducts generated from products of lipid oxidation: focus on HNE and one. Drug Metab Rev. 2006;38:651–675. doi: 10.1080/03602530600959508. [DOI] [PubMed] [Google Scholar]
- 6.Kurtz AJ, Lloyd RS. 1,N2-deoxyguanosine adducts of acrolein, crotonaldehyde, and trans-4-hydroxynonenal cross-link to peptides via Schiff base linkage. J Biol Chem. 2003;278:5970–5976. doi: 10.1074/jbc.M212012200. [DOI] [PubMed] [Google Scholar]
- 7.LoPachin RM, Gavin T, Petersen DR, Barber DS. Molecular mechanisms of 4-hydroxy-2-nonenal and acrolein toxicity: nucleophilic targets and adduct formation. Chem Res Toxicol. 2009;22:1499–1508. doi: 10.1021/tx900147g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Doorn JA, Petersen DR. Covalent modification of amino acid nucleophiles by the lipid peroxidation products 4-hydroxy-2-nonenal and 4-oxo-2-nonenal. Chem Res Toxicol. 2002;15:1445–1450. doi: 10.1021/tx025590o. [DOI] [PubMed] [Google Scholar]
- 9.Petersen DR, Doorn JA. Reactions of 4-hydroxynonenal with proteins and cellular targets. Free Radic Biol Med. 2004;37:937–945. doi: 10.1016/j.freeradbiomed.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 10.Uchida K, Stadtman ER. Covalent attachment of 4-hydroxynonenal to glyceraldehyde-3-phosphate dehydrogenase. A possible involvement of intra- and intermolecular cross-linking reaction. J Biol Chem. 1993;268:6388–6393. [PubMed] [Google Scholar]
- 11.Friguet B, Stadtman ER, Szweda LI. Modification of glucose-6-phosphate dehydrogenase by 4-hydroxy-2-nonenal. Formation of cross-linked protein that inhibits the multicatalytic protease. J Biol Chem. 1994;269:21639–21643. [PubMed] [Google Scholar]
- 12.Stewart BJ, Doorn JA, Petersen DR. Residue-specific adduction of tubulin by 4-hydroxynonenal and 4-oxononenal causes cross-linking and inhibits polymerization. Chem Res Toxicol. 2007;20:1111–1119. doi: 10.1021/tx700106v. [DOI] [PubMed] [Google Scholar]
- 13.Carbone DL, Doorn JA, Kiebler Z, Petersen DR. Cysteine modification by lipid peroxidation products inhibits protein disulfide isomerase. Chem Res Toxicol. 2005;18:1324–1331. doi: 10.1021/tx050078z. [DOI] [PubMed] [Google Scholar]
- 14.Vander Jagt DL, Hunsaker LA, Vander Jagt TJ, Gomez MS, Gonzales DM, Deck LM, Royer RE. Inactivation of glutathione reductase by 4-hydroxynonenal and other endogenous aldehydes. Biochem Pharmacol. 1997;53:1133–1140. doi: 10.1016/s0006-2952(97)00090-7. [DOI] [PubMed] [Google Scholar]
- 15.Mitchell AE, Morin D, Lame MW, Jones AD. Purification, mass spectrometric characterization, and covalent modification of murine glutathione S-transferases. Chem Res Toxicol. 1995;8:1054–1062. doi: 10.1021/tx00050a009. [DOI] [PubMed] [Google Scholar]
- 16.Ishii T, Tatsuda E, Kumazawa S, Nakayama T, Uchida K. Molecular basis of enzyme inactivation by an endogenous electrophile 4-hydroxy-2-nonenal: identification of modification sites in glyceraldehyde-3-phosphate dehydrogenase. Biochemistry. 2003;42:3474–3480. doi: 10.1021/bi027172o. [DOI] [PubMed] [Google Scholar]
- 17.Sampey BP, Carbone DL, Doorn JA, Drechsel DA, Petersen DR. 4-Hydroxy-2-nonenal adduction of extracellular signal-regulated kinase (Erk) and the inhibition of hepatocyte Erk-Est-like protein-1-activating protein-1 signal transduction. Mol Pharmacol. 2007;71:871–883. doi: 10.1124/mol.106.029686. [DOI] [PubMed] [Google Scholar]
- 18.Roede JR, Carbone DL, Doorn JA, Kirichenko OV, Reigan P, Petersen DR. In vitro and in silico characterization of peroxiredoxin 6 modified by 4-hydroxynonenal and 4-oxononenal. Chem Res Toxicol. 2008;21:2289–2299. doi: 10.1021/tx800244u. [DOI] [PubMed] [Google Scholar]
- 19.Stewart BJ, Roede JR, Doorn JA, Petersen DR. Lipid aldehyde-mediated cross-linking of apolipoprotein B-100 inhibits secretion from HepG2 cells. Biochim Biophys Acta. 2009;1791:772–780. doi: 10.1016/j.bbalip.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu W, Kato M, Akhand AA, Hayakawa A, Suzuki H, Miyata T, Kurokawa K, Hotta Y, Ishikawa N, Nakashima I. 4-hydroxynonenal induces a cellular redox status-related activation of the caspase cascade for apoptotic cell death. J Cell Sci. 2000;113:635–641. doi: 10.1242/jcs.113.4.635. [DOI] [PubMed] [Google Scholar]
- 21.Raza H, John A, Brown EM, Benedict S, Kambal A. Alterations in mitochondrial respiratory functions, redox metabolism and apoptosis by oxidant 4-hydroxynonenal and antioxidants curcumin and melatonin in PC12 cells. Toxicol Appl Pharmacol. 2008;226:161–168. doi: 10.1016/j.taap.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 22.Berhane K, Widersten M, Engstrom A, Kozarich JW, Mannervik B. Detoxication of base propenals and other alpha, beta-unsaturated aldehyde products of radical reactions and lipid peroxidation by human glutathione transferases. Proc Natl Acad Sci U S A. 1994;91:1480–1484. doi: 10.1073/pnas.91.4.1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukuda A, Osawa T, Hitomi K, Uchida K. 4-Hydroxy-2-nonenal cytotoxicity in renal proximal tubular cells: protein modification and redox alteration. Arch Biochem Biophys. 1996;333:419–426. doi: 10.1006/abbi.1996.0410. [DOI] [PubMed] [Google Scholar]
- 24.Sekura R, Meister A. γ-Glutamylcysteine synthetase. J Biol Chem. 1977;252:2599–2605. [PubMed] [Google Scholar]
- 25.Griffith OW, Mulcahy RT. The enzymes of glutathione synthesis: gamma-glutamylcysteine synthetase. Adv Enzymol Relat Areas Mol Biol. 1999;73:209–267. doi: 10.1002/9780470123195.ch7. [DOI] [PubMed] [Google Scholar]
- 26.Chen Y, Shertzer HG, Schneider SN, Nebert DW, Dalton TP. Glutamate cysteine ligase catalysis: dependence on ATP and modifier subunit for regulation of tissue glutathione levels. J Biol Chem. 2005;280:33766–33774. doi: 10.1074/jbc.M504604200. [DOI] [PubMed] [Google Scholar]
- 27.Tu Z, Anders MW. Expression and characterization of human glutamate-cysteine ligase. Arch Biochem Biophys. 1998;354:247–254. doi: 10.1006/abbi.1998.0676. [DOI] [PubMed] [Google Scholar]
- 28.Yang Y, Chen Y, Johansson E, Schneider SN, Shertzer HG, Nebert DW, Dalton TP. Interaction between the catalytic and modifier subunits of glutamate-cysteine ligase. Biochem Pharmacol. 2007 doi: 10.1016/j.bcp.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 29.Dickinson DA, Levonen AL, Moellering DR, Arnold EK, Zhang H, Darley-Usmar VM, Forman HJ. Human glutamate cysteine ligase gene regulation through the electrophile response element. Free Radic Biol Med. 2004;37:1152–1159. doi: 10.1016/j.freeradbiomed.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 30.Lu SC. Regulation of glutathione synthesis. Mol Aspects Med. 2009;30:42–59. doi: 10.1016/j.mam.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Forman HJ, Dickinson DA, Iles KE. HNE--signaling pathways leading to its elimination. Mol Aspects Med. 2003;24:189–194. doi: 10.1016/s0098-2997(03)00013-x. [DOI] [PubMed] [Google Scholar]
- 32.Liu RM, Gao L, Choi J, Forman HJ. gamma-glutamylcysteine synthetase: mRNA stabilization and independent subunit transcription by 4-hydroxy-2-nonenal. Am J Physiol. 1998;275:L861–869. doi: 10.1152/ajplung.1998.275.5.L861. [DOI] [PubMed] [Google Scholar]
- 33.Liu RM, Borok Z, Forman HJ. 4-Hydroxy-2-nonenal increases gamma-glutamylcysteine synthetase gene expression in alveolar epithelial cells. Am J Respir Cell Mol Biol. 2001;24:499–505. doi: 10.1165/ajrcmb.24.4.4307. [DOI] [PubMed] [Google Scholar]
- 34.Dickinson DA, Iles KE, Watanabe N, Iwamoto T, Zhang H, Krzywanski DM, Forman HJ. 4-hydroxynonenal induces glutamate cysteine ligase through JNK in HBE1 cells. Free Radic Biol Med. 2002;33:974. doi: 10.1016/s0891-5849(02)00991-7. [DOI] [PubMed] [Google Scholar]
- 35.Krzywanski DM, Dickinson DA, Iles KE, Wigley AF, Franklin CC, Liu RM, Kavanagh TJ, Forman HJ. Variable regulation of glutamate cysteine ligase subunit proteins affects glutathione biosynthesis in response to oxidative stress. Arch Biochem Biophys. 2004;423:116–125. doi: 10.1016/j.abb.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 36.Iles KE, Liu RM. Mechanisms of glutamate cysteine ligase (GCL) induction by 4-hydroxynonenal. Free Radic Biol Med. 2005;38:547–556. doi: 10.1016/j.freeradbiomed.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 37.Franklin CC, Backos DS, Mohar I, White CC, Forman HJ, Kavanagh TJ. Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol Aspects Med. 2009;30:86–98. doi: 10.1016/j.mam.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ochi T. Hydrogen peroxide increases the activity of gamma-glutamylcysteine synthetase in cultured Chinese hamster V79 cells. Arch Toxicol. 1995;70:96–103. doi: 10.1007/BF02733669. [DOI] [PubMed] [Google Scholar]
- 39.Ochi T. Menadione causes increases in the level of glutathione and in the activity of gamma-glutamylcysteine synthetase in cultured Chinese hamster V79 cells. Toxicology. 1996;112:45–55. doi: 10.1016/0300-483x(96)03348-3. [DOI] [PubMed] [Google Scholar]
- 40.Toroser D, Yarian CS, Orr WC, Sohal RS. Mechanisms of gamma-glutamylcysteine ligase regulation. Biochim Biophys Acta. 2006;1760:233–244. doi: 10.1016/j.bbagen.2005.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tu Z, Anders MW. Identification of an important cysteine residue in human glutamate- cysteine ligase catalytic subunit by site-directed mutagenesis. Biochem J. 1998;336:675–680. doi: 10.1042/bj3360675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang LS. The functional involvement of Lys-38 in the heavy subunit of rat kidney gamma-glutamylcysteine synthetase: chemical modification and mutagenesis studies. J Protein Chem. 1996;15:321–326. doi: 10.1007/BF01887121. [DOI] [PubMed] [Google Scholar]
- 43.Seelig GF, Simondsen RP, Meister A. Reversible dissociation of g-glutamylcysteine synthetase into two subunits. J Biol Chem. 1984;259:9345–9347. [PubMed] [Google Scholar]
- 44.Krejsa CM, Franklin CC, White CC, Ledbetter JA, Schieven GL, Kavanagh TJ. Rapid activation of glutamate cysteine ligase following oxidative stress. J Biol Chem. 2010;285:16116–16124. doi: 10.1074/jbc.M110.116210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang CS, Moore WR, Meister A. On the active site thiol of gamma-glutamylcysteine synthetase: relationships to catalysis, inhibition, and regulation. Proc Natl Acad Sci U S A. 1988;85:2464–2468. doi: 10.1073/pnas.85.8.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun W-M, Huang Z-Z, Lu SC. Regulation of γ-glutamylcysteine synthetase by protein phosphorylation. Biochem J. 1996;320:321–328. doi: 10.1042/bj3200321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Franklin CC, Krejsa CM, Pierce RH, White CC, Fausto N, Kavanagh TJ. Caspase-3-Dependent Cleavage of the Glutamate-L-Cysteine Ligase Catalytic Subunit during Apoptotic Cell Death. Am J Pathol. 2002;160:1887–1894. doi: 10.1016/S0002-9440(10)61135-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hartley DP, Kroll DJ, Petersen DR. Prooxidant-initiated lipid peroxidation in isolated rat hepatocytes: detection of 4-hydroxynonenal- and malondialdehyde-protein adducts. Chem Res Toxicol. 1997;10:895–905. doi: 10.1021/tx960181b. [DOI] [PubMed] [Google Scholar]
- 49.Backos DS, Brocker CN, Franklin CC. Manipulation of cellular GSH biosynthetic capacity via TAT-mediated protein transduction of wild-type or a dominant-negative mutant of glutamate cysteine ligase alters cell sensitivity to oxidant-induced cytotoxicity. Toxicol Appl Pharmacol. 2010;243:35–45. doi: 10.1016/j.taap.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.White CC, Viernes H, Krejsa CM, Botta D, Kavanagh TJ. Fluorescence-based microtiter plate assay for glutamate-cysteine ligase activity. Anal Biochem. 2003;318:175–180. doi: 10.1016/s0003-2697(03)00143-x. [DOI] [PubMed] [Google Scholar]
- 51.Pieper U, Eswar N, Braberg H, Madhusudhan MS, Davis FP, Stuart AC, Mirkovic N, Rossi A, Marti-Renom MA, Fiser A, Webb B, Greenblatt D, Huang CC, Ferrin TE, Sali A. MODBASE, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res. 2004;32:D217–222. doi: 10.1093/nar/gkh095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pedretti A, Villa L, Vistoli G. VEGA--an open platform to develop chemo-bio-informatics applications, using plug-in architecture and script programming. J Comput Aided Mol Des. 2004;18:167–173. doi: 10.1023/b:jcam.0000035186.90683.f2. [DOI] [PubMed] [Google Scholar]
- 53.Li H, Robertson AD, Jensen JH. Very fast empirical prediction and rationalization of protein pKa values. Proteins. 2005;61:704–721. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 54.Ausiello G, Cesareni G, Helmer-Citterich M. ESCHER: a new docking procedure applied to the reconstruction of protein tertiary structure. Proteins. 1997;28:556–567. [PubMed] [Google Scholar]
- 55.Ray S, Misso NL, Lenzo JC, Robinson C, Thompson PJ. Gamma-glutamylcysteine synthetase activity in human lung epithelial (A549) cells: factors influencing its measurement. Free Radic Biol Med. 1999;27:1346–1356. doi: 10.1016/s0891-5849(99)00182-3. [DOI] [PubMed] [Google Scholar]
- 56.Poli G, Schaur RJ, Siems WG, Leonarduzzi G. 4-hydroxynonenal: a membrane lipid oxidation product of medicinal interest. Med Res Rev. 2008;28:569–631. doi: 10.1002/med.20117. [DOI] [PubMed] [Google Scholar]
- 57.Nadkarni DV, Sayre LM. Structural definition of early lysine and histidine adduction chemistry of 4-hydroxynonenal. Chem Res Toxicol. 1995;8:284–291. doi: 10.1021/tx00044a014. [DOI] [PubMed] [Google Scholar]
- 58.Biterova EI, Barycki JJ. Mechanistic details of glutathione biosynthesis revealed by crystal structures of Saccharomyces cerevisiae glutamate cysteine ligase. J Biol Chem. 2009;284:32700–32708. doi: 10.1074/jbc.M109.025114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fraser JA, Saunders RD, McLellan LI. Drosophila melanogaster glutamate-cysteine ligase activity is regulated by a modifier subunit with a mechanism of action similar to that of the mammalian form. J Biol Chem. 2002;277:1158–1165. doi: 10.1074/jbc.M106683200. [DOI] [PubMed] [Google Scholar]
- 60.Solis WA, Dalton TP, Dieter MZ, Freshwater S, Harrer JM, He L, Shertzer HG, Nebert DW. Glutamate-cysteine ligase modifier subunit: mouse Gclm gene structure and regulation by agents that cause oxidative stress. Biochem Pharmacol. 2002;63:1739–1754. doi: 10.1016/s0006-2952(02)00897-3. [DOI] [PubMed] [Google Scholar]
- 61.Shi MM, Iwamoto T, Forman HJ. gamma-Glutamylcysteine synthetase and GSH increase in quinone-induced oxidative stress in BPAEC. Am J Physiol. 1994;267:L414–421. doi: 10.1152/ajplung.1994.267.4.L414. [DOI] [PubMed] [Google Scholar]
- 62.Liu RM, Gao L, Choi J, Forman HJ. γ-glutamylcysteine synthetase: mRNA stabilization and independent subunit transcription by 4-hydroxy-2-nonenal. Am J Physiol. 1998;275:L861–869. doi: 10.1152/ajplung.1998.275.5.L861. [DOI] [PubMed] [Google Scholar]
- 63.Carbone DL, Doorn JA, Kiebler Z, Sampey BP, Petersen DR. Inhibition of Hsp72-mediated protein refolding by 4-hydroxy-2-nonenal. Chem Res Toxicol. 2004;17:1459–1467. doi: 10.1021/tx049838g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang CS, Chang LS, Anderson ME, Meister A. Catalytic and regulatory properties of the heavy subunit of rat kidney gamma-glutamylcysteine synthetase. J Biol Chem. 1993;268:19675–19680. [PubMed] [Google Scholar]
- 65.Lee JI, Kang J, Stipanuk MH. Differential regulation of glutamate-cysteine ligase subunit expression and increased holoenzyme formation in response to cysteine deprivation. Biochem J. 2006;393:181–190. doi: 10.1042/BJ20051111. [DOI] [PMC free article] [PubMed] [Google Scholar]