

Synopsis
T cell receptor (TCR) recognition of antigenic peptides bound and presented by major histocompatibility complex (MHC) molecules forms the basis of the cellular immune response to pathogens and cancer. TCRs bind peptide/MHC molecules weakly and with fast kinetics, features which have hindered detailed biophysical studies of these interactions. Modified peptides resulting in enhanced TCR binding could help overcome these challenges. Further, there is considerable interest in using modified peptides with enhanced TCR binding as the basis for clinical vaccines. Here, we studied how fluorine substitutions in an antigenic peptide can selectively impact TCR recognition. Using a structure-guided design approach, we found that fluorination of the HTLV-1 Tax11-19 peptide (Tax) enhanced binding by the Tax-specific TCR A6, yet weakened binding by the Tax-specific TCR B7. The changes in affinity were consistent with crystallographic structures and fluorine chemistry, and with A6, independent of other substitutions in the interface. Peptide fluorination thus provides a means to selectively modulate TCR binding affinity without significantly perturbing peptide composition or structure. Lastly, in probing the mechanism of fluorine’s effect on TCR binding, our data were most consistent with fluorine’s unique “polar hydrophobicity,” a finding which should impact other attempts to alter molecular recognition with fluorine.
Keywords: T cell receptor, peptide-MHC, fluorine, vaccine design, structure, thermodynamics
Introduction
T cell receptor (TCR) recognition of peptides bound and presented by class I or class II major histocompatibility complex (MHC) molecules forms the basis of the cellular immune response to pathogens and cancer. Interactions between TCRs and peptide/MHC complexes (pMHC) also shape the T cell repertoire as it develops and are responsible for maintenance of the T cell repertoire. Due to the central role these interactions play in cellular immunity, there is considerable interest in the biophysical properties of TCR-pMHC interactions. Yet generally speaking, the soluble ectodomains of TCRs bind pMHC with weak-to-moderate affinities and fast kinetics, properties which have complicated in-depth physical studies of TCR-pMHC interactions. To help overcome this, various investigators have engineered high affinity TCR molecules using molecular evolution or computational design techniques [1-3]. However, the engineered molecules all have multiple mutations in one or more of the TCR complementary-determining-region (CDR) loops, which can complicate extrapolation of results from binding studies performed with the evolved molecules to the wild-type molecules.
We have been pursuing a different approach to overcome the limitation of weak TCR binding affinities, aiming to enhance TCR binding by generating minimally modified peptides that incorporate non-standard amino acid side chains. Our rationale is that although peptide modifications still perturb recognition, compared to molecular evolution, more carefully controlled variations can be introduced, the consequences of which are more easily accounted for.
Peptides that specifically enhance TCR binding may also be of interest in the design of clinical vaccines based on cellular immunity, as enhancing TCR affinity via peptide modifications has been discussed as a means to help break immunological tolerance or otherwise overcome the poor antigenicity of various tumor or viral antigens [4, 5]. In these cases, ideal modifications will influence TCR affinity in a selective manner so that affinity is enhanced only against a specific receptor (or class of receptors), thus avoiding unwanted and potentially dangerous cross-reactivity. Modifications should have little or no effect on peptide conformation, and ideally, concepts of both positive and negative design principles could be employed, such that affinity is raised with one set of receptors but weakened with others.
Here, we explored the use of fluorinated peptide variants as a means to alter TCR recognition in a minimally perturbing yet highly selective manner. Fluorine substitutions are often used in medicinal chemistry to enhance ligand binding affinities [6], and have recently been used to probe immune recognition [7]. We reasoned that by combining selective peptide fluorination with insight from crystallographic structures, we could target structural properties in interfaces between TCRs and their pMHC ligands, simultaneously carrying out both positive and negative design. We modified the HTLV-1 Tax11-19 peptide (LLFGYPVYV; referred to as Tax), which when presented by the class I MHC HLA-A*0201 (HLA-A2) is recognized by the A6 and B7 TCRs as a strong agonist [8, 9]. Despite differences in CDR loop composition and surface chemistry, A6 and B7 bind Tax/HLA-A2 with nearly identical affinities, kinetics, and structural topologies [10]. By fluorinating the central tyrosine (Tyr5) of the Tax peptide, we were able to take advantage of the differences between A6 and B7 and generate peptides that enhance affinity with A6, but weaken affinity with B7. The enhancements with A6 were achieved exclusively through decreases in the TCR dissociation rate and were independent of other substitutions in the interface. Overall, the data demonstrate how subtle variations in peptide composition, and fluorine substitutions in particular, can be used to selectively modulate TCR binding affinity.
Lastly, determination of crystallographic structures of TCR-peptide/HLA-A2 ternary complexes and binding thermodynamics allowed us to probe how fluorination modulates TCR binding affinity. As recently reviewed [6], due to fluorine’s unusual chemistry and a lack of detailed studies with fluorinated compounds, fluorine’s effects on the affinity of biomolecular interactions is poorly understood. Our structural and thermodynamic results indicated a unique “polar hydrophobicity” mechanism [11], as neither electrostatics nor hydrophobicity alone could explain the results.
Experimental
Proteins and peptides
Soluble versions of the A6 and A6c134 TCRs and the HLA-A2 ectodomains were refolded from bacterially expressed inclusion bodies and purified using ion-exchange and size-exclusion chromatography as previously described [10]. Fmoc protected fluorinated L-phenylalanine derivatives for incorporation at position 5 of the Tax peptide were purchased from Anaspec and used without modification. Peptides were generated in-house via solid state synthesis using an ABI 433A instrument. Peptide identity and purity were confirmed via LC/MS. Peptide nomenclature in Figure 2 is based on standard ring numbering. The two peptides that resulted in highest affinity with the A6 TCR contained 4-fluoro-phenylalanine at position 5 and 3,4-di-fluoro-phenylalanine at position 5. As diagrammed in Figure 2, these peptides are referred to as Tax-Y5F4F and Tax-Y5F34FF, respectively.
Figure 2.
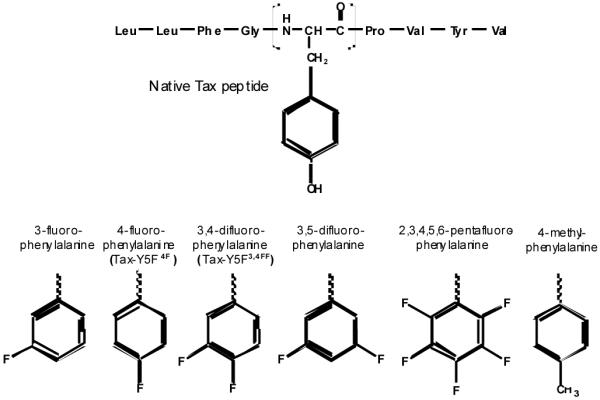
Peptide variants studied. The native Tax peptide with tyrosine at position 5 is shown in the top panel. The bottom panel shows the phenylalanine variants incorporated into at position 5.
Surface plasmon resonance
Surface plasmon resonance experiments were performed using a Biacore 3000 instrument as previously described [10]. Briefly, the TCR was coupled to a standard CM5 sensor surface using amine coupling. All injections were double-referenced using a blank flow cell and a buffer injection. Conditions were 10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.005% surfactant P-20, pH 7.4, 25 °C. Data were processed, and for dissociation rates, fit using Biaevaluation 4.1 (Biacore, Inc). Steady-state experiments were fit using custom routines in Origin 7.5 (OriginLab, Inc). For all titrations with affinities > 10 μM, the activity of the sensor surface was determined independently using the native Tax/HLA-A2 or Tax-Y5F34FF/HLA-A2 ligand. This value was then fixed in the analysis of the weaker-binding ligands, increasing the accuracy of the fitted affinity. Kinetic data were collected at the maximum flow rate of 100 μL/min. Typical surface capacities were ~500 RU. Kinetic data were fit globally to a single exponential decay function to obtain the dissociation rate koff. The association rate kon was determined from the ratio of koff to KD. Error propagation was performed using standard error propagation methods [12].
Isothermal titration calorimetry
Titration calorimetry experiments were performed using a Microcal VP-ITC as previously described [13], with the TCR in the cell and the pMHC in the syringe. Buffer conditions were 20 mM HEPES, 150 mM NaCl, pH 7.4 (room temperature pH was adjusted so that the desired value of 7.4 was maintained across the entire temperature range). Protein concentrations were 20 μM TCR in the cell and 150 μM pMHC in the syringe. Data were processed using the Microcal routines in Origin and analyzed using in-house custom fitting routines [13]. Data analysis incorporated the use of a baseline offset, obviating the need to perform separate dilution injections.
X-ray crystallography
X-ray crystallography was performed as previously described [14]. Crystals of the A6-Tax-Y5F4F/HLA-A2 and A6-Y5F34FF/HLA-A2 complexes were grown from 15% PEG4000, 0.2M MgCl2 buffered with 0.1 M Tris at pH 8.5. Cryo-protection consisted of mother liquor supplemented with 25% glycerol. Diffraction data were collected at Argonne National Laboratories at the indicated beamlines. Data reduction, structure solution, refinement, and structure validation were performed as previously described [14]. The search model for molecular replacement with the ternary complexes was PDB entry 2GJ6 [14], with the coordinates for the peptides and CDR loops removed. Electrostatic potentials for Fig. 1 were calculated with DelPhi as incorporated into Accelrys Discovery Studio using default parameters. Solvent accessible surfaces were drawn with a 1.4 Å radius probe.
Figure 1.
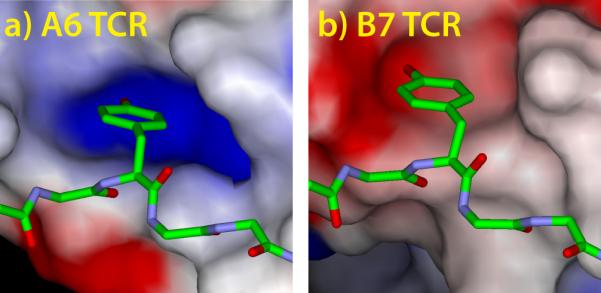
The pocket that accommodates Tyr5 of the Tax peptide is oppositely charged in the A6 TCR (A) and the B7 TCR (B). Electrostatic surface potentials were calculated as described in the Experimental section and are colored red to blue, from −10 kT to +10 kT.
Results and Discussion
Fluorinated Tax peptide variants result in enhanced TCR affinity with the A6 but not B7 T cell receptor
A prominent feature of the A6 and B7 TCRs is a pocket formed by the hypervariable CDR3 loops of the α and β chains. In both the A6 and B7 ternary complexes with Tax/HLA-A2, this pocket accommodates the central tyrosine (Tyr5) of the peptide [9, 15]. With A6, the pocket is capped by Arg95 of CDR3β, which introduces positive charge density and acts as a hydrogen bond donor to the Tyr5 hydroxyl (Fig. 1A). With B7, the pocket is capped by Asp30 of CDR1α, which introduces negative charge density and acts as a hydrogen bond acceptor from the Tyr5 hydroxyl (Fig. 1B). In designing peptide variants, we reasoned that due to fluorine’s strong electronegativity, addition of fluorine atoms to the aromatic ring of position 5 would enhance binding by A6 yet weaken binding by B7.
The fluorinated peptide variants examined were all commercially available derivatives of phenylalanine, and included 3-fluoro-phenylalanine, 4-fluoro-phenylalanine, 3,4-difluoro-phenylalanine, 3,5-difluorophenylalanine, and 2,3,4,5-penta-fluoro-phenylalanine (Fig. 2). Binding affinities with the A6 and B7 TCRs were measured at 25 °C in a steady-state surface plasmon resonance assay.
Binding data for the various fluorinated peptide variants are shown in Fig. 3 and tabulated in Table 1. We found that placing fluorine at either position 3 or 4 of the phenylalanine ring resulted in enhanced affinity with the A6 TCR. Fluorine at position 4 resulted in an approximately 3-fold stronger TCR binding affinity (ΔΔG° = −0.7 kcal/mol), whereas fluorine at position 3 resulted in an approximately 2-fold stronger TCR binding affinity (ΔΔG° = −0.3 kcal/mol). The effects of substitutions at positions 3 and 4 were additive, such that substituting 3,4-difluoro-phenylalanine for tyrosine at position 5 in the Tax peptide resulted in an approximate 5-fold enhancement in A6 binding affinity, from 2.1 μM to 0.46 μM (ΔΔG° = −0.9 kcal/mol). None of the fluorinated peptides resulted in an affinity enhancement with the B7 TCR, instead weakening binding by as much as 14-fold. No binding with either receptor was seen with the 2,3,4,5-penta-fluoro-phenylalanine variant using peptide/HLA-A2 concentrations as high as 85 μM. Thus, in accordance with the crystallographic structures and basic fluorine chemistry, fluorination of position 5 of the Tax peptide resulted in selective modulation of TCR binding affinity.
Figure 3.

Equilibrium binding data for A6 (A) and B7 (B) TCR recognition of the position 5 variant Tax peptides presented by HLA-A2. Data were collected at 25 °C and processed as described in the Experimental section. For all titrations with affinities > 10 μM, the activity of the sensor surface was determined independently using the native Tax/HLA-A2 or Tax-Y5F34FF/HLA-A2 ligand. Measured affinities are reported in Table 1. (C, D) Fitted curves from the panels in A and B plotted as a logarithmic function of pMHC concentration, illustrating the change in affinity with different peptide variants.
Table 1.
A6 and B7 TCR binding affinities for modified Tax peptides presented by HLA-A2
| Position 5 amino acid | A6 TCR | B7 TCR | ||
|---|---|---|---|---|
| K D | ΔΔG° a | K D | ΔΔG° a | |
| Tyrosine (native Tax peptide) | 2.1 ± 0.4 | 1.2 ± 0.2 | ||
| 2,3,4,5,6-penta-fluoro-phenylalanine | n.d. b | n.d. b | ||
| 3,5-di-fluoro-phenylalanine | 2.8 ± 0.3 | +0.2 ± 0.1 | 28 ± 2 | +1.9 ± 0.1 |
| 3-fluoro-phenylalanine | 1.2 ± 0.2 | −0.3 ± 0.1 | 3.1 ± 0.9 | +0.6 ± 0.2 |
| 4-fluoro-phenylalanine (Y5F4F) | 0.64 ± 0.02 | −0.7 ± 0.1 | 16 ± 1 | +1.5 ± 0.1 |
| 3,4-di-fluoro-phenylalanine (Y5F34FF) | 0.46 ± 0.02 | −0.9 ± 0.1 | 17 ± 2 | +1.6 ± 0.1 |
| 4-methyl-phenylalanine | 10 ± 2 | +0.9 ± 0.2 | > 80 | |
Difference in binding free energy between derivatized peptides and the native Tax peptide.
No binding detected using ligand concentrations as high as 85 μM.
Higher affinity with the A6 TCR is achieved exclusively by decreases in TCR dissociation rate and cannot be attributed solely to hydrophobicity
From the initial set of peptides examined, we selected the 4-fluoro-phenylalanine and 3,4-difluoro-phenylalanine Tax variants for further study, as these resulted in the strongest affinity gain with the A6 TCR. As shown in Figure 2, these peptides are further referred to as Tax-Y5F4F for the 4-fluoro-phenylalanine variant and Tax-Y5F34FF for the 3,4-difluoro-phenylalanine variant.
Figure 4 shows the results of kinetic surface plasmon resonance experiments for A6 binding HLA-A2 presenting Tax-Y5F4F and Tax-Y5F34FF; data are tabulated in Table 2. Both peptide variants resulted in a decreased dissociation rate (or increased TCR-pMHC half-life) for the A6 TCR. Indeed, for both peptides, the enhancement in receptor binding affinity was entirely due to a decrease in the receptor off-rate: the incorporation of 4-fluoro-phenylalanine into the Tax peptide (affinity enhancement of three-fold) decreased the dissociation rate three-fold (from 0.11 s−1 to 0.039 s−1), whereas incorporation of 3,4-difluoro-phenylalanine (affinity enhancement of five-fold) decreased the dissociation rate five-fold (from 0.11 s−1 to 0.024 s−1). In terms of the half-life of the TCR-pMHC complexes, the Tax-Y5F4F and Tax-Y5F34FF peptides increase the value from 6.3 seconds to 18 and 30 seconds, respectively.
Figure 4.

Kinetic data for A6 TCR recognition of the Tax-Y5F4F (panel A) and Tax-Y5F34FF (panel B) peptides presented by HLA-A2. After correction for bulk refractive index shifts, dissociation phases were fit globally to a single exponential decay function to yield koff (gray lines in panels A and B). Panel C shows normalized kinetic data for A6 recognition of the native Tax, Tax-Y5F4F, and Tax-Y5F34FF ligands, highlighting the incremental decreases in dissociation rate with fluorination of position 5. Measured kinetics are reported in Table 2.
Table 2.
A6 TCR binding kinetics with the native, Tax-Y5F4F and Tax-Y5F34FF peptides
| Peptide | kon (M−1 s−1)a | koff(s−1)b | t1/2 (s) |
|---|---|---|---|
| Native Tax | 5.2 (± 0.1) × 104 | 0.11 ± 0.01 | 6.3 ± 0.2 |
| Tax-Y5F4F | 6.0 (± 0.2) × 104 | 3.85 (± 0.01) × 10−2 | 18.0 ± 0.1 |
| Tax-Y5F34FF | 5.1 (± 0.2) × 104 | 2.40 (± 0.02) × 10−2 | 28.9 ± 0.2 |
kon calculated from koff/ KD.
Averages and standard deviations of six independent measurements.
In addition to possessing high electronegativity, fluorine is also hydrophobic. To explore the role of hydrophobicity in the enhanced binding affinity with the A6 TCR, we examined the effect of substituting the position 5 side chain of the Tax peptide with 4-methyl-phenylalanine (i.e., the tyrosine hydroxyl replaced with a methyl group). As seen in Table 1, the affinity of the A6 TCR for the 4-methyl-phenylalanine peptide was weakened compared to either the native peptide or 4-fluoro-phenylalanine, indicating that simply increasing hydrophobicity cannot account for the results seen with the fluorinated peptides, and that the affinity modulation achieved with the Tax-Y5F4F and Tax-Y5F34FF peptides involves fluorine’s unique chemistry.
The A6 TCR recognizes fluorinated Tax variants with more favorable entropy changes and moderate heat capacity changes
We next examined the thermodynamic basis for the A6 TCR’s enhanced affinity towards the Tax-Y5F4F and Tax-Y5F34FF peptides using isothermal titration calorimetry. For both peptides, titrations of the A6 TCR with peptide/HLA-A2 at 25 °C yielded a very weak signal, indicative of weak binding enthalpies below the calorimeter’s sensitivity. As titrations of the A6 TCR with native Tax/HLA-A2 at the same conditions and with the same concentrations resulted in a binding enthalpy of −3.4 kcal/mol [13], the weak signal with the fluorinated peptides indicates that the A6 TCR binds the fluorinated Tax variants with a less favorable binding enthalpy, and that the affinity enhancement due to fluorination is entropic in nature.
Although the weak signal at 25 °C could potentially have been overcome by performing the titrations with higher protein concentrations, because of the already large sample requirements for calorimetry, to obtain better thermodynamic data we instead repeated the titrations at 4 °C. Our reasoning was that the binding heat capacity change would amplify the signal through the fundamental relationship between enthalpy, heat capacity, and temperature (ΔH°T2 = ΔH°T1 + ΔCp[T2 − T1]). Under these conditions, we obtained high quality data that fit well to single-site binding isotherms (Fig. 5A-B). Again taking advantage of the heat capacity change, we observed similarly high quality data performing the titrations at 37 °C (Fig. 5C-D). At both temperatures, the A6 TCR recognized the fluorinated peptides with a less favorable ΔH° compared to recognition of the native peptide [13], supporting our conclusions drawn from the 25 °C titrations. Entropy changes at both temperatures were more favorable than with the native peptide, and the trends in ΔH° and ΔS° were amplified with increasing extent of fluorination (Table 3). The enhanced affinity of the A6 TCR resulting from fluorination at position 5 of the Tax peptide thus results from favorable gains in entropy, offset by unfavorable losses in enthalpy.
Figure 5.
Isothermal calorimetric titrations of the A6 TCR with the Tax-Y5F4F and Tax-Y5F34FF ligands at 4 °C and 37 °C. Thermodynamic parameters are reported in Table 3. At 4 °C, recognition of both peptides is enthalpically opposed and entropically favored, with increasing fluorination resulting in a greater enthalpic penalty. At 37 °C, binding is both enthalpically and entropically driven, yet increasing fluorination again weakens the binding enthalpy change.
Table 3.
Calorimetrically determined binding thermodynamics for A6 recognition of Tax-peptide variants.a
| Peptide | Δ H ° | Δ S ° | Δ G ° | KD | Δ H ° | Δ S ° | Δ G ° | KD |
|---|---|---|---|---|---|---|---|---|
| 4 °C | 37 °C | |||||||
| Native Tax b | 4.4 ± 0.2 | 43 ± 2 | −7.7 ± 0.2 | 0.88 ± 0.09 | −7.1 ± 0.1 | 4.7 ± 0.4 | −8.59 ± 0.04 | 0.9 ± 0.1 |
| Tax-Y5F4F | 6.0 ± 0.1 | 51 ± 1 | −8.2 ± 0.1 | 0.37 ± 0.05 | −6.4 ± 0.2 | 10 ± 1 | −9.5 ± 0.1 | 0.20 ± 0.03 |
| Tax-Y5F34FF | 7.5 ± 0.3 | 58 ± 1 | −8.7 ± 0.1 | 0.14 ± 0.03 | −5.4 ± 0.1 | 14 ± 1 | −9.7 ± 0.1 | 0.16 ± 0.02 |
Units of kcal/mol for ΔH° and ΔG° and cal/mol/K for ΔS°. KD values are in μM.
Data for native Tax from Armstrong et al. (2008); 4 °C data calculated from propagation of globally fitted parameters [13].
The availability of enthalpy measurements at 4 °C and 37 °C, together with the very weak binding enthalpies at 25 °C, allowed us to estimate binding heat capacity changes near −0.4 kcal/mol/K for A6 recognition of both Tax-Y5F4F/HLA-A2 and Tax-Y5F34FF/HLA-A2. This value is identical within error to the value of −0.39 kcal/mol/K observed for A6 recognition of the native peptide under the same conditions [13].
Structures of the A6 TCR bound to the Tax-Y5F4F and Tax-Y5F34FF ligands
To examine the structural consequences of recognition of the fluorinated peptides, we next crystallized and determined the structures of the A6 TCR bound to the Tax-Y5F4F/HLA-A2 and Tax-Y5F34FF/HLA-A2 complexes. The complexes crystallized in the same space group and with similar unit cell dimensions as other A6–peptide/HLA-A2 complexes [14-16]. The two structures were solved using molecular replacement, with the structure of A6 bound to the Tax-Y5K-IBA/HLA-A2 ligand as a search model [14], with the coordinates for the CDR loops and the peptide excluded. Crystallization and refinement statistics are presented in Table 4. Structural overviews and electron density images are provided as supporting information.
Table 4.
X-ray Data and Refinement Statistics
| Complex | A6–Tax-Y5F4F/HLA-A2 | A6–Tax-Y5F34FF/HLA-A2 |
| PDB ID | 3D39 | 3D3V |
| Source | APS 23ID | APS 19BM |
| Space group | C2 | C2 |
| Unit Cell | ||
| a (Å) | 224.3 | 224.5 |
| b (Å) | 48.3 | 48.5 |
| c (Å) | 93.2 | 93.8 |
| β (°) | 90.5 | 90.6 |
| Molecules/a.u. | 1 | 1 |
| Resolution (Å) | 20 – 2.8 | 20 – 2.8 |
| Total number of reflections | 22591 | 24935 |
| Mosaicity (°) | 0.54 | 0.46 |
| Completeness (%) | 92.8 (96) a | 99.6 (96.9) |
| I/σ | 18.6 (2.8) | 26.9 (2.2) |
| Rmerge (%) | 10.1 (28.1) | 6.5 (41) |
| Average redundancy | 3.4 (2.8) | 3.6 (3.2) |
| Rwork (%, no. reflections) | 20.1 (21425) | 22.0 (23658) |
| Rfree, (%, no. reflections) | 26.9 (1166) | 27.7 (1277) |
| Average B factor (Å2) | 66.4 | 63.7 |
| Ramachandran plot | ||
| Most favored (%) | 88.8 | 84.1 |
| Allowed (%) | 10.8 | 14.5 |
| Generously allowed (%) | 0.3 | 1.2 |
| RMS deviations from ideality | ||
| Bonds (Å) | 0.012 | 0.015 |
| Angles (°) | 1.587 | 1.749 |
| Coordinate errorb (Å) | 0.35 | 0.42 |
Numbers in parenthesis refer to the highest resolution shell
Mean estimate based on maximum likelihood methods
Topologically, the two structures were identical to the structure with the native Tax peptide. The Tax-Y5F4F and Tax-Y5F34FF structures superimpose onto the native structure with RMSD values of 0.65 and 0.68 Å, respectively, and the two structures themselves superimpose with RMSD values of 0.40 Å (superimpositions are for the backbone atoms of the two variable domains, the peptide, and the peptide binding domain). Excluding position 5, all atoms of the Tax-Y5F4F and Tax-Y5F34FF peptides superimpose on the native Tax peptide with RMSD values of 0.32 and 0.35 Å; the value for the two modified peptides is 0.22 Å.
In both structures, however, there are small changes in the region accommodating the position 5 side chain (Fig. 6). In the structure with the Tax-Y5F4F peptide, these changes are limited to small movements necessary to accommodate the new chemical environment in the position 5 pocket (Fig. 6A). These motions include a 106° rotation of the χ1 angle of Ser31α (in the CDR1α loop), which moves the serine side chain oxygen 1.6 Å further away from the fluorine atom compared to the serine’s position in the native structure, resulting in a fluorine – oxygen distance of 3.8 Å (the distance would be 2.2 Å if the serine torsion had not rotated). Surprisingly, Arg95β of the CDR3β loop is translated 1.1 Å away from its position in the native structure, placing its guanidinium group 4.7 Å away from the fluorine atom – the distance would be 3.2 Å if the arginine position had not changed. There are some small changes in the CDR3β loop, mostly stemming from ϕ/ψ bond rotations in the Gly-Gly-Ala motif of residues 99β-101β. A consequence of these rotations is a 3.1 Å displacement of the Gly100β carbonyl oxygen. The reason for these structural changes is unclear, as there are no steric overlaps that would otherwise necessitate them. Likely they are a consequence of the new chemical environment forcing an unexpected positional change in Arg95β that propagates further down the main chain.
Figure 6.

Interfacial details from the structures of the A6–Tax-Y5F4F/HLA-A2 and A6–Tax-Y5F34FF/HLA-A2 structures. A) Cross-eyed stereo comparison of the interfaces with the Tax-Y5F4F peptide (cyan, yellow) and the native Tax peptide (pink, blue). The shifts in Arg95 of CDR3β and Ser31α are apparent, as is the slight rearrangement of the CDR3β backbone. B) Cross-eyed stereo comparison of the interfaces with the Tax-Y5F34FF peptide (cyan, yellow) and the native Tax peptide (pink, blue). The dual conformations of the modified position 5 side chain are apparent, as is the larger shift in CDR3β. C) Alternate view of the Tax-Y5F34FF interface, showing the relationship between Arg102β and Glu154 of the HLA-A2 α2 helix (green).
In the structure with the doubly fluorinated Tax-Y5F34FF peptide, the position five side chain could be clearly refined in two positions, differing by a 185° rotation around χ2 and a 26° rotation in χ1 (Fig. 6B). In this structure, there are also changes in the position of Ser31α and Arg95β similar to those in the structure with the singly fluorinated peptide (although the rotation of the Ser31α χ1 is smaller at only 67°). However, in the doubly fluorinated structure the ϕ/ψ angle changes in the CDR3β loop are larger than in the singly fluorinated structure, resulting in a 3.7 Å displacement in the Gly101β α carbon. To avoid an overlap with the main chain, the solvent-exposed side chain of Arg101β occupies a different position, 2.8 Å away from the side chain of Glu154 of the HLA-A2 α2 helix (Fig. 6C). As in the singly-fluorinated Tax-Y5F4F structure, there is no apparent reason for this shift; this observation may reflect an intrinsic flexibility of this region of the loop, amplified due to the altered chemistry within the position 5 pocket. Note that it is not unusual for the CDR3β loops of TCRs, and of the A6 TCR in particular, to shift dramatically upon recognition of ligand [14, 17].
With the A6 TCR, the Y5F34FF substitution in the Tax peptide is independent of other alterations in the interface
One use for peptide modifications that enhance TCR binding is to study the consequences of interfacial modifications that weaken binding to levels difficult to measure. Such use requires that the various modifications act independently of each other. To investigate whether the Y5F34FF substitution is independent of other changes in the interface, we combined it with two other substitutions in the Tax peptide, proline 6 → alanine (P6A) and tyrosine 8 → phenylalanine (Y8F). If the Y5F34FF substitution is independent of these changes, the loss in affinity with the P6A and Y8F substitutions should be the same with doubly-fluorinated phenylalanine at position 5 as it is with the native tyrosine at position 5.
Using surface plasmon resonance, we found with the A6 TCR that the P6A substitution in the Tax-Y5F34FF peptide weakened A6 TCR binding affinity from 0.46 to 25 μM, amounting to an increase in binding free energy (ΔΔG°) of 2.4 ± 0.1 kcal/mol. This increase was identical to that seen when the P6A substitution is introduced into the native Tax peptide (a change in KD from 2.1 to 120 μM, or a ΔΔG° of 2.4 ± 0.2 kcal/mol [18]). With the Y8F substitution in the Tax-Y5F34FF peptide, we observed a drop in binding affinity from 0.46 to 10.5 μM, amounting to a ΔΔG° of 1.8 ± 0.1 kcal/mol. Again, this increase was identical to that seen when Y8F is introduced into the native Tax peptide (a change in KD from 2.1 to 42 μM, or a ΔΔG° of 1.7 ± 0.2 kcal/mol). Thus, the Y8F substitution is independent of the P6A and Y8F substitutions (binding data for these additional substituted peptides are in the supporting information).
We also examined the effect of the Y5F34FF substitution on the binding of the high affinity A6 variant A6c134, which substitutes the sequence MSAQ for GSSR in the CDR3β loop [2]. With isothermal titration calorimetry, we measured an affinity of 8 nM for A6c134 recognition of native Tax, and 3 nM for recognition of Tax-Y5F34FF (Fig. 7). The effect of the Y5F34FF substitution on the binding of the A6c134 variant thus amounted to a ΔΔG° of −0.7 ± 0.3 kcal/mol, close to the value of −0.9 ± 0.1 kcal/mol measured for wild-type A6. As with the wild-type receptor, the affinity gain due to fluorination was entropically driven and enthalpically opposed. The Y5F34FF substitution is thus independent of the four mutations that define the A6c134 variant.
Figure 7.
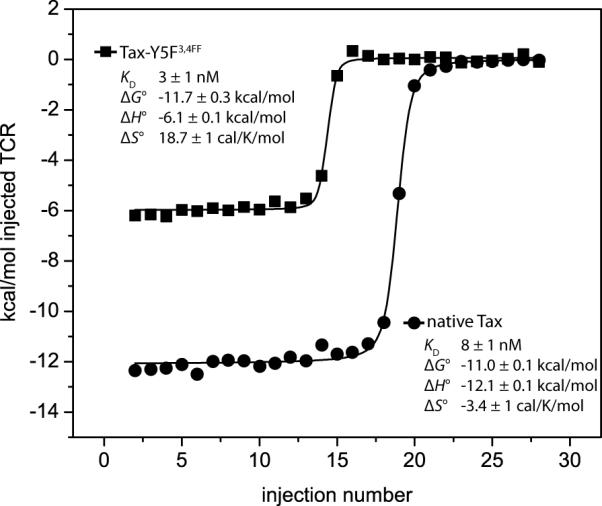
Isothermal calorimetric titration of the high affinity A6 TCR variant A6c134 with the native Tax and Tax-Y5F34FF ligands. The affinity enhancement (ΔΔG°) due to fluorination is similar to that seen with the wild-type receptor, and as with the wild-type receptor, affinity enhancement is entropically driven and enthalpically opposed.
The mechanism of affinity modulation with fluorinated Tax peptides
Although fluorine is commonly used in medicinal chemistry to enhance ligand binding, the mechanisms by which fluorine influences binding affinity are poorly understood. Fluorine is hydrophobic and highly electronegative, yet it only rarely participates as a hydrogen bond acceptor due to its low polarizability [19-21]. Instead, fluorine interacts with full or partial positive charges via dipolar interactions, although the strength of these are subject to some debate [11]. A recent survey of complexes between proteins and fluorinated ligands in the Protein Data Bank highlighted the propensity for fluorine to interact with the guanidinium group of arginine [6]. Indeed, such an interaction was part of our design scheme, as in the A6 TCR, the pocket which accommodates tyrosine 5 of the peptide is capped by Arg95 of CDR3β. The enhanced binding seen with the A6 TCR, and the corresponding reduced binding with the B7 TCR (whose pocket is capped by a negatively charged aspartic acid) support the assignment of the enhanced affinity to electrostatic considerations.
Yet are electrostatics the only reason for the affinity changes with the A6 and B7 TCRs? The data argue against this: in the X-ray structures of A6 with the singly fluorinated Tax-Y5F4F and doubly fluorinated Tax-Y5F34FF peptides, Arg95 of the A6 CDR3β loop has moved away from the fluorine atoms. While it could be that this movement is necessary to optimize the distance for a favorable electrostatic interaction, the binding thermodynamics are likewise inconsistent with a purely electrostatic mechanism. For both the Tax-Y5F4F and Tax-Y5F34FF peptides, the affinity gain is entropically driven and enthalpically opposed: a purely electrostatic mechanism would be expected to be enthalpically favored. Although the binding thermodynamics must also include the energetics associated with the conformational changes seen in each structure, and these could mask the energetics associated with burial of the fluorine atoms, the enthalpic penalties and entropic gains due to fluorination of the peptide are large and additive with the degree of fluorination at both 4 °C and 37 °C. This suggests that the hydrophobic nature of fluorine is also a significant contributor to the affinity gain with both peptides.
Although hydrophobicity thus seems involved, a classical hydrophobic mechanism as seen with hydrocarbons would necessitate an increase in the magnitude of the binding heat capacity change due to the effects of hydrocarbons on the hydrogen bonding pattern in bulk water [22]. However, as noted above this is not observed: the binding ΔCp for A6 recognition of both the singly and doubly fluorinated peptides is identical within error to that observed with the native peptide. Importantly though, the hydrophobicity of fluorocarbons and hydrocarbons arise from different physical mechanisms, as evidenced by the limited miscibility of the two classes of compounds. Indeed, classical hydrophobicity cannot account for the affinity changes, as replacing Tyr5 with 4-methyl-phenylalanine resulted in weaker binding of the A6 TCR.
We thus conclude that the A6 and B7 affinity modulation with the fluorinated peptides is due largely to the unique “polar hydrophobicity” of fluorine [11]. Accordingly, with the A6 TCR, burial of the fluorinated side chain in the Tyr5 pocket results in a favorable electrostatic interaction with Arg95β, offsetting the arginine’s desolvation penalty. Further gains in affinity are provided by the hydrophobic nature of fluorine, and compared to the native tyrosine, the lack of a desolvation penalty for the ring hydroxyl. The small structural differences between the complexes with native Tax and the fluorinated variants must also play a role, although given the measured binding thermodynamics and the additive effects of fluorination, we judge these effects to be secondary to the direct effects from the fluorine atoms. With the B7 TCR, hydrophobicity and the lack of a desolvation from the ring hydroxyl are unable to offset the electrostatic repulsion between Asp30α and the electron-rich fluorine atoms, resulting in a loss in TCR binding affinity.
Concluding remarks
In conclusion, we have demonstrated how fluorine substitutions can be used to minimally modify antigenic peptides in order to selectively modulate TCR binding. Such modifications will be useful in the design of peptides that boost TCR affinity, in turn facilitating experiments that are difficult or impossible because of the weak affinities TCRs maintain towards their natural pMHC ligands. They may also be useful in the design of candidates for clinical vaccines based on cellular immunity, where enhanced TCR affinity can be desirable for breaking immunological tolerance or otherwise overcoming the poor antigenicity of native antigens. The ability to incorporate both positive and negative design principles with fluorine substitutions is particularly attractive for the latter case. As demonstrated here, regions in TCR binding sites of positive electrostatic potential can be targeted to enhance affinity, whereas regions with negative electrostatic potential can be targeted to weaken affinity. Lastly, the assignment of the mechanism of affinity enhancement with the A6 TCR to fluorine’s unique “polar hydrophobicity” fills a gap in the experimental data of how fluorine can be used to modulate biomolecular interactions, and should impact other attempts to alter molecular recognition via fluorine substitutions.
Supplementary Material
Acknowledgements
We thank Alison Wojnarowicz and Emily Doan for outstanding technical assistance and Prof. Başar Bilgiçer for helpful comments on the manuscript. The authors declare no conflicts of interest.
Funding
Supported in part by grant R01GM067079 from the National Institute of General Medical Sciences, National Institutes of Health, USA. OYB was supported by a fellowship from the Walther Cancer Research Center. KHP was supported by the Notre Dame Chemistry-Biochemistry-Biology Interface training program, funded by T32GM075762 from the National Institute of General Medical Sciences, National Institutes of Health, USA. Results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center and GM/CA – CAT at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. GM/CA – CAT has been funded in whole or in part with Federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Science (Y1-GM-1104).
Abbreviations
- TCR
T cell receptor
- MHC
major histocompatibility complex
- CDR
complementary determining region
- pMHC
peptide/MHC complex
References
- 1.Holler PD, Holman PO, Shusta EV, O’Herrin S, Wittrup KD, Kranz DM. In vitro evolution of a T cell receptor with high affinity for peptide/MHC. Proc. Natl. Acad. Sci. U. S. A. 2000;97:5387–5392. doi: 10.1073/pnas.080078297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li Y, Moysey R, Molloy PE, Vuidepot A-L, Mahon T, Baston E, Dunn S, Liddy N, Jacob J, Jakobsen BK, Boulter JM. Directed evolution of human T-cell receptors with picomolar affinities by phage display. Nat. Biotech. 2005;23:349–354. doi: 10.1038/nbt1070. [DOI] [PubMed] [Google Scholar]
- 3.Haidar JN, Pierce B, Yu Y, Tong W, Li M, Weng Z. Structure-based design of a T-cell receptor leads to nearly 100-fold improvement in binding affinity for pepMHC. Proteins. 2009;74:948–960. doi: 10.1002/prot.22203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Slansky JE, Rattis FM, Boyd LF, Fahmy T, Jaffee EM, Schneck JP, Margulies DH, Pardoll DM. Enhanced antigen-specific antitumor immunity with altered peptide ligands that stabilize the MHC-peptide-TCR complex. Immunity. 2000;13:529–538. doi: 10.1016/s1074-7613(00)00052-2. [DOI] [PubMed] [Google Scholar]
- 5.McMahan RH, Slansky JE. Mobilizing the low-avidity T cell repertoire to kill tumors. Sem. Can. Biol. 2007;17:317–329. doi: 10.1016/j.semcancer.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muller K, Faeh C, Diederich F. Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 7.Gomez-Nunez M, Haro KJ, Dao T, Chau D, Won A, Escobar-Alvarez S, Zakhaleva V, Korontsvit T, Gin DY, Scheinberg DA. Non-Natural and Photo-Reactive Amino Acids as Biochemical Probes of Immune Function. PLoS ONE. 2008;3:e3938. doi: 10.1371/journal.pone.0003938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Utz U, Banks D, Jacobson S, Biddison WE. Analysis of the T-cell receptor repertoire of human T-cell leukemia virus type 1 (HTLV-1) Tax-specific CD8+ cytotoxic T lymphocytes from patients with HTLV-1-associated disease: evidence for oligoclonal expansion. J. Virol. 1996;70:843–851. doi: 10.1128/jvi.70.2.843-851.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding YH, Smith KJ, Garboczi DN, Utz U, Biddison WE, Wiley DC. Two human T cell receptors bind in a similar diagonal mode to the HLA-A2/Tax peptide complex using different TCR amino acids. Immunity. 1998;8:403–411. doi: 10.1016/s1074-7613(00)80546-4. [DOI] [PubMed] [Google Scholar]
- 10.Davis-Harrison RL, Armstrong KM, Baker BM. Two Different T Cell Receptors use Different Thermodynamic Strategies to Recognize the Same Peptide/MHC Ligand. J. Mol. Biol. 2005;346:533–550. doi: 10.1016/j.jmb.2004.11.063. [DOI] [PubMed] [Google Scholar]
- 11.Biffinger JC, Kim HW, DiMagno SG. The Polar Hydrophobicity of Fluorinated Compounds. ChemBioChem. 2004;5:622–627. doi: 10.1002/cbic.200300910. [DOI] [PubMed] [Google Scholar]
- 12.Bevington PR, Robinson DK. Data reduction and error analysis for the physical sciences. McGraw-Hill; New York: 1992. [Google Scholar]
- 13.Armstrong KM, Baker BM. A Comprehensive Calorimetric Investigation of an Entropically Driven T Cell Receptor-Peptide/Major Histocompatibility Complex Interaction. Biophys. J. 2007;93:597–609. doi: 10.1529/biophysj.107.104570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gagnon SJ, Borbulevych OY, Davis-Harrison RL, Turner RV, Damirjian M, Wojnarowicz A, Biddison WE, Baker BM. T Cell Receptor Recognition via Cooperative Conformational Plasticity. J. Mol. Biol. 2006;363:228–243. doi: 10.1016/j.jmb.2006.08.045. [DOI] [PubMed] [Google Scholar]
- 15.Garboczi DN, Ghosh P, Utz U, Fan QR, Biddison WE, Wiley DC. Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature. 1996;384:134–141. doi: 10.1038/384134a0. [DOI] [PubMed] [Google Scholar]
- 16.Ding YH, Baker BM, Garboczi DN, Biddison WE, Wiley DC. Four A6-TCR/peptide/HLA-A2 structures that generate very different T cell signals are nearly identical. Immunity. 1999;11:45–56. doi: 10.1016/s1074-7613(00)80080-1. [DOI] [PubMed] [Google Scholar]
- 17.Armstrong KM, Piepenbrink KH, Baker BM. Conformational changes and flexibility in T-cell receptor recognition of peptide-MHC complexes. Biochem. J. 2008;415:183–196. doi: 10.1042/BJ20080850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baker BM, Gagnon SJ, Biddison WE, Wiley DC. Conversion of a T Cell Antagonist into an Agonist by Repairing a Defect in the TCR/Peptide/MHC Interface. Implications for TCR Signaling. Immunity. 2000;13:475–484. doi: 10.1016/s1074-7613(00)00047-9. [DOI] [PubMed] [Google Scholar]
- 19.Dunitz JD. Organic Fluorine: Odd Man Out. ChemBioChem. 2004;5:614–621. doi: 10.1002/cbic.200300801. [DOI] [PubMed] [Google Scholar]
- 20.Dunitz JD, Taylor R. Organic Fluorine Hardly Ever Accepts Hydrogen Bonds. Chemistry - A European Journal. 1997;3:89–98. [Google Scholar]
- 21.Howard JAK, Hoy VJ, O’Hagan D, Smith GT. How good is fluorine as a hydrogen bond acceptor? Tetrahedron. 1996;52:12613–12622. [Google Scholar]
- 22.Sharp KA, Madan B. Hydrophobic effect, water structure, and heat capacity changes. J. Phys. Chem. B. 1997;101:4343–4348. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.