

Abstract
Glutathione peroxidase 3 (GPX3) is one of the key enzymes in the cellular defense against oxidative stress and the hepatocyte growth factor receptor, (MET) has been suggested to be influenced by the GPX3 gene expression. In a previous microarray study performed by our group, Gpx3 was identified as a potential biomarker for rat endometrial adenocarcinoma (EAC), since the expression was highly downregulated in rat EAC tumors. Herein, we have investigated the mRNA expression and Gpx3 and Met in rat EAC by real time quantitative PCR (qPCR), and the methylation status of Gpx3. In addition we have examined the expression of GPX3 and MET in 30 human EACs of different FIGO grades and 20 benign endometrial tissues. We found that the expression of GPX3 was uniformly down regulated in both rat and human EAC, regardless of tumor grade or histopathological subtype, implying that the down-regulation is an early event in EAC. The rate of Gpx3 promoter methylation reaches 91%, where biallelic methylation was present in 90% of the methylated tumors. The expression of the Met oncogene was slightly upregulated in EACs that showed loss of expression of Gpx3, but no tumor suppressor activity of Gpx3/GPX3 was detected. Preliminary results also suggest that the production of H2O2 is higher in rat endometrial tumors with down-regulated Gpx3 expression. A likely consequence of loss of GPX3 protein function would be a higher amount of ROS in the cancer cell environment. Thus, the results suggest important clinical implications of the GPX3 expression in EAC, both as a molecular biomarker for EAC and as a potential target for therapeutic interventions.
Background
Endometrial carcinoma (EC) is the most common gynecological malignancy observed in the western society with an incidence rate of approximately 15-20 per 100.000 women per year. Endometrioid adenocarcinoma (EAC) arises from cells that form the glands in the endometrium. It is the most prevalent subtype constituting approximately 80% of all endometrial cancers. Endometroid andenocarcinoma can be histologically graded according to the FIGO system (International Federation of Gynecology and Obstretics) or classified as low grade or high grade by an alternative architectural binary grading system. The disease may either be an estrogen-dependent low-grade endometroid variant (type I) or a non-estrogen-dependent high grade variant (type II). As expected, estrogen-dependent endometroid carcinomas preferentially affect women in the pre- or peri-menopausal phase, whereas type II EACs usually develops in older post-menopausal women. Type II tumors are typically of high-grade endometroid adenocarcinomas, papillary serous or clear cell types, and generally carry a poor prognosis [1-5].
Due to the complexity of cancer etiology caused by the genetic heterogeneity present in the human population and the influences of environmental factors, it can be advantageous to turn to inbred animal models. In the present and previous works, we have used tumor material from crosses including the BDII/Han rat model, where more than 90% of the female virgins of the BDII inbred rat strain spontaneously develop endometrial cancer during their life time. These tumors are hormone dependent ECs and thus represent spontaneous hormonal carcinogenesis [6-8].
In a recent comprehensive microarray study of endometrial cancer cell lines, we found that the expressions of 354 genes were significantly altered relative to normal/pre-malignant endometrium. When applying traditional statistical analyses and gene classification analysis on the microarray data (Waikato environment of knowledge analysis, Weka), we could identify a three-gene signature (Gpx3, Bgn and Tgfb3), that might have important implications in EAC carcinogenesis [9]. It was also revealed that Gpx3 displayed the most significantly altered gene expression in comparisons among endometrial tumors and normal/pre-malignant endometrium.
GPX3 constitutes the backbone of the cellular antioxidant defense system, together with Superoxide Dismutase (SOD) and Catalase (CAT)[10]. GPX3 catalyses the reduction of peroxides and protects cells against oxidative damage. The decreased mRNA expression of Gpx3 in rat endometrial tumors might result in an impaired defense against endogenous and exogenous genotoxic compounds, which could potentially lead to an increased mutation rate including genes involved in carcinogenesis. The gene expression of GPX3 in human has previously been shown to be silenced in prostate cancer, ovarian clear cell adenocarcinoma, gastric carcinoma and in Barret's disease by epigenetic mechanisms, such as hyper-methylation [11-16]. Furthermore, Yu, et al. [17-23], suggested that GPX3 also contains tumor suppressor activity by, directly or in-directly, regulating cell growth and proliferation through unknown mechanisms. GPX3 may influence the expression of MET (mesenchymal-epithelial transition factor) that encodes a tyrosine kinase receptor for hepatocyte growth factor (HGF). Abnormal expression/activation of the MET receptor has been reported in numerous human cancer diseases [17-23].
The aim of this study was to investigate the mechanisms underlying the down-regulation of Gpx3 and potential implications in EAC carcinogenesis. The production of hydrogen peroxide in the endometrial tumors displaying loss of Gpx3 expression as well as in the endometrial samples with normal/high expression was measured. Since GPX3 has been suggested to exhibit tumor suppressor activity, that could regulate the transcription of the oncogene, Met, we estimated the correlation of the expression between Met and Gpx3 by employing a real time RT-PCR expression study. In order to verify the results from the BDII endometrial cancer rat model in human, we also examined the mRNA expression of GPX3 and MET in human endometroid tumor samples of FIGO grade I, II and III and benign samples.
Results
QPCR expression analysis of Gpx3 and Met
The statistical analyses applied for comparing replicates, revealed no significant differences among the replicates in either of the data sets. An average of the replicates Ct value were used in the following calculations of the relative quantitative gene expression, the delta-delta Ct value.
The majority of the rat tumors displayed an almost total loss of expression of Gpx3 whereas the non-/pre-malignant endometrial samples displayed a normal/high expression, as also shown by the highly significant difference among malignant and premalignant/normal samples, (p < 0.001, Figure 1, Table 1). The mRNA expression of GPX3 in the human material assessed in 30 human EACs in FIGO grade I-III (10 tumors from each grade), and 21 benign endometrial samples differed significantly between normal and malignant tissues (P < 0.001), but no differences among the different groups of malignant tumors were seen (P > 0.5) (Table 2).
Figure 1.

Gene expression of of Gpx3/GPX3 and Met/MET in rat and human. (a) Gpx3 expression in rat endometrial adenocarcinoma and pre-malignant endometrial cell lines. (b) GPX3 gene expression in human endometrial adenocarcinomas of benign samples and FIGO grade I-III tumor samples. (c) Gene expression of Met in rat endometrial adenocarcinoma and pre-malignant endometrial cell lines. (d) MET gene expression in human endometrial adenocarcinomas of benign samples and FIGO grade I-III tumor samples. The median in each group is represented by a horizontal line that indicates the mean delta delta Ct value in each group.
Table 1.
Overview of the rat material and the tests performed in the study.
| Sample | Cell type | Q-PCR Gpx3 |
Q-PCR Met |
Methylation | Demethylation | H2O2 | FISH |
|---|---|---|---|---|---|---|---|
| NUT43 | NME | X | X | X | X | ||
| NUT56 | NME | X | X | X | X | ||
| NUT6 | EAC | X | X | X | |||
| NUT12 | EAC | X | X | X | X | X | |
| NUT31 | EAC | X | X | X | |||
| NUT39 | EAC | X | X | X | |||
| NUT46 | EAC | X | X | X | |||
| NUT50 | EAC | X | X | X | |||
| NUT51 | EAC | X | X | X | |||
| NUT52 | EAC | X | X | X | |||
| NUT81 | EAC | X | X | X | X | ||
| NUT84 | EAC | X | X | X | X | ||
| NUT127 | EAC | X | X | X | |||
| RUT5 | EAC | X | |||||
| RUT12 | EAC | X | X | X | |||
| RUT13 | EAC | X | X | X | |||
| RUT30 | EAC | X | X | X |
Table 2.
Overview of the human material and the tests performed in the study
| Sample | Tumor grade | qPCR GPX3 | qPCR MET |
|---|---|---|---|
| Lung | normal | X | X |
| 1-20 | benign | X | X |
| 21-30 | I | X | X |
| 31-40 | II | X | X |
| 41-50 | III | X | X |
In the rat cell lines, the expression of the Met gene was slightly higher among the malignant cell lines (P = 0.054). In the human material the expression of MET was significantly lower among the malign tumors (P < 0.001), but there were no differences between FIGO grades (P > 0.5) (Figure 1).
Epigenetic inactivation of the Gpx3 gene
The methylation status of the Gpx3 promoter region on bisulfite treated DNA was examined with methylation specific primers (MSP) in methylation-specific PCR of the inbred rat strain BDII, one NME cell line and 15 EAC cell lines (Table 3). The NME cell line was not methylated, while in the BDII rat strain and in 14 out of 15 EAC cell lines (93%), the Gpx3 promoter region was hypermethylated of (Table 4).
Table 3.
Methylation specific primer (MSP) pairs.
| Primer pairs | Forward primer | Reverse primer | Fragment size (bp) |
|---|---|---|---|
| Methylated | GTTGTTATTGGTTAGGAAGTTTTCG | GCGTCTTAAAATAACCACCGTC | 114 |
| Unmethylated | TTGTTATTGGTTAGGAAGTTTTTGG | AACACATCTTAAAATAACCACCATC | 116 |
| Actb | GGAAATCGTGCGTGACATTA | AGGAAGGAAGGCTGGAAGAG | 183 |
Methylation specific primer (MSP) pairs for Gpx3 used in the methylation specific PCR. The gene Actb was used as control.
Table 4.
Methylation status of the Gpx3 promotor region in the rat EAC cell lines.
| Methylation status | |||
|---|---|---|---|
| Sample | Cell type | Methylated | Unmethylated |
| BDII | Strain DNA | M+/M+ | |
| NUT43 | NME | M-/M- | |
| NUT56 | NME | -------- | -------- |
| NUT6 | EAC | M+/M+ | |
| NUT12 | EAC | M+/M+ | |
| NUT31 | EAC | M+/M+ | |
| NUT39 | EAC | M+/M- | |
| NUT46 | EAC | M+/M+ | |
| NUT50 | EAC | M+/M+ | |
| NUT51 | EAC | M+/M+ | |
| NUT52 | EAC | M+/M+ | |
| NUT81 | EAC | M+/M+ | |
| NUT84 | EAC | M-/M- | |
| NUT127 | EAC | M+/M+ | |
| RUT5 | EAC | M+/M+ | |
| RUT12 | EAC | M+/M+ | |
| RUT13 | EAC | M+/M+ | |
| RUT30 | EAC | M+/M+ | |
* M+/M+ diallelic methylation, M+/M- monoallelic methylation,
M-/M- unmethylated, ------ no results available
Demethylation of the Gpx3 gene
Two of the tumor cell lines (NUT12 and NUT81) that displayed Gpx3 promoter biallelic hypermethylation were randomly selected for treatment with the demethylating agent 5-aza-2´-deoxy-cy-tidine (5Aza-dC) in combination with the histone deacetylace inhibitor, trichostatin A (TSA) (Figure 2). As is shown, the Gpx3 mRNA expression was fully restored after the demethylation treatment.
Figure 2.
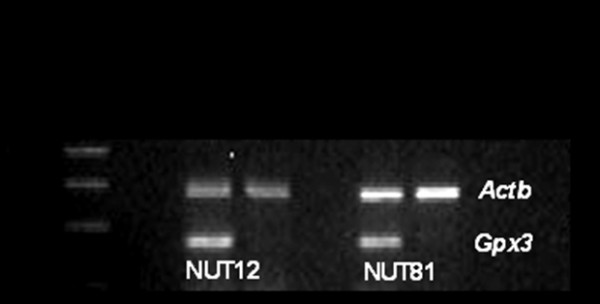
Demethylation of endometrial tumor cell lines (NUT12 and NUT81) induced activation of Gpx3 transcription. The cells were treated with the demethylating agent 5-aza-2´-deoxycytidine (5Aza-dC) in combination with trichostatin A (TSA), a specific inhibitor of histone deacetylase.
Reactive oxidative species (ROS) in cancer cells
The production of hydrogen peroxide in one rat endometrial tumor cell line (NUT12) with loss of expression of Gpx3 and in two non-malignant endometrial cell lines (NUT43 and NUT56) with Gpx3 expression was measured. A higher generation of hydrogen peroxide was produced in the tumor cell line than in the premalignant cell lines (Figure 3).
Figure 3.

Measurements of hydrogen peroxide produced in one endometrial tumor with loss of expression of Gpx3 and in pre-malignant samples (NUT43 and NUT56) with Gpx3 expression.
Deletion of the gene Gpx3 in the rat EAC tumor NUT84
FISH was performed on the cell line where Gpx3 was unmethylated, but with a decreased gene expression of Gpx3. The Gpx3 probe was generated from an NME cell line (NUT43). Six primer pairs representing all exons of Gpx3 were designed and used in PCR (Table 5). The PCR products were subsequently pooled, amplified and fluorescently labeled in DOP-PCR. As a positive control, we used a probe generated from a RNO2 BAC. The result of the FISH experiment showed that Gpx3 was deleted in the NUT84 cell line.
Table 5.
Primers used for the construction of the Gpx 3 specific probe.
| Primer | Sequence | Product size (bp) |
|
|---|---|---|---|
| Exon 1 | F | TTCTCCCCAAAACCACTGAG | |
| R | CCCTTCTCTCCCTCCTAAGC | 583 | |
| Exon 2 | F | GTGGTCCCATGATGCTCTCT | |
| R | GGGTTCAAGATTTGGGTGTG | 592 | |
| Exon 3 | F | CTGTAGCAGCCATCCAACTG | |
| R | ACCTTGTTCTGTCCGTCACC | 588 | |
| Exon 4 | F | GAAGAGACAGGCTGGGTGAT | |
| R | CCCAAAGAGACCACCATCTC | 589 | |
| Exon 5 | F | TCCATGTCAGCCACTCACTC | |
| R | GAAGTTGTAGGCCCTGAGA | 594 | |
| Exon 6 | F | TAAGACTGATGCCCCCTCAC | |
| R | AGGTTTGAGCAGGACCATTG | 587 |
Discussion
Gpx3 was identified as a potential molecular biomarker for rat EAC in a previous microarray study [9]. In this study, we sought to confirm the previously demonstrated down-regulation of Gpx3 expression from the microarray study, of a selection of previously used rat endometrial tumors (Table 1).
As indicated in the previous study, the rat tumors as well as the human tumors indeed displayed an almost total loss of expression of Gpx3, whereas the non-/pre-malignant endometrial samples displayed a normal/high expression, p < 0.001 (Figure 1). The mRNA expression of GPX3 in the human material was measured in 30 human EACs in FIGO grade I-III (10 tumors from each grade), and 21 benign endometrial samples using qPCR (Table 2). Regardless of tumor grade, the expression of GPX3 was low in all tumors, whereas the benign endometrial tissues exhibited a relatively high expression. Thus, in comparisons between tumor FIGO grades we found no significant difference (p > 0.5), while between malignant tumors and benign tissues, we found significant differences, (p < 0.001) (Figure 1). These observations are in agreement with the down-regulated expression of Gpx3 in the rat EACs described above. Accordingly, the dramatic loss of Gpx3 expression in almost all rat EAC samples and the consistently low expression of GPX3 in human EAC, suggest that the down-regulation of Gpx3 expression is an early event in EAC carcinogenesis.
In a study performed by Yu et al. [17-23], it was suggested that GPX3 has tumor suppressor activity as they could show that induced over-expression of GPX3 in prostate cancer cell lines decreased invasiveness, anchorage independent growth and colony formation. Moreover, xenografted prostate cancer cells expressing GPX3, showed reduction of tumor size, elimination of metastasis and reduction of animal death. Their findings also suggest that the GPX3 tumor suppressor activity seems to involve transcriptional regulation of the tumor oncogene, MET [12]. MET activation by HGF ligand binding, initiates a whole spectrum of biological activities, including growth promotion, motility and invasiveness [24]. Multiple signaling transduction pathways are induced by MET engagement, including; the MAPK pathway, the RAS pathway, the P13K pathway, the STAT pathway, the beta catenin pathway and the Notch pathway [25]. To investigate whether Gpx3/GPX3 regulates the expression of Met/MET in the rat EAC and human tumor material, we performed qPCR on Met as well. The expression of Met in the rat tumors was slightly higher in the endometrial tumors with a loss of Gpx3 expression. Applying Pearsson´s correlation test, no significant correlation between the Gpx3 and Met gene expression was detected in the rat tumor material. Thus, in the tumor materials included in this study, we could not confirm any tumor suppressor activity of Gpx3. In the human material highly significant differences in the expression of MET between normal and tumor tissues were found (P < 0.001), but not in the way as would be expected from previous studies, where it was suggested that down regulation of GPX3 causes up-regulation of MET in some cancer types [17-23]. Here, when GPX3 was down regulated, also the MET gene was down regulated, which was also confirmed by the positive correlation displayed by Pearsson´s correlation test (0.67, P < 0.001). Thus, in this study we could not detect any tumor suppressor activity of GPX3 in MET/HGF mediated pathways as suggested by Yu, et al. [17-23].
The GPX3 gene was shown to display epigenetic inactivation in prostate cancer, ovarian clear cell adenocarcinoma, gastric carcinoma and Barret's disease [11-16]. We therefore investigated the methylation status in the BDII inbred rat strain, the EAC cell lines with confirmed decreased mRNA expression (n = 14) in the NME cell lines with a confirmed normal expression of Gpx3 (n = 2) (Figure 1). Hypermethylation of the Gpx3 promoter region was found in 14 EAC cell lines. Thirteen of the EAC samples the Gpx3 promoter were biallelically methylated (Table 4), One tumor, NUT39, showed monoallelic methylation and Gpx3 was therefore not as much down-regulated in this tumor as in the tumors with biallelic hypermethylation, which was also confirmed in the expression analysis (Table 4). The premalignant samples (NME), with an up-regulated expression of Gpx3, were not methylated.
Two of the tumor cell lines, NUT12 and NUT81, that displayed biallelic hypermethylation of the Gpx3 promoter, were selected for treatment with the demethylating agent 5-aza-2´-deoxy-cy-tidine (5Aza-dC) in combination with the histone deacetylace inhibitor, trichostatin A (TSA). The Gpx3 mRNA expression was fully restored after the demethylation treatment (Figure 2). Hence, these results confirm that promoter methylation plays an essential role in silencing of the Gpx3 expression.
Cancer cells are constantly exposed to oxidative stress, and it has been shown that human tumor cell lines generate reactive oxidative species (ROS) to a much higher extent than do non-transformed cell lines [26]. When we measured the production of hydrogen peroxide in two rat endometrial tumor cell lines with loss of expression of Gpx3 and in one premalignant/normal endometrial cell line with normal Gpx3 expression, we found that a higher generation of hydrogen peroxide was produced in the tumor cell lines than in the premalignant cell line (Figure 3). The loss of the protective properties of Gpx3 most likely makes the endometrial cells more vulnerable to ROS damage and genome instability. These findings suggest that the GPX3 function is impaired in endometroid adenocarcinoma, and a likely consequence is an increased amount of hydrogen peroxide and other reactive oxidative species (ROS). Clearly, more tumor cell lines have to be investigated and further functional analyses are required to elucidate the role of ROS in EAC.
In our study Gpx3 was strongly down regulated in NUT84. From the result of the methylation studies of this cell line it became clear to us that the loss of expression did not depend on epigenetic inactivation of the gene (Table 4). However, the down-regulation may be explained by other mechanisms, such as structural aberrations at the site of the Gpx3 locus. In fact in a previous study a chromosomal deletion has been observed in the region of RNO10 where Gpx3 is located, (RNO10q22, 40.3 Mb) which might explain the low expression of the gene [27]. Consequently, we decided to perform FISH on that cell line with a Gpx3 probe that was generated from an NME cell line (NUT43) with normal expression of Gpx3. From the results of the FISH experiment, we could determine that Gpx3 was included in the deletion of that was previously detected [27], which explains the decreased expression of Gpx3 in NUT84. As the BDII rat strain did not exhibit a deletion in the region including Gpx3 most probably the deletion is an event that has occurred during the tumor development.
Conclusion
We have found a consistent down-regulated expression of Gpx3 in both rat and human EACs. The limited expression of Gpx3 in the rat cell lines was correlated to biallelic hyper-methylation of the Gpx3 promoter region. Demethylation of the genomes resulted in a restored expression, suggesting that the hypermethylation is responsible for the down-regulation of Gpx3. From the FISH images we could confirm that the Gpx3 gene was deleted in one of the tumors that was down regulated, but not methylated (NUT84). In previous studies in human prostate cancer, it was suggested that GPX3 exhibits tumor suppressor activity by transcriptional regulation of the oncogene MET [11-16]. We could not confirm any such tumor suppressor activity of GPX3/Gpx3 either in human or rat endometrial tumors.
It has been proposed that ROS overproduction is required for hypoxic activation of HIF-1 [28] and the results from the preliminary experiments in this study indicate that the GPX3 function is impaired also in endometroid adenocarcinoma. To conclude, the results presented here propose that there are important clinical implications of GPX3 expression in EAC, both as an important molecular biomarker for EAC and as a potential target for therapeutic interventions.
Materials and methods
Animal crosses and tumor material
The animal material was derived from crosses between BDII/Han females and males from two non-susceptible rat strains, BN/Han and SPRDCu3/Han, where at first an F1 progeny was produced. Subsequently, an F2 offspring by brother/sister mating of the F1 progeny, and a backcross progeny (N1), by crossing the F1 males to BDII females, were produced. The female progeny was palpated twice each week for identification of uterine tumors. Animals suspected to have tumors were euthanized and the tumor tissue surgically removed, subjected to pathological characterization and subsequently used to establishment of cell cultures [29]. In this study we have investigated cell lines established from tissues pathologically classified as endometrial adenocarcinomas (EAC) and from tissues of normal/pre-malignant endometrium (NME). RUT cell lines originate from tumors developed in the F1 and F2 progeny and NUT cell lines originate from the tumors in the backcross progeny (Table 1).
In vitro cell culture conditions
Primary cell cultures established from the EAC tumors were propagated in Dulbecco's modified Eagle medium, supplemented with 100 IU/100 μg/ml penicillin/streptomycin, L-glutamine, MEM amino acids, MEM Non Essential Amino acids, MEM Vitamins solution and 10% heat-inactivated fetal bovine serum, for 3-5 passages in order to obtain the required amount of cells. The NME cell lines were cultured under the same conditions, but in medium containing 20% fetal bovine serum. The cells were grown at 37°C in an atmosphere of 95% humidity and 5% CO2 and harvested by trypsinizination at a confluence of 80-90% (approx 1x106 cells).
RNA extraction
Total RNA was extracted from the harvested cells of the different endometrial rat cell lines with a KingFisher mL Instrument (Thermo Electron Corporation, USA) according to the manufacturer's protocol (MagAttract Tissue Mini M48 Kit, Qiagen). RNA was spectrophotometrically quantified (NanoDrop technologies, USA).
Quantitative PCR (qPCR) of Gpx3 and Met in cell lines from adenocarcinomas (EAC) and normal/premalignant tissue (NME)
A total of 14 EAC and 2 NME cell lines were used in qPCR analysis with GAPDH as an endogenous control and Universal Rat Reference RNA, Agilent Technologies, Inc as a calibrator (Table 1). RT-PCR was performed using High Capacity cDNA Reverse Transcription Kit according to the manufacturer's protocol (Applied Bio-systems). Template cDNA was added to TaqMan Universal Master Mix (AB; Applied Biosystems, Foster City, CA, USA) in a 12.5 μl reaction with specific pre-designed probes for the Gpx3 and Met (Applied Biosystems). Reactions were performed in duplicates and threshold cycle number was averaged. Relative gene expression quantification was calculated according to the comparative Ct method using GAPDH as an endogenous control and Universal Rat reference RNA (Stratagene) as calibrator. The relative quantitative gene expression, were determined as follows: 2-(Ct sample-Ct calibrator), where Ct values of the calibrator and sample are determined by subtracting the Ct value of the target gene from the value of the GAPDH gene.
Bisulfite treatment and methylation-specific PCR
One μg DNA from the susceptible rat strain BDII, 15 EAC and 2 NME cell lines were denatured, sodium bisulfite treated and purified using Epitect Bisulfite Kit according to the manufacturer's protocol (Qiagen) (Table 1). As a positive control, DNA from an endometrial cell line (NUT43) with a normal expression of Gpx3, and thus unmethylated was treated with methylase and subsequently treated with sodium bisulfite. The modified tumor/control DNA was used as template for methylation-specific PCR. Methylation specific primers (MSP) were designed using the publicly available MethPrimer program http://www.urogene.org/methprimer/[30]. The bisulphite modification of DNA converts unmethylated cytosines to uracils, whereas methylated cytosines will remain unchanged. Bisulphite treated DNA was amplified with either methylation specific or un-methylation specific primer sets. PCR was carried out in a final 25 μl volume containing 50 ng of template DNA The mixture was heated at 94°C for 1 min and then subjected to 35 cycles of 94°C, 55°C and 72°C and a final extension at 72°C for 7 min. The PCR product was analyzed on a 2% agarose gel with appropriate size marker and the absence or presence of PCR product were detected.
5-aza-2´-deoxycytidine and trichostatin A (TSA) treatment in rat endometrial tumor cell lines
Two EAC cell lines (NUT12 and NUT81) with confirmed biallelic methylated promoter status of Gpx3 were treated with 5-aza-2´-deoxycytidine and trichostatin A (Sigma). Cells were grown in a medium containing 2.5 μM 5-aza-cytidine for 96 hours, with the medium and drug being replaced every 24 hours and the addition of 300 nM TSA was added for the last 16 hours. After 96 hours, the drugs were removed and total RNA for Gpx3 RT-PCR expression analysis was extracted using AllPrep RNA/DNA Mini Kit according to the manufacturer's protocol (Qiagen).
Hydrogen peroxide measurements
The intra- and extra-cellular amount of hydrogen peroxide in the endometrial cell lines were measured using the Amplex® Red Peroxide/Peroxidase Hydrogen assay kit according to the manufacturer's protocol (Molecular Probes, Invitrogen). The three cell lines investigated (NUT12, NUT43, NUT56) were seeded to a 96 wells plate (Corning) with an initial number of 5000 cells/well (Table 1). In each well, the amount of hydrogen peroxide was measured post 72 hours of incubation, as described above. Each cell line was replicated 20 times. The cells were lysated by adding RIPA buffer (25 mM TRIS-HCl pH 7.6, 150 mM NaCl, 1% deoxycholate, 0, 1% SDS and 1% NP40) followed by incubation on ice for 30 minutes. Intra- and extra-cellular H2O2 concentrations were assessed by pooling 50 μl of cell lysate with 50 μl of used cell culture media.
Development of FISH probes for Gpx3
DNA from the pre-malignant cell line, NUT43, with a normal expression of Gpx3 was used to generate a probe that represented only the Gpx3 gene. Six primer pairs, specific for the Gpx3 gene, with a product size of approximately 600 base pair each (Table 5), were designed by using the Primer 3 program available on the internet: http://fokker.wi.mit.edu/primer3/input.htm. Amplification was performed by PCR and carried out in a final 25 μl volume containing 100 ng of template DNA. The mixture was heated at 94°C for 1 min and then subjected to 35 cycles of 94°C, 58°C and 72°C and a final extension at 72°C for 7 min. Sizes of the PCR products were determined on a 2% agarose gel with an appropriate size marker. The PCR products were then purified by Mini Elute PCR purification kit (QIAGEN) and the concentration of DNA was measured by NanoDrop (NanoDrop Technologies, USA) amplified product. The amplified sequences were then pooled and fluorescently labeled by dNTP in DOP-PCR and subsequently used as a Gpx3 specific probe in FISH.
One probe, which was used as positive control, was developed from a RNO2 BAC clone (CH230-397A17 from BACPAC Resources Center, Oakland, California). The BAC DNA was amplified by DOP-PCR as follows. The reactions were performed in a final volume of 25 μl and with a BAC DNA concentration of 20 ng/μl. The mixture was heated at 94°C for 1 min and then subjected to 35 cycles of 94°C, 55°C and 72°C and a final extension at 72°C for 7 min. The product was verified on a 2% agarose gel with an appropriate size marker. For FISH, the Nick Translation kit from Abbott molecular was used according to the manufactures protocol.
Human material
A total of 30 EACs in FIGO grade I-III (10 tumors from each grade embedded in archival formalin fixed paraffin (FFPE) were used in the study. Apart from the endometrial tumors, 21 benign endometrial tissues were collected, and reference material from lung was used in the normalization process (Table 2). All samples were anonymous. A pathologist marked the tumor area in samples in the hematoxylin and eosin slide. Using a Tissue Micro Array-equipment (Pathology Devices), 3-4 cores (∅0.6 mm) of tumor tissue was punched out from the paraffin block. After standard tissue sample deparaffinization using xylene and alcohols, samples were lyzed in a Tris-chloride, EDTA, sodium dodecyl sulfate (SDS) and proteinase K containing buffer. RNA was then extracted and used for the real time qPCR.
Quantitative PCR (qPCR) of GPX3 and MET in FIGO grade I-III human EACs
Total RNA was extracted and used for qPCR according to the same procedure as for the rat samples.
Statistical analysis
For statistical evaluations of Ct values for differences among replicates we applied paired samples t-test and for comparisons of normal and malignant tissues independent sample t-test was applied. (PASW Statistics 18, SPSS Inc, Chicago, USA). In both tests the null hypotheses were assuming no differences between replicates, and no differences between tissue types respectively. The Pearson correlation test was performed to check for correlation between the expression of Gpx3 and Met. The significance levels were set to P < 0.5 in all statistical tests.
List of abbreviations
EAC: endometrial adenocarcinoma; NME: normal/pre-malignant endometrium; NUT: backcross, rat uterine tumor; RUT: rat uterine tumor
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SK and EF contributed with all original ideas, designed all studies, performed the data analysis and drafted the manuscript. SK and EF carried out the methylation specific PCR and Q-PCR. SK and JC performed the hydrogen-peroxide assays and the de-methylation study. EF, GH and MK were responsible for the human endometrial study. KKL supervised the project and contributed with ideas and took part in the preparation of the manuscript. All authors have read and approved the final version of the manuscript.
Contributor Information
Eva Falck, Email: eva.falck@his.se.
Sandra Karlsson, Email: sandra.karlsson@his.se.
Jessica Carlsson,, Email: jessica.carlsson@his.se.
Gisela Helenius, Email: gisela.helenius@orebroll.se.
Mats Karlsson, Email: mats.karlsson3@orebroll.se.
Karin Klinga-Levan, Email: karin.klinga.levan@his.se.
Acknowledgements
This work was supported by the Swedish National Research School in Bioinformatics and Genomics, Erik Philip-Sörensen Foundation, and the Nilsson-Ehle foundation. We are grateful to Karin Lilja for excellent technical assistance.
References
- Prat J. Prognostic parameters of endometrial carcinoma. Hum Pathol. 2004;35(6):649–662. doi: 10.1016/j.humpath.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Bokhman JV. Two pathogenetic types of endometrial carcinoma. Gynecologic Oncology. 1983;15:10–17. doi: 10.1016/0090-8258(83)90111-7. [DOI] [PubMed] [Google Scholar]
- Deligdisch L, Holinka CF. Endometrial carcinoma: two diseases? Cancer Detection and Prevention. 1987;10(3-4):237–246. [PubMed] [Google Scholar]
- Lax SF, Kurman RJ. A dualistic model for endometrial carcinogenesis based on immunohistochemical and molecular genetic analyses. Verh Dtsch Ges Pathol. 1997;81:228–232. [PubMed] [Google Scholar]
- Greven KM, Corn BW. Endometrial cancer. Curr Probl Cancer. 1997;21(2):65–127. doi: 10.1016/S0147-0272(97)80002-5. [DOI] [PubMed] [Google Scholar]
- Deerberg FKJ. Endometrial Carcinoma in BDII/Han rats: model of a spontaneous hormone-dependent tumor. J Natl Cancer Inst. 1987;78:1245–1251. [PubMed] [Google Scholar]
- Kaspareit-Rittinghausen J, Deerberg F, Rapp K. Mortality and incidence of spontaneous neoplasms in BDII/Han rats. Z Versuchstierkd. 1987;30(5-6):209–216. [PubMed] [Google Scholar]
- Vollmer G. Endometrial cancer: experimental models useful for studies on molecular aspects of endometrial cancer and carcinogenesis. Endocr Relat Cancer. 2003;10(1):23–42. doi: 10.1677/erc.0.0100023. [DOI] [PubMed] [Google Scholar]
- Karlsson S, Olsson B, Klinga-Levan K. Gene expression profiling predicts a three-gene expression signature of endometrial adenocarcinoma in a rat model. Cancer Cell Int. 2009;9:12. doi: 10.1186/1475-2867-9-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson JOG, Sjöstedt A, Wahlström J, Axelsson KL, Andersson RGG. Cyclic guanosine monophosphate metabolism in human amnion cells trisomic for chromosome 21. Biology of the Neonate. 1990;57:343–348. doi: 10.1159/000243211. [DOI] [PubMed] [Google Scholar]
- Saga Y, Ohwada M, Suzuki M, Konno R, Kigawa J, Ueno S, Mano H. Glutathione peroxidase 3 is a candidate mechanism of anticancer drug resistance of ovarian clear cell adenocarcinoma. Oncol Rep. 2008;20(6):1299–1303. [PubMed] [Google Scholar]
- Yu YP, Yu G, Tseng G, Cieply K, Nelson J, Defrances M, Zarnegar R, Michalopoulos G, Luo JH. Glutathione peroxidase 3, deleted or methylated in prostate cancer, suppresses prostate cancer growth and metastasis. Cancer Res. 2007;67(17):8043–8050. doi: 10.1158/0008-5472.CAN-07-0648. [DOI] [PubMed] [Google Scholar]
- Lodygin D, Epanchintsev A, Menssen A, Diebold J, Hermeking H. Functional epigenomics identifies genes frequently silenced in prostate cancer. Cancer Res. 2005;65(10):4218–4227. doi: 10.1158/0008-5472.CAN-04-4407. [DOI] [PubMed] [Google Scholar]
- Zhang X, Yang JJ, Kim YS, Kim KY, Ahn WS, Yang S. An 8-gene signature, including methylated and down-regulated glutathione peroxidase 3, of gastric cancer. Int J Oncol. 2010;36(2):405–414. [PubMed] [Google Scholar]
- Jee CD, Kim MA, Jung EJ, Kim J, Kim WH. Identification of genes epigenetically silenced by CpG methylation in human gastric carcinoma. Eur J Cancer. 2009;45(7):1282–1293. doi: 10.1016/j.ejca.2008.12.027. [DOI] [PubMed] [Google Scholar]
- Lee OJ, Schneider-Stock R, McChesney PA, Kuester D, Roessner A, Vieth M, Moskaluk CA, El-Rifai W. Hypermethylation and loss of expression of glutathione peroxidase-3 in Barrett's tumorigenesis. Neoplasia. 2005;7(9):854–861. doi: 10.1593/neo.05328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4(12):915–925. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- Rong S, Segal S, Anver M, Resau JH, Vande Woude GF. Invasiveness and metastasis of NIH 3T3 cells induced by Met-hepatocyte growth factor/scatter factor autocrine stimulation. Proc Natl Acad Sci USA. 1994;91(11):4731–4735. doi: 10.1073/pnas.91.11.4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taulli R, Scuoppo C, Bersani F, Accornero P, Forni PE, Miretti S, Grinza A, Allegra P, Schmitt-Ney M, Crepaldi T. et al. Validation of met as a therapeutic target in alveolar and embryonal rhabdomyosarcoma. Cancer Res. 2006;66(9):4742–4749. doi: 10.1158/0008-5472.CAN-05-4292. [DOI] [PubMed] [Google Scholar]
- Lutterbach B, Zeng Q, Davis LJ, Hatch H, Hang G, Kohl NE, Gibbs JB, Pan BS. Lung cancer cell lines harboring MET gene amplification are dependent on Met for growth and survival. Cancer Res. 2007;67(5):2081–2088. doi: 10.1158/0008-5472.CAN-06-3495. [DOI] [PubMed] [Google Scholar]
- Takayama H, LaRochelle WJ, Sharp R, Otsuka T, Kriebel P, Anver M, Aaronson SA, Merlino G. Diverse tumorigenesis associated with aberrant development in mice overexpressing hepatocyte growth factor/scatter factor. Proc Natl Acad Sci USA. 1997;94(2):701–706. doi: 10.1073/pnas.94.2.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Ferrell LD, Faouzi S, Maher JJ, Bishop JM. Activation of the Met receptor by cell attachment induces and sustains hepatocellular carcinomas in transgenic mice. J Cell Biol. 2001;153(5):1023–1034. doi: 10.1083/jcb.153.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt L, Junker K, Nakaigawa N, Kinjerski T, Weirich G, Miller M, Lubensky I, Neumann HP, Brauch H, Decker J. et al. Novel mutations of the MET proto-oncogene in papillary renal carcinomas. Oncogene. 1999;18(14):2343–2350. doi: 10.1038/sj.onc.1202547. [DOI] [PubMed] [Google Scholar]
- Bottaro DP, Rubin JS, Faletto DL, Chan AM-L, Kmiecik TE, Vande Woude GF, Aaronson SA. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251:802–804. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- Gentile A, Trusolino L, Comoglio PM. The Met tyrosine kinase receptor in development and cancer. Cancer Metastasis Rev. 2008;27(1):85–94. doi: 10.1007/s10555-007-9107-6. [DOI] [PubMed] [Google Scholar]
- Szatrowski TP, Nathan CF. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991;51(3):794–798. [PubMed] [Google Scholar]
- Nordlander C, Karlsson S, Karlsson A, Sjoling A, Winnes M, Klinga-Levan K, Behboudi A. Analysis of chromosome 10 aberrations in rat endometrial cancer-evidence for a tumor suppressor locus distal to Tp53. Int J Cancer. 2007;120(7):1472–1481. doi: 10.1002/ijc.22533. [DOI] [PubMed] [Google Scholar]
- Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA. 1998;95(20):11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helou K, Walentinsson A, Beckmann B, Johansson Å, Hedrich HJ, Szpirer C, Klinga-Levan K, Levan G. Analysis of genetic changes in rat endometrial carcinomas by means of comparative genome hybridization. Cancer Genet Cytogenet. 2001;172(2):118–127. doi: 10.1016/S0165-4608(00)00435-0. [DOI] [PubMed] [Google Scholar]
- Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18(11):1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]