

Abstract
The von Hippel–Lindau tumor suppressor protein (VHL), when mutated and inactivated, has been associated with renal and CNS cancer development. VHL normally plays an important role in targeting for degradation of the HIF-1α (hypoxia inducible factor-1α) transcription factor, a primary positive regulator of vascular endothelial growth factor (VEGF) production. In this report we demonstrate that VHL destabilization can be induced by Src kinase and may be involved in other cancers, including breast cancer. We have found that elevated Src can trigger a drastic reduction in VHL stability even under normoxic conditions, through phosphorylation of VHL tyrosine residue 185, leading to ubiquitination and proteasome-mediated degradation of VHL. The Src-induced degradation of VHL protein leads to increased HIF-1α levels and transcriptional activity and increased VEGF production. In this manner, Src regulation of VHL protein stability may play an important role in promoting VEGF expression, tumor angiogenesis, and cancer progression.
Keywords: VHL, tumor suppressor, stability, Src, proto-oncogene, oncogene
Introduction
Src is a cytoplasmic tyrosine kinase that can contribute to cancer development and progression, when mutated or overexpressed. Src protein levels and/or Src protein kinase activity are frequently elevated in human cancers and cell lines, including those of the breast,1,2 colon,3-6 brain,7,8 and pancreas.9,10 Our laboratory has previously reported elevated levels of endogenous Src activity in breast and colon cancer cells, ranging from 1.7- to 17.3-fold,2,6 and that Src kinase can be activated by high levels of the protein tyrosine phosphatase PTP1B in breast and colon cancer cells.2,6,11 Furthermore, Src has been shown to be activated by a mutation resulting in truncation of the Src protein in a subset of advanced metastatic colon carcinomas.5 Elevated Src activity has been reported to upregulate vascular endothelial growth factor (VEGF) production in several normal and tumorigenic cells types.12-14 Src has also been shown to be required for VEGF-mediated vascular permeability.15 These and other findings indicate that one means through which Src can promote tumor development is by stimulation of angiogenesis.
VEGF production is normally regulated by changes in tissue oxygenation. Under normoxic conditions, the von Hippel–Lindau protein (VHL, an E3 ligase) is capable of physically binding to and triggering proteasome-mediated destruction of a proline-hydroxylated form of hypoxia inducible factor-1α (HIF-1α), the regulatable component of the HIF-1 transcription factor heterodimer. Therefore, in the presence of VHL, HIF-1α is targeted for proteasome-mediated degradation.16-18 However, under hypoxic conditions, such as those existing within interior regions of solid tumors, hydroxylation of HIF-1α is inhibited, thus preventing binding of VHL to HIF-1α and preventing HIF-1α degradation in these tumor cells. The HIF-1 transcription factor is then able to recognize the VEGF promoter and upregulate VEGF transcription. VEGF produced and secreted from the tumor cells is able to bind to cell surface receptors on endothelial cells, stimulating endothelial cell proliferation and angiogenesis and contributing to tumor progression and metastasis.19,20
Importantly, functional inactivation of the VHL tumor suppressor protein through germline mutations has been documented in highly vascular tumors that overproduce VEGF, such as renal carcinomas, phaeochromocytomas, and central nervous system hemangioblastomas.21-26 In this article, we report that elevated Src activity in breast cancer and other cell lines can drastically reduce VHL stability and half-life under normoxic conditions. Specifically, elevated Src activity can mediate increased tyrosine phosphorylation of VHL and ubiquitination of VHL, which promotes degradation of VHL in 26S proteasomes. Our results demonstrate that a dramatic reduction of VHL levels mediated by Src can contribute to increased cellular synthesis of VEGF, via increased levels of HIF-1α protein. Overall, we have found an important mechanism of Src-dependent destabilization of VHL protein that is likely to be involved in promoting tumor angiogenesis and tumorigenesis.
Results
Elevated Src activity is associated with increased VEGF production, reduced VHL protein, and increased phosphorylated VHL in breast carcinoma cell lines
Src has been implicated as an important signaling component in hypoxia-induced upregulation of VEGF.12-14 In addition, Src activation appears to play a role in VEGF production and vascularization by some colon and ovarian cancer cells.13,14,27 When we examined several breast carcinoma cell lines that have previously been reported by our lab to exhibit high levels of endogenous Src activity,2,11 we found that they also produced higher levels of VEGF, relative to the nontumorigenic breast epithelial cell line MCF-10A, even under normoxic conditions (20% O2; Fig. 1A). We also found that some breast carcinoma cells lines, such as SK-BR-3 and Hs 578T that exhibited elevated levels of endogenous Src activity,2 had lower levels of endogenous VHL and higher levels of tyrosine phosphorylated VHL (Fig. 1B). Thus, these results suggested that high Src activity was correlated with decreased VHL protein levels.
Figure 1.
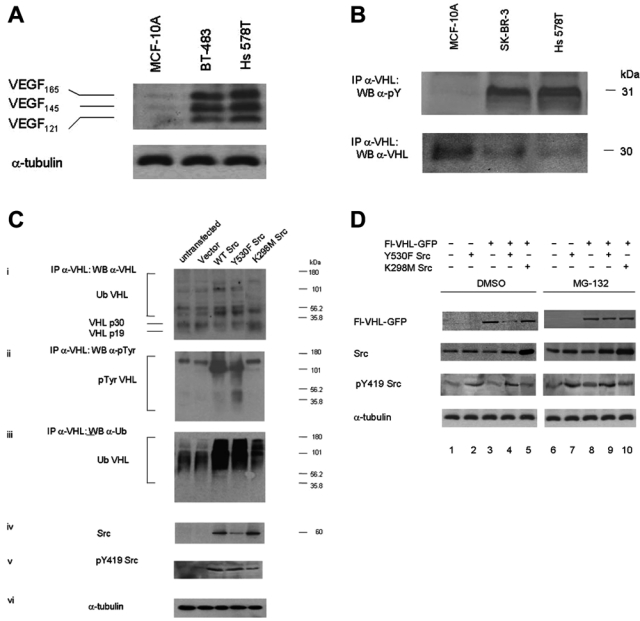
Elevated Src activity is associated with reduced von Hippel–Lindau (VHL) protein and increased tyrosine phosphorylated and ubiquitinated VHL. (A) MCF-10A are nontumorigenic breast epithelial cells, and BT-483 and Hs 578T are breast carcinoma cells. Conditioned media samples were collected from the different cell lines and were analyzed by Western blotting for vascular endothelial growth factor (VEGF) protein using anti-VEGF polyclonal antibody. Conditioned media volumes assayed were adjusted for differences in the cell numbers of the cultures from which the samples were collected. The results are representative of 2 independent experiments. (B) MCF-10A, SK-BR-3, and Hs 578T cell lysates were prepared in 2% sodium dodecyl sulfate (SDS), 50 mM Tris (pH 6.8) lysis buffer and boiled. Endogenous VHL was isolated by immunoprecipitation with anti-VHL polyclonal antibody, followed by Western blotting for phosphotyrosine and endogenous VHL. The results are representative of 2 independent experiments. (C) HEK 293T cells were transfected with the indicated plasmids. Forty-eight hours posttransfection, the cells were treated with 25 µM MG-132 for 16 h. Cell lysates were prepared in 2% SDS, 50 mM Tris (pH 6.8) lysis buffer and boiled. Endogenous VHL was then isolated by immunoprecipitation with anti-VHL polyclonal antibody, followed by immunoblotting for (i) VHL, (ii) phosphotyrosine, and (iii) ubiquitin. (iv) Total Src, (v) pY419 Src, and (vi) α-tubulin protein levels were detected by Western blotting. The results are representative of 3 independent experiments. (D) HEK 293T cells were transfected with the indicated plasmids. Forty-eight hours posttransfection, the cells were treated with either DMSO (lanes 1-5) or 25 µM MG-132 for 16 h (lanes 6-10). The cells were then lysed, and Western blotting was performed for Flag, Src, pY419 Src, and α-tubulin. The results are representative of 3 independent experiments.
Elevated Src activity is associated with reduced VHL protein and increased tyrosine phosphorylated and ubiquitinated VHL
In order to study the effect of elevated Src activity on VHL protein levels, we transiently transfected Src into HEK 293T cells. We found that both elevated wild-type Src levels (WT Src) as well as elevated constitutively active Src levels (Y530F Src) were associated with reduced endogenous VHL protein levels, concomitant with increased tyrosine phosphorylation of VHL by Src (Fig. 1Cii, lanes 3 and 4) and increased polyubiquitinated VHL (Fig. 1Ciii; lanes 3 and 4). In further experiments, we found that the 26S proteosome inhibitor MG132 could protect exogenously expressed VHL from degradation induced by elevated Src levels (Fig. 1D, lane 9). This finding suggests that the Src-mediated reduction in VHL protein levels is likely due to proteasome-dependent degradation. We have also found that activated Src did not downregulate transcription of VHL mRNA, based on RT-PCR experiments (unpublished observations).
Knockdown or inhibition of Src increases VHL protein levels
In order to confirm that Src was involved in destabilizing VHL, we used siRNA to knock down Src levels. In these experiments, we observed 70% knockdown in Src protein levels, compared to Src protein levels in cells transfected with control, nontargeting siRNA in HEK 293T as well as in SK-BR-3 breast cancer cells (Fig. 2 Ai and Bii; Fig. 2C). Knockdown of Src was associated with 4-fold increases of VHL protein levels (Fig. 2 Ai, Bi; Fig. 2C). Also, there was decreased phosphorylation of VHL accompanying increased endogenous p30 and p19 VHL protein levels (Fig. 2Ai). siRNA targeting of VHL was also used to confirm the identity of the p30 and p19 VHL proteins. Moreover, consistent with our earlier data, transfection of Src-specific siRNA was associated with a 60% decrease in VEGF protein (Fig. 2C). Furthermore, treatment of NIH 3T3 cells with the Src inhibitor PP2 effectively decreased Src activity, as reflected in lower Y419 phosphorylation, and was associated with increased stability of VHL protein, along with lower levels of ubiquitinated VHL (Supplementary Fig. S1). Thus, these results support a significant role for Src and Src activity in downregulating VHL protein levels that can ultimately upregulate VEGF (summarized in the model in Fig. 3).
Figure 2.
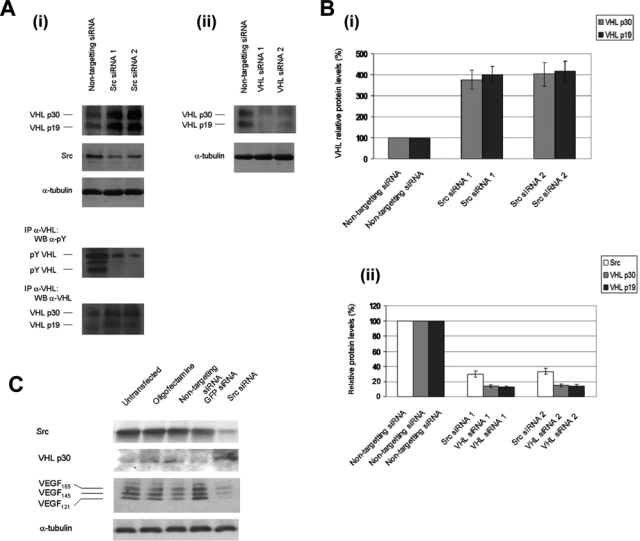
siRNA knockdown of Src increases von Hippel–Lindau (VHL) protein levels. (A) (i, ii) HEK 293T cells were transfected with nontargeting control siRNA, Src siRNA, or VHL siRNA. Forty-eight hours posttransfection, cell lysates were prepared in 2% sodium dodecyl sulfate (SDS), 50 mM Tris (pH 6.8) lysis buffer and boiled, and Western blotting was conducted for endogenous VHL, Src, and α-tubulin. Endogenous VHL protein was also isolated by immunoprecipitation with anti-VHL polyclonal antibody, followed by immunoblotting for phosphotyrosine and VHL. (B) (i) Quantitation of the VHL protein levels of the results from part A. (ii) Quantitation of the knockdown of VHL and Src protein levels of the results from part A. Densitometry was performed using ImageQuant TL software from Amersham Biosciences (Piscataway, NJ). Values were normalized relative to the values for α-tubulin protein levels. Results are representative of n = 3. (C) SK-BR-3 breast cancer cells were transfected with oligofectamine alone (no siRNA), nontargeting control siRNA, green fluorescent protein (GFP) siRNA, or Src siRNA. Conditioned media samples were collected 48 h after transfection and were analyzed by Western blotting for vascular endothelial growth factor (VEGF) using anti-VEGF polyclonal antibody. The conditioned media volumes assayed were corrected for differences in the cell numbers of the cell cultures from which the samples were collected. Western blotting was also conducted on cell lysates using anti-Src monoclonal antibody, anti-VHL polyclonal antibody, and anti-α-tubulin monoclonal antibody. The results are representative of 2 independent experiments.
Figure 3.
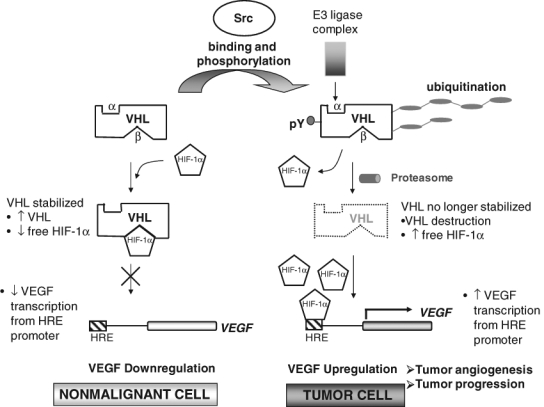
Model of Src downregulation of von Hippel–Lindau (VHL) tumor suppressor protein and promotion of tumor angiogenesis and tumor progression. Src-directed posttranslational tyrosine phosphorylation modification of VHL allows tagging and recruitment of VHL for proteolysis by an E3 ligase complex. Also, posttranslational modification of VHL may displace VHL from the VHL E3 ligase complex (thereby facilitating ubiquitin tagging and recruitment of VHL for proteolysis by E3 ligase complexes and/or facilitating VHL protein instability). Consequently, increased levels of HIF-1α upregulate vascular endothelial growth factor (VEGF) production and ultimately increase tumor angiogenesis and the potential for tumor progression.
Elevated Src activity decreases the protein half-life of VHL
We conducted cycloheximide block experiments in order to study the effect of elevated Src levels on VHL half-life. When HEK 293T cells were transfected with empty vector (pCI) and VHL expression plasmid (Fl-VHL-GFP), the exogenously expressed VHL was very stable and exhibited a half-life of greater than 16 h (Fig. 4Ai, left panel; Fig. 4Aii). In contrast, when cells were cotransfected with constitutively active Src and Fl-VHL-GFP, VHL exhibited a drastically shortened half-life of approximately 5 h (Fig. 4Ai, middle panel; Fig. 4Aii). Interestingly, when the cells were transfected with kinase dead Src and Fl-VHL-GFP, VHL exhibited a half-life of about 12 h (Fig. 4Ai, right panel; Fig. 4Aii). The intermediate level of VHL half-life reduction observed in the presence of kinase dead Src suggested that there might also be a kinase-independent function of Src that could play a partial role in VHL downregulation. 35S metabolic labeling-chase experiments also confirmed that elevated Src activity was correlated with a shortened half-life of VHL (Fig. 4B). Similar reductions in VHL protein stability were also observed for endogenous VHL in HEK 293T cells. When HEK 293T cells were transfected with constitutively active Src, levels of endogenous VHL p30 and p19 were reduced down to about 50% within 6 to 8 h (Fig. 4C).
Figure 4.
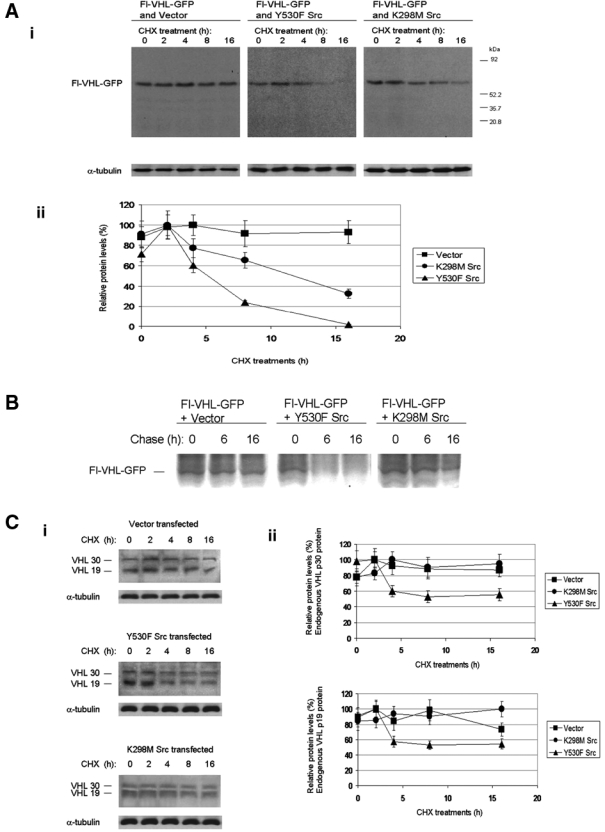
Src activity decreases the protein half-life of von Hippel–Lindau (VHL) protein. (A) (i) HEK 293T cells were transfected with the indicated plasmids. Forty-eight hours posttransfection, the cells were treated with 100 µM final concentration of cycloheximide for 0 h, 2 h, 4 h, 8 h, or 16 h. Western blotting was then conducted for Flag and α-tubulin. (ii) Quantitation of the results from part Ai. VHL protein levels were normalized to α-tubulin. The darkest band density for each trial set was assigned a protein level of 100%, and the other protein levels were plotted relative to 100%. The results are the mean of 3 independent experiments. (B) HEK 293T cells transfected with the indicated plasmids. The cells were then subjected to 35S metabolic labeling in vivo (of intact cells) and chase assays. Fl-VHL-GFP was isolated by immunoprecipitation with anti-VHL antibody. The immune complexes were then resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and detection was conducted by autoradiography. The results are representative of 3 independent experiments. (C) (i) HEK 293T cells were transfected with the indicated plasmids. Forty-eight hours posttransfection, the cells were treated with 100 µM final concentration of cycloheximide for 0 h, 2 h, 4 h, 8 h, or 16 h. Western blotting was then conducted for endogenous VHL and α-tubulin. (ii) Quantitation of the results from part Ci. The results are the mean of 3 independent experiments.
Elevated Src activity downregulates intracellular VHL protein levels
Fluorescence microscopy was also used to demonstrate the effects of Src on VHL stability. Cellular levels of VHL could be detected by the signal produced by the green fluorescent protein (GFP)–tagged VHL epifluorescence when introduced into the RCC 786-O cell line (a renal cell carcinoma cell line lacking endogenous wild-type VHL, as previously described28). Cells cotransfected with Fl-VHL-GFP and activated Src exhibited a 66% decrease in intensity of the green fluorescent signal, in comparison to control cells cotransfected with Flag-VHL-GFP and empty vector (Fig. 5A). In contrast, cells cotransfected with Flag-VHL-GFP and kinase dead Src exhibited only a small 22% decrease in green fluorescent signal (Fig. 5A). These results could also suggest that the kinase dead form of Src may have a small destabilizing effect on VHL protein. However, the greatest effect was that of activated Src on decreased Fl-VHL-GFP protein levels.
Figure 5.
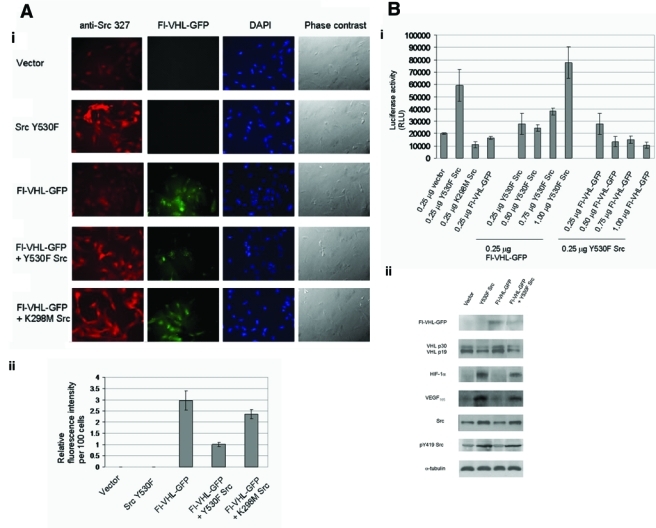
Src activity downregulates the intracellular fluorescence of green fluorescent protein (GFP)–tagged von Hippel–Lindau (VHL) protein, and Src destabilization of VHL upregulates HIF-1α. (A) (i) RCC 786-O renal carcinoma cells were seeded on glass coverslips in 6-well plates. The cells were transfected with the indicated plasmids for 24 h, and the cover slips were then processed for fluorescence microscopy. Anti-Src monoclonal antibody was used to probe for Src. Fl-VHL-GFP was visualized by GFP fluorescence. (ii) Intensities of Fl-VHL-GFP fluorescence were measured using ImageJ image data analysis software. Fl-VHL-GFP fluorescence intensities per 300 cells were measured for each transfection, and these raw fluorescence intensity values were normalized to intensity values per 100 cells. The fluorescence intensity values per 100 cells were then standardized relative to a value of 1 per 100 cells assigned to the fluorescence intensity for the Fl-VHL-GFP and Y530F Src cotransfection condition. The graph charts the mean ± 1 SD from 3 independent experiments done in triplicate. (B) (i) HEK 293T cells were transfected with the indicated plasmids along with HREx3-luciferase reporter plasmid (containing 3 hypoxia responsive element [HRE]) binding sites for HIF-1α). Forty-eight hours posttransfection, the cells were lysed, and the lysates were subjected to luciferase activity assays (Promega, Madison, WI). The graph charts the mean ± 1 SD from 3 independent experiments. (ii) HEK 293T cells were transfected with the indicated plasmids. Forty-eight hours posttransfection, the cells were then lysed, and Western blotting was performed for Flag, endogenous VHL, HIF-1α, vascular endothelial growth factor (VEGF), Src, pY419 Src, and α-tubulin. The results are representative of 3 independent experiments.
Src destabilization of VHL upregulates HIF-1α activity
In order to determine effects of Src-mediated destabilization of VHL protein on levels of HIF-1α, we used assays involving a luciferase reporter plasmid that contained 3 hypoxia responsive element (HRE) binding sites for HIF-1α. As seen in Figure 5Bi, activated Src upregulated HRE-luciferase reporter activity by 3.6-fold (Fig. 5Bi, left group). Src kinase activity was required for this upregulation, as it was not observed with the kinase dead Src mutant (Fig. 5Bi, left group). We further performed reciprocal dose-response experiments by introducing graduated amounts of activated Src plasmid or Fl-VHL-GFP plasmid, respectively, into HEK 293T cells (Fig. 5Bi, middle and right groups). Introduction of increasing amounts of activated Src plasmid was able to override the dampening effects of Fl-VHL-GFP plasmid (Fig. 5Bi, middle group). In contrast, in reverse dose-response experiments, with the introduction of increasing amounts of Fl-VHL-GFP plasmid in the presence of constant amounts of activated Src plasmid, the HRE-luciferase reporter activity decreased to subbasal levels (Fig. 5Bi, right group). Immunoblot analyses further supported this interrelationship between Src and VHL (Fig. 5Bii). Taken together, these results and our previous results indicate that Src activation promotes destabilization of VHL and also causes increased levels of HIF-1α and HIF-1α activity, as well as increased levels of VEGF.
Src destabilization of VHL upregulates tubulogenesis in vitro.
Tubulogenesis in vitro can be used as a means to monitor proangiogenic activity. We conducted endothelial tube formation assays to study the effects of Src-directed destabilization of VHL protein on endothelial tube formation, following previously published protocols.29,30 In this assay, endothelial cells will form capillary networks when seeded on Matrigel in response to angiogenic factors such as VEGF that are present in conditioned media. The extent and lengths of endothelial tube formation are indicators of the angiogenic potential. As seen in Figure 6A, significant endothelial tube formation occurred in the presence of conditioned medium from HEK 293T cells that had been transfected with activated Src. In contrast, the least amount of tube formation was observed with conditioned medium from cells transfected with a control vector alone or with Fl-VHL-GFP alone, and endothelial tube formation was also reduced when Fl-VHL-GFP was cotransfected with activated Src.
Figure 6.
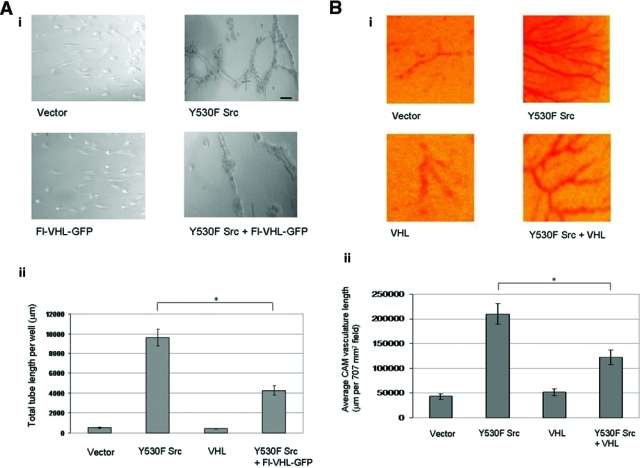
Src destabilization of von Hippel–Lindau (VHL) protein upregulates tubulogenesis in vitro and chick embryo chorioallantoic membrane blood vessel growth. (A) (i) Photographs of endothelial tube formation assays, on conditioned media collected from HEK 293T cells transfected with the indicated plasmids 48 h posttransfection. (ii) Quantification of capillary network formation was done by measuring total tube length per well using ImageJ image data analysis software. Scale bar: 100 µm. The graph charts the averages ± 1 SD from 3 separate transfections done in duplicate. Statistical significance was determined by Student t test; *P < 0.05. (B) (i) Photographs of developing chick chorioallantoic membranes (CAMs), implanted with gelatin sponges loaded with HEK 293T cells transfected with the indicated plasmids. (ii) Quantitation of angiogenesis was done by measuring the length of vasculature per 707 mm2 field, using ImageJ image data analysis software. Four fields per CAM were measured for 9 CAMs per transfection condition. The graph charts the averages ± 1 SD. Statistical significance was determined by Student t test; *P < 0.05.
Src destabilization of VHL upregulates chick embryo chorioallantoic membrane blood vessel growth
As another readout for the effects of Src destruction of VHL tumor suppressor protein on angiogenesis, we performed chorioallantoic membrane assays. In this assay, blood vasculature of the chick chorioallantoic membrane (CAM) grows in response to growth signals received from cells loaded onto gelatin sponges placed onto the CAM, as previously described.31 The average lengths of vasculature per field are indicators of angiogenic potential. As shown in Figure 6B, extensive angiogenesis occurred in the presence of HEK 293T cells that had been transfected with activated Src. In contrast, the least angiogenesis occurred in cells transfected with a control vector alone or with Flag-VHL-GFP alone, and angiogenesis was also reduced when Fl-VHL-GFP was cotransfected with activated Src.
Y185 is a major phosphorylation site on VHL, and Y185F mutation confers increased protein stability to VHL protein
In vitro kinase reactions with purified baculovirus expressed human Src kinase and GST-VHL wild-type (GST-VHL WT) were performed in an attempt to determine the site(s) on VHL phosphorylated by Src kinase. Phosphorylation of GST-VHL occurred only in the presence of Src, and control GST alone was not phosphorylated by purified Src kinase (Fig. 7A). These results indicated that Src is able to directly phosphorylate VHL. Phospho-GST-VHL produced in this manner was isolated and purified by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Tyrosine phosphorylation on Src phosphorylated GST-VHL was mapped through mass spectrometric analysis performed by the proteomics center at the University of Victoria. This analysis indicated that Src was able to phosphorylate VHL tyrosine residue 185 (Fig. 7B and Supplementary Fig. S2). This residue closely matches the consensus sequence for Src phosphorylation and lies within the α domain of VHL.32 As Y185 was the only phosphorylation site detected in this analysis, it is likely to be the major Src phosphorylation site in VHL.
Figure 7.
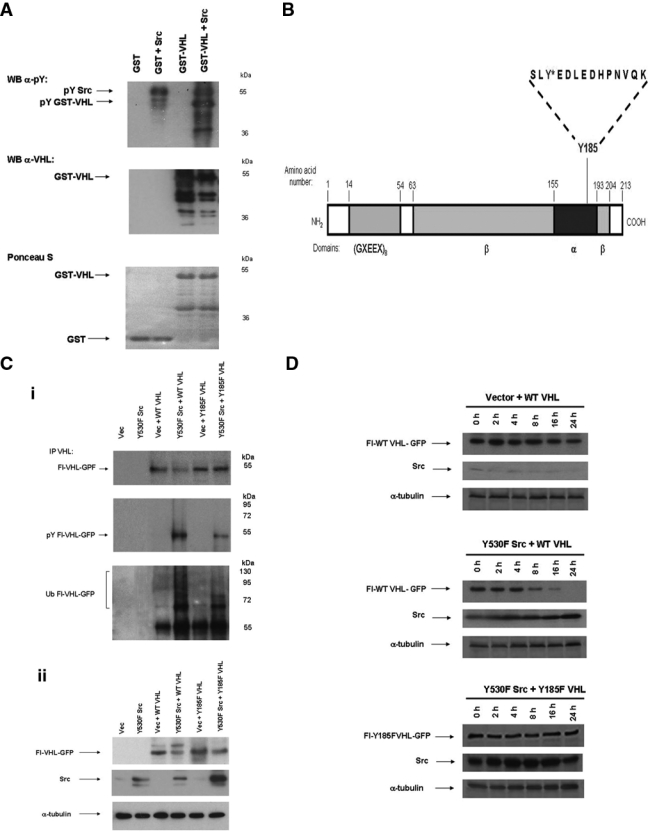
Y185 is a major phosphorylation site on von Hippel–Lindau (VHL) protein, and Y185F mutation confers increased protein stability to VHL protein. (A) In vitro kinase phosphorylation reactions were conducted on purified glutathione S-transferase (GST) and GST-VHL (500 pmol each) in the absence and presence of purified human baculovirus-expressed Src kinase (available from Millipore [Billerica, MA], product #14-117). The reaction samples were stopped by boiling in Laemmli sample buffer, resolved on 10% acrylamide sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel, and transferred to nitrocellulose. Immunoblotting was then conducted for phosphotyrosine and VHL. Ponceau S staining of the nitrocellulose was also performed. The results are representative of 3 independent experiments. (B) The major site of tyrosine phosphorylation on pVHL (Y185) is shown here marked with an asterisk. Tyrosine phosphorylation on GST-VHL phosphorylated in vitro using purified Src kinase was mapped through mass spectrometric analysis involving a combination of methods, including matrix-assisted laser desorption/ionization time of flight (MALDI-TOF), electrospray ionization, and tandem mass spectrometry (MS/MS) fragmentation/sequence determination (performed by the Proteomics Centre at the University of Victoria, BC). See Supplementary Fig. S2 for additional details. (C) (i) HEK 293T cells were transfected with the indicated plasmids. Forty-eight hours posttransfection, cell lysates were prepared in 2% SDS, 50 mM Tris (pH 6.8) lysis buffer and boiled. Exogenous VHL was isolated by immunoprecipitation with anti-VHL polyclonal antibody, followed by Western blotting for VHL, phosphotyrosine, or ubiquitin. (ii) Western blotting on whole-cell lysate was also conducted for VHL, Src, and α-tubulin. The results are representative of 3 independent experiments. (D) HEK 293T cells were transfected with the indicated plasmids. Forty-eight hours posttransfection, the cells were treated with 100 µM final concentration of cycloheximide for 0 h, 2 h, 4 h, 8 h, 16 h, or 24 h. Western blotting was then conducted on whole-cell lysate for VHL, Src, and α-tubulin. The results are representative of 3 independent experiments.
When we coexpressed constitutively active Src with wild-type VHL (WT VHL) or with a Y185F point mutant of VHL (Y185F VHL), we found that Y185F VHL protein was more stable than WT VHL (Fig. 7 C and D). These results also showed that there was decreased tyrosine phosphorylation and ubiquitination when mutant Y185F VHL was coexpressed with activated Src in vivo (in intact cells; Fig. 7Ci). In contrast, in vivo tyrosine phosphorylation and ubiquitination on VHL was markedly increased when WT VHL was coexpressed with activated Src (Fig. 7Ci). Cycloheximide treatment experiments to measure protein stability further showed that Y185F VHL was resistant to Src-mediated degradation, relative to WT VHL (Fig. 7D). In total, the data indicated that Y185F mutation of VHL protein confers resistance to Src-directed degradation.
Discussion
Both Src protein and Src tyrosine kinase activity are frequently elevated in human cancers and cancer cell lines, and current evidence supports a role for Src in the development, growth, progression, and metastasis of a number of human cancers, including those of the breast,1,2 colon,3-6 brain,7,8 and pancreas.33,34 In this study, we have found that elevated Src activity upregulates VEGF production in breast cancer and other cell types, and this can occur independent of hypoxia. Because VEGF is a key regulator of angiogenesis, these results suggest that, in tumors expressing elevated levels of Src kinase, it is likely that VEGF production and tumor angiogenesis can be stimulated before the onset of tumor hypoxia, in a manner that is independent of tumor size. This would contribute to both the establishment and growth of the primary tumor, as well as aid in tumor metastases even in relatively small, early stage tumors.
It appears that one mechanism by which Src causes the upregulation of VEGF in our cell models is through enhanced degradation of VHL, as measured by both a reduction in VHL protein levels and a reduction in its half-life. We noticed that the reduction in VHL protein was accompanied by increased tyrosine phosphorylation and ubiquitination of VHL, suggesting this as a possible cause of the increased rate of VHL degradation. Src has been previously demonstrated to regulate other cellular proteins through phosphorylation and ubiquitin-mediated degradation. Src promotes disruption of cell-cell adhesions in epithelial cells through tyrosine phosphorylation, leading to ubiquitination and endocytosis of E-cadherin complexes.35 This process is thought to be mediated by binding of the Hakai E3 ligase to tyrosine phosphorylated E-cadherin. In a different study, Src activation and tyrosine phosphorylation of c-Cbl E3 ligase causes Cbl to undergo self-ubiquitination and proteasome-mediated destruction.36 It may be possible that either the Hakai and c-Cbl E3 ligases are involved in Src-directed degradation of VHL, and we are currently examining these possibilities.
Destruction of VHL and upregulation of VEGF has also been attributed to contributing to malignant phenotype of some liver and gastric cancers.37 In this study, it appeared that an E2 ubiquitin-conjugating enzyme (E2-EPF UCP) could form a complex with VHL and could catalyze ubiquitination of VHL, promoting the destruction of VHL via the 26S proteasome. The authors did not examine if there were specific upstream factors that have cooperated to facilitate the association between VHL and the E2 enzyme, and it is interesting to speculate that Src may play some role.
Accompanying the ubiquitination and degradation of VHL upon Src overexpression, we demonstrated that VHL also becomes tyrosine phosphorylated, suggesting that this phosphorylation plays a role in the aforementioned events. We went further to show that Src directly phosphorylates VHL and have identified this newly characterized phosphorylation site as tyrosine 185 within the α-domain of VHL. The α-domain is involved in the formation of the complex of proteins responsible for ubiquitin-mediated protein degradation, including elongin C. Mutants of the VHL protein that we generated by mutating this tyrosine to phenylalanine cannot be phosphorylated on this site and have increased intracellular stability upon Src overexpression, strongly supporting the importance of this site in regulating some aspects of VHL stability. Phosphorylation on tyrosine 185 of VHL might affect VHL stability by disrupting protein complex formation at the α-domain, thereby targeting VHL for proteolysis by other E3 ligase complexes or by VHL self-ubiquitination. Src binding to VHL might also contribute to displacing VHL from assembly with stabilizing VHL E3 ligase complex proteins (such as elongin C and cullin 2) to further facilitate VHL destabilization. Some support for this last possibility comes from our observation that in the presence of kinase dead Src, in some experiments we observed an intermediate level of VHL destabilization, suggesting that there may also be a kinase-independent function of Src that could play a partial role in VHL downregulation. Kinase dead Src binding to VHL may directly destabilize the VHL protein or may disrupt VHL association with proteins that would otherwise stabilize VHL. Kinase-independent Src functions have also been reported independently by other labs.38,39
Although we have shown that increased degradation of VHL appears to be a major mechanism by which Src regulates VEGF levels in our model systems, other mechanisms of Src-induced VEGF upregulation have also been reported. One report communicated that Src can also regulate VEGF production by promoting elevated HIF-1α expression in NIH3T3 cells, the colon cancer line HT29, and the osteosarcoma cell line Saos-2 through increased cap-dependent translation of HIF-1α.40 Another report showed Src involvement in both constitutive and EGF-induced upregulation of VEGF through activation of phosphatidylinositol 3′-kinase (PI3K)–AKT and p38 mitogen-activated protein kinase (MAPK) pathways in pancreatic cancer cell lines.41 Thus, it appears that several mechanisms may mediate Src-induced upregulation of VEGF from tumor cells. We have not examined if more than one of these pathways are functioning to regulate VEGF levels in each of our model systems, so we are currently unable to predict whether several of these pathways are functional in a single cell type or whether different cell types each possess single regulatory mechanisms for VEGF production.
In summary, we describe a key Src-VHL pathway that is likely to be involved in tumor angiogenesis and tumor progression. We found that Src promotes the destabilization of the VHL tumor suppressor protein, which ultimately facilitates Src induction of VEGF production from cells. This mechanism of Src-dependent destruction of VHL provides novel insights into our understanding of interplay between an oncogenic protein and tumor suppressor and reveals a new mechanism of Src kinase function in targeting a tumor suppressor protein. Determining the relative contributions of the components of the Src oncoprotein-VHL tumor suppressor mechanism will be undoubtedly important in adding to our understanding of the molecular bases of cancer.
Materials and Methods
Cell Culture
The mammalian cell lines, unless otherwise stated, were obtained from American Type Culture Collection (ATCC, Manassas, VA) and were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum and antibiotics (100 units/mL penicillin, 100 µg/mL streptomycin, and 0.25 µg/mL amphotericin) (Gibco Invitrogen) at 37°C in 5% CO2. Human mammary epithelial control cell lines MCF-10-A cells were maintained in DMEM/Ham’s F12 (1:1, Gibco Invitrogen) supplemented with 5% horse serum (Sigma, St. Louis, MO), 2 mM L-glutamine (Gibco Invitrogen), 20 ng/mL epidermal growth factor (Gibco Invitrogen), 100 ng/mL cholera toxin (Calbiochem, San Diego, CA), 0.01 mg/mL insulin (Sigma), 500 ng/mL hydrocortisone (Sigma), and antibiotics (100 units/mL penicillin, 100 µg/mL streptomycin, and 0.25 µg/mL amphotericin) (Gibco Invitrogen).
Materials
Anti-Src mouse monoclonal antibody 327 was purified from hybridomas provided by Dr. Joan Brugge (Harvard). Anti-VHL (FL 1-181) rabbit polyclonal antibody and anti-VEGF (A-20) rabbit polyclonal antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-VHL (1g32) mouse monoclonal antibody was purchased from BD Biosciences Pharmingen (San Diego, CA). Anti-Flag M2 mouse monoclonal antibody was purchased from Sigma. Anti-HIF-1α mouse monoclonal antibody was purchased from BD Biosciences Transduction Laboratories. Anti-α-tubulin (DM1A, CP06) mouse monoclonal antibody was purchased from Calbiochem. Antiphosphotyrosine (4G10) mouse monoclonal antibody was provided by Dr. Stephen Robbins. Antiubiquitin (Ubi-1) mouse monoclonal antibody was purchased from Zymed Invitrogen (Carlsbad, CA). MG-132 and PP2 were purchased from Calbiochem. Cycloheximide was purchased from Sigma. Purified baculovirus-expressed human Src kinase was prepared in our lab (available from Millipore, Billerica, MA, product 14-117).
Plasmids
Active Src mutant pCI-CMVp-Y530F and the kinase dead Src pCI-CMVp-K298M were generated in our laboratory using QuickChange® site-directed mutagenesis (Stratagene, La Jolla, CA). The Flag-tagged VHL-GFP expression plasmid (pcDNA3.1-CMVp-Flag-VHL-GFP) and Flag-GFP-GFP control expression plasmid (pcDNA3.1-CMVp-Flag-GFP-GFP) were kindly provided by Dr. Stephen Lee (University of Ottawa) and have been described previously.42 The GST-VHL (glutathione S-transferase–VHL) fusion construct was generated by subcloning VHL cDNA from pcDNA3.1-CMVp-Flag-VHL-GFP into pGEX-4T-3 vector (AMRAD Corporation, Melbourne, Australia). Mutant Y185F VHL was generated using QuickChange® site-directed mutagenesis (Stratagene). HREx3-Luciferase reporter construct (pGL3-HREx3-SV40p-Luciferase) was a gift from Dr. Roland Wenger (University of Zurich). The LNCX-β-galactosidase expression construct (LNCX-CMVp-β-galactosidase) was from Clontech (Mountain View, CA).
Transfections
Transfections were conducted as described previously.6 Forty-eight hours posttransfection, the cells were lysed in 1% NP-40 lysis buffer (150 mM NaCl, 50 mM Tris [pH 7.5], 1% Nonidet P-40, 2 mM EDTA, 50 µg/mL leupeptin, 10 µg/mL aprotinin, 200 µM sodium orthovanadate, 4 mg/mL p-nitrophenyl phosphate).
Immunoprecipitations
Immunoprecipitations were conducted as detailed previously.43 Immunoprecipitations were performed by adding 3 µg of antibody per 1-mg cell extract sample. In the case of immunoprecipitations to detect ubiquitination and phosphorylation, immunoprecipitations were conducted on boiled SDS lysates (2% SDS, 50 mM Tris [pH 6.8] lysis buffer).
Cycloheximide Treatment Half-Life Experiments
Cells were seeded on 60-mm plates and subjected to appropriate transfections. Forty-eight hours posttransfection, the cells were treated with 100 µM final concentration of cycloheximide (Sigma) for 0 h, 2 h, 4 h, 8 h, 16 h, or 24 h. The cells were then lysed in 1% NP-40 lysis buffer and subjected to SDS-PAGE and Western blotting.
35S Metabolic Labeling and Chase Experiments
Cells were starved in methionine (Met)- and cysteine (Cys)–free medium for 2 h, then labeled in Met- and Cys-free DMEM medium (Gibco Invitrogen) containing 100 µCi/mL 35S-Met and 35S-Cys Promix cell-labeling mix (Amersham Biosciences) for 16 h to maximize labeled 35S-Met and 35S-Cys incorporation. After labeling, the cells were then washed 2 times and chased with complete DMEM containing 10% fetal bovine serum and unlabeled Met and Cys at 20-fold excess relative to the normal concentration in DMEM for the indicated times. Cell lysates were prepared in 1% NP-40 lysis buffer, and the cell lysates were immunoprecipitated with anti-VHL polyclonal antibody. The immune complexes were then resolved by SDS-PAGE. Subsequently, the gels were fixed, treated with ENHANCE solution (Dupont NEN, Boston, MA) according to the manufacturer’s instructions, dried, and subjected to autoradiography.
Fluorescence Microscopy
Immunostaining was conducted as detailed previously.44 Immunostaining for Src was performed for 1 h in a humidified chamber, using anti-Src 327 monoclonal antibody (1:100 dilution) in 1% bovine serum albumin (BSA) in phosphate-buffered saline (PBS). Flag-VHL-GFP was detected by GFP fluorescence. Immunofluorescence was then visualized with 100x objective using a conventional Zeiss microscope. Photos were taken using a Photometrics CoolSNAPcl (Roper Scientific, Tucson, AZ) camera and RS Image software (Roper Scientific).
siRNA Experiments
Unless otherwise stated, siRNAs were constructed from complementary strands of 21 nucleotide single-stranded RNAs synthesized by Dr. Richard Pon (University of Calgary Oligonucleotide Synthesis Facility). GFP siRNA was purchased from Xeragon (Valencia, CA). siRNA transfections were conducted as described previously.6
Luciferase Assays
Luciferase assays were conducted using the HREx3-Luciferase reporter plasmid as described previously.45
Endothelial Tube Formation Assays
The immortalized human endothelial cell line, EA.hy926, was a gift from Dr. Cora-Jean S. Edgell (University of North Carolina). This cell line maintains angiogenic behavior and shows sustained expression of many differentiated functions of endothelium.46,47 Endothelial tube formation assays were performed as detailed in published protocols.29,30 In summary, HEK 293T cells were transfected with the plasmids of interest. Forty-eight hours posttransfection, conditioned media from each transfection condition were collected for endothelial tube formation assays. EA.hy926 endothelial cells were then seeded at 4 × 103 per well of a 96-well tissue culture plate onto the surface of 100-µL polymerized ECM matrix (extracellular matrix from Engelbreth Holm-Swarm mouse sarcoma, BD Biosciences) per well. Then, 100 µL of conditioned media from each transfection condition was then overlaid onto each well of endothelial cells seeded on the ECM matrix. After a 16-h incubation at 37°C, the extent of formation of capillary networks for each condition was photographed under an inverted light microscope at 100x magnification. Quantification of capillary network formation was done by measurement of total tube length per well using ImageJ image data analysis software.
CAM Assays
Fertilized chicken eggs (Ijtsma Farms, Airdire, Alberta) were incubated at 37°C under conditions of constant humidity. CAM assays were conducted as described previously.31 On embryonic day 4, the fertilized chicken eggs were cracked, and chick embryos with intact yolks were placed into 100 × 20-mm culture dishes and returned to incubation. On embryonic day 6, 3 × 105 HEK 293T cells from each transfection condition were loaded onto 5-mm3 gelatin sponges (Johnson & Johnson, New Brunswick, NJ) and implanted onto the surface of the developing CAM eggs. The CAM eggs were then returned to incubation. On embryonic day 8, CAM vasculature was digitally photographed. Quantitation of angiogenesis was done by measuring length of vasculature per 707 mm2 field, using ImageJ image data analysis software. Four fields per CAM were measured for 9 CAMs per transfection condition.
Supplementary Material
Acknowledgments
The authors are grateful to Stephen Lee for the Flag-VHL-GFP expression vector, Roland Wenger for the HREx3-Luc reporter plasmid, Joan Brugge for the anti-Src 327 monoclonal hybridoma, Steve Robbins for 4G10 antibody, and Cora-Jean S. Edgell for the EA.hy926 endothelial cell line. They are grateful to Shirin Bonni, Marvin Fritzler, Dallan Young, Michael Ohh, Derek Smith, and Kamala Patel for helpful discussions. They also thank Laura Robson and Rebecca Leong-Quong for their technical assistance.
Footnotes
The authors declared no potential conflicts of interest with respect to the authorship and/or publication of this article.
This work was supported by grants to DJF from the Canadian Breast Cancer Foundation (Prairies/Northwest Territories Chapter), the Alberta Cancer Board, the Canadian Breast Cancer Research Alliance, and the Canadian Institutes of Health Research.
Supplementary material for this article is available on the Genes & Cancer Web site (http://ganc.sagepub.com/supplemental).
References
- 1. Verbeek BS, Vroom TM, Adriaansen-Slot SS, Ottenhoff-Kalff AE, Geertzema JG, Hennipman A, et al. c-Src protein expression is increased in human breast cancer: an immunohistochemical and biochemical analysis. J Pathol 1996;180(4):383-8 [DOI] [PubMed] [Google Scholar]
- 2. Egan C, Pang A, Durda D, Cheng HC, Wang JH, Fujita DJ. Activation of Src in human breast tumor cell lines: elevated levels of phosphotyrosine phosphatase activity that preferentially recognizes the Src carboxy terminal negative regulatory tyrosine 530. Oncogene 1999;18(5):1227-37 [DOI] [PubMed] [Google Scholar]
- 3. Bolen JB, Veillette A, Schwartz AM, DeSeau V, Rosen N. Analysis of pp60c-src in human colon carcinoma and normal human colon mucosal cells. Oncogene Res 1987;1(2):149-68 [PubMed] [Google Scholar]
- 4. Cartwright CA, Meisler AI, Eckhart W. Activation of the pp60c-src protein kinase is an early event in colonic carcinogenesis. Proc Natl Acad Sci USA 1990;87(2):558-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Irby RB, Mao W, Coppola D, Kang J, Loubeau JM, Trudeau W, et al. Activating SRC mutation in a subset of advanced human colon cancers. Nat Genet 1999;21(2):187-90 [DOI] [PubMed] [Google Scholar]
- 6. Zhu S, Bjorge JD, Fujita DJ. PTP1B contributes to the oncogenic properties of colon cancer cells through Src activation. Cancer Res 2007;67(21):10129-37 [DOI] [PubMed] [Google Scholar]
- 7. Ding Q, Stewart J, Jr, Olman MA, Klobe MR, Gladson CL. The pattern of enhancement of Src kinase activity on platelet-derived growth factor stimulation of glioblastoma cells is affected by the integrin engaged. J Biol Chem 2003;278(41):39882-91 [DOI] [PubMed] [Google Scholar]
- 8. Weissenberger J, Loeffler S, Kappeler A, Kopf M, Lukes A, Afanasieva TA, et al. IL-6 is required for glioma development in a mouse model. Oncogene 2004;23(19):3308-16 [DOI] [PubMed] [Google Scholar]
- 9. Visser CJ, Rijksen G, Woutersen RA, de Weger RA. Increased immunoreactivity and protein tyrosine kinase activity of the protooncogene pp60c-src in preneoplastic lesions in rat pancreas. Lab Invest 1996;74(1):2-11 [PubMed] [Google Scholar]
- 10. Lutz MP, Esser IB, Flossmann-Kast BB, Vogelmann R, Luhrs H, Friess H, et al. Overexpression and activation of the tyrosine kinase Src in human pancreatic carcinoma. Biochem Biophys Res Commun 1998;243(2):503-8 [DOI] [PubMed] [Google Scholar]
- 11. Bjorge JD, Pang A, Fujita DJ. Identification of protein-tyrosine phosphatase 1B as the major tyrosine phosphatase activity capable of dephosphorylating and activating c-Src in several human breast cancer cell lines. J Biol Chem 2000;275(52):41439-46 [DOI] [PubMed] [Google Scholar]
- 12. Mukhopadhyay D, Tsiokas L, Zhou XM, Foster D, Brugge JS, Sukhatme VP. Hypoxic induction of human vascular endothelial growth factor expression through c-Src activation. Nature 1995;375(6532):577-81 [DOI] [PubMed] [Google Scholar]
- 13. Ellis LM, Staley CA, Liu W, Fleming RY, Parikh NU, Bucana CD, et al. Down-regulation of vascular endothelial growth factor in a human colon carcinoma cell line transfected with an antisense expression vector specific for c-src. J Biol Chem 1998;273(2):1052-7 [DOI] [PubMed] [Google Scholar]
- 14. Wiener JR, Nakano K, Kruzelock RP, Bucana CD, Bast RC, Jr, Gallick GE. Decreased Src tyrosine kinase activity inhibits malignant human ovarian cancer tumor growth in a nude mouse model. Clin Cancer Res 1999;5(8):2164-70 [PubMed] [Google Scholar]
- 15. Weis S, Shintani S, Weber A, Kirchmair R, Wood M, Cravens A, et al. Src blockade stabilizes a Flk/cadherin complex, reducing edema and tissue injury following myocardial infarction. J Clin Invest 2004;113(6):885-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Semenza GL. Expression of hypoxia-inducible factor 1: mechanisms and consequences. Biochem Pharmacol 2000;59(1):47-53 [DOI] [PubMed] [Google Scholar]
- 17. Guillemin K, Krasnow MA. The hypoxic response: huffing and HIFing. Cell 1997;89(1):9-12 [DOI] [PubMed] [Google Scholar]
- 18. Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol 1992;12(12):5447-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cavallaro U, Christofori G. Molecular mechanisms of tumor angiogenesis and tumor progression. J Neurooncol 2000;50(1-2):63-70 [DOI] [PubMed] [Google Scholar]
- 20. Neufeld G, Kessler O, Vadasz Z, Gluzman-Poltorak Z. The contribution of proangiogenic factors to the progression of malignant disease: role of vascular endothelial growth factor and its receptors. Surg Oncol Clin North Am 2001;10(2):339-56, ix [PubMed] [Google Scholar]
- 21. Kaelin WG., Jr The von Hippel–Lindau tumor suppressor protein and clear cell renal carcinoma. Clin Cancer Res 2007;13(pt 2):680s-4s [DOI] [PubMed] [Google Scholar]
- 22. Kaelin WG., Jr Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer 2002;2(9):673-82 [DOI] [PubMed] [Google Scholar]
- 23. Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML, et al. Identification of the von Hippel–Lindau disease tumor suppressor gene. Science 1993;260(5112):1317-20 [DOI] [PubMed] [Google Scholar]
- 24. Chen F, Kishida T, Yao M, Hustad T, Glavac D, Dean M, et al. Germline mutations in the von Hippel–Lindau disease tumor suppressor gene: correlations with phenotype. Hum Mutat 1995;5(1):66-75 [DOI] [PubMed] [Google Scholar]
- 25. Zbar B, Kishida T, Chen F, Schmidt L, Maher ER, Richards FM, et al. Germline mutations in the Von Hippel–Lindau disease (VHL) gene in families from North America, Europe, and Japan. Hum Mutat 1996;8(4):348-57 [DOI] [PubMed] [Google Scholar]
- 26. Neumann HP, Bender BU. Genotype-phenotype correlations in von Hippel–Lindau disease. J Intern Med 1998;243(6):541-5 [DOI] [PubMed] [Google Scholar]
- 27. Fleming RY, Ellis LM, Parikh NU, Liu W, Staley CA, Gallick GE. Regulation of vascular endothelial growth factor expression in human colon carcinoma cells by activity of src kinase. Surgery 1997;122(2):501-7 [DOI] [PubMed] [Google Scholar]
- 28. Pause A, Lee S, Lonergan KM, Klausner RD. The von Hippel–Lindau tumor suppressor gene is required for cell cycle exit upon serum withdrawal. Proc Natl Acad Sci USA 1998;95(3):993-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Troyanovsky B, Levchenko T, Mansson G, Matvijenko O, Holmgren L. Angiomotin: an angiostatin binding protein that regulates endothelial cell migration and tube formation. J Cell Biol 2001;152(6):1247-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Potente M, Urbich C, Sasaki KI, Hofmann WK, Heeschen C, Aicher A, et al. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J Clin Invest 2005;115(9):2382-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kwon M, Yoon CS, Fitzpatrick S, Kassam G, Graham KS, Young MK, et al. p22 is a novel plasminogen fragment with antiangiogenic activity. Biochemistry 2001;40(44):13246-53 [DOI] [PubMed] [Google Scholar]
- 32. Songyang Z, Carraway KL, III, Eck MJ, Harrison SC, Feldman RA, Mohammadi M, et al. Catalytic specificity of protein-tyrosine kinases is critical for selective signalling. Nature 1995;373(6514):536-9 [DOI] [PubMed] [Google Scholar]
- 33. Visser CJ, Rijksen G, Woutersen RA, de Weger RA. Increased immunoreactivity and protein tyrosine kinase activity of the protooncogene pp60c-src in preneoplastic lesions in rat pancreas. Lab Invest 1996;74(1):2-11 [PubMed] [Google Scholar]
- 34. Lutz MP, Esser IB, Flossmann-Kast BB, Vogelmann R, Luhrs H, Friess H, et al. Overexpression and activation of the tyrosine kinase Src in human pancreatic carcinoma. Biochem Biophys Res Commun 1998;243(2):503-8 [DOI] [PubMed] [Google Scholar]
- 35. Fujita Y, Krause G, Scheffner M, Zechner D, Leddy HE, Behrens J, et al. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol 2002;4(3):222-31 [DOI] [PubMed] [Google Scholar]
- 36. Bao J, Gur G, Yarden Y. Src promotes destruction of c-Cbl: implications for oncogenic synergy between Src and growth factor receptors. Proc Natl Acad Sci USA 2003;100(5):2438-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jung CR, Hwang KS, Yoo J, Cho WK, Kim JM, Kim WH, et al. E2-EPF UCP targets pVHL for degradation and associates with tumor growth and metastasis. Nat Med 2006;12(7):809-16 [DOI] [PubMed] [Google Scholar]
- 38. Schwartzberg PL, Xing L, Hoffmann O, Lowell CA, Garrett L, Boyce BF, et al. Rescue of osteoclast function by transgenic expression of kinase-deficient Src in src-/- mutant mice. Genes Dev 1997;11(21):2835-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kaplan KB, Swedlow JR, Morgan DO, Varmus HE. c-Src enhances the spreading of src-/- fibroblasts on fibronectin by a kinase-independent mechanism. Genes Dev 1995;9(12):1505-17 [DOI] [PubMed] [Google Scholar]
- 40. Karni R, Dor Y, Keshet E, Meyuhas O, Levitzki A. Activated pp60c-Src leads to elevated hypoxia-inducible factor (HIF)-1alpha expression under normoxia. J Biol Chem 2002;277(45):42919-25 [DOI] [PubMed] [Google Scholar]
- 41. Summy JM, Trevino JG, Baker CH, Gallick GE. c-Src regulates constitutive and EGF-mediated VEGF expression in pancreatic tumor cells through activation of phosphatidyl inositol-3 kinase and p38 MAPK. Pancreas 2005;31(3):263-74 [DOI] [PubMed] [Google Scholar]
- 42. Groulx I, Bonicalzi ME, Lee S. Ran-mediated nuclear export of the von Hippel–Lindau tumor suppressor protein occurs independently of its assembly with cullin-2. J Biol Chem 2000;275(12):8991-9000 [DOI] [PubMed] [Google Scholar]
- 43. Chou MT, Wang J, Fujita DJ. Src kinase becomes preferentially associated with the VEGFR, KDR/Flk-1, following VEGF stimulation of vascular endothelial cells. BMC Biochem 2002;3(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jakymiw A, Raharjo E, Rattner JB, Eystathioy T, Chan EK, Fujita DJ. Identification and characterization of a novel Golgi protein, golgin-67. J Biol Chem 2000;275(6):4137-44 [DOI] [PubMed] [Google Scholar]
- 45. Babic I, Cherry E, Fujita DJ. SUMO modification of Sam68 enhances its ability to repress cyclin D1 expression and inhibits its ability to induce apoptosis. Oncogene 2006;25(36):4955-64 [DOI] [PubMed] [Google Scholar]
- 46. Edgell CJ, McDonald CC, Graham JB. Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc Natl Acad Sci USA 1983;80(12):3734-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ferri C, Pittoni V, Piccoli A, Laurenti O, Cassone MR, Bellini C, et al. Insulin stimulates endothelin-1 secretion from human endothelial cells and modulates its circulating levels in vivo. J Clin Endocrinol Metab 1995;80(3):829-35 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.