

Abstract
Respiratory viral infections cause significant morbidity and increase the risk for chronic pulmonary graft-versus-host disease (GVHD) after hematopoietic cell transplantation (HCT). Our overall hypothesis is that local innate immune activation potentiates adaptive alloimmunity. In this study, we hypothesized that a viral pathogen-associated molecular pattern (PAMP) alone can potentiate pulmonary GVHD after allogeneic HCT. We, therefore, examined the effect of pulmonary exposure to polyinosinic:polycytidylic acid (poly I:C), a viral mimetic that activates innate immunity, in an established murine HCT model. Poly I:C induced a marked pulmonary T cell response in allogeneic HCT mice as compared to syngeneic HCT, with increased CD4+ cells in the lung fluid and tissue. This lymphocytic inflammation persisted at 2 weeks post poly I:C exposure in allogeneic mice and was associated with CD3+ cell infiltration into the bronchiolar epithelium and features of epithelial injury. In vitro, poly I:C enhanced allospecific proliferation in a mixed lymphocyte reaction. In vivo, poly I:C exposure was associated with an early increase in pulmonary monocyte recruitment and activation as well as a decrease in CD4+FOXP3+ regulatory T cells in allogeneic mice as compared to syngeneic. In contrast, intrapulmonary poly I:C did not alter the extent of systemic GVHD in either syngeneic or allogeneic mice. Collectively, our results suggest that local activation of pulmonary innate immunity by a viral molecular pattern represents a novel pathway that contributes to pulmonary GVHD after allogeneic HCT, through a mechanism that includes increased recruitment and maturation of intrapulmonary monocytes.
Keywords: poly I:C, pulmonary graft-versus-host disease, allogeneic, lymphocytic bronchiolitis, respiratory viral infection, monocytes
1. INTRODUCTION
Graft-versus-host disease (GVHD) is the primary obstacle to successful long-term outcomes after hematopoietic cell transplantation (HCT) (1-3). While GVHD affects multiple organ systems, pulmonary manifestations carry significant morbidity and mortality and are increasingly recognized due to the improving short-term HCT survival (4-8). Histologically, pulmonary GVHD is associated with lymphocytic bronchiolitis (LB) and, in its chronic form, bronchiolitis obliterans (BO) (9, 10). Lymphocytic bronchiolitis and BO are also observed following lung transplantation and, due to immunological similarities between the two transplant settings, likely occurs as a result of similar mechanisms (11-13). Chronic pulmonary GVHD is generally progressive, responds poorly to medical intervention, and is associated with mortality as high as 50% at 5 years (14).
Respiratory viral infections (RVIs) are an increasingly recognized risk factor for chronic pulmonary GVHD amongst allogeneic HCT recipients (15, 16). A recent prospective study of pediatric allogeneic HCT patients found that RVIs were predictive of eventual pulmonary GVHD development (5). Similarly, RVIs have also been implicated in the development of acute and chronic rejection after human lung transplantation (17, 18). In general these processes are thought to be mediated by adaptive immune responses to the virus through mechanisms such as epitope spreading or T-cell cross-reactivity.
Our group, however, has developed the novel hypothesis that local activation of pulmonary innate pattern recognition receptors critically regulates alloimmune lung disease. In support of this idea, studies by our group and others have shown that polymorphisms in genes of innate pattern recognition pathways modulate allorecognition in various transplant settings. Specifically, polymorphic variation in lipopolysaccharide (LPS)-binding proteins has been shown to influence the development of chronic airway disease after human HCT (19). Similarly, we have demonstrated that Toll-like receptor-4 (TLR4) and CD14 polymorphisms that exacerbate or attenuate the innate response to LPS are associated with increased or decreased rates of human lung allograft rejection, respectively (20-22). In addition, we have further established that local pulmonary LPS treatment potentiates lymphocytic lung inflammation as a manifestation of pulmonary GVHD after murine HCT (23). Further studies in non-pulmonary transplantation, particularly in models of costimulatory blockade-induced tolerance, also support the importance of innate immune activation through TLR3, TLR7, and TLR9 in allograft rejection (24-26).
2. OBJECTIVE
In the current study, we sought to extend our previous results and test the hypothesis that that viral innate immune activation alone could potentiate pulmonary GVHD after allogeneic HCT. If correct, our hypothesis would identify a novel mechanism by which viral infections could enhance alloimmune lung injury, independent of an established antiviral adaptive immune response, a finding clinically relevant to HCT or lung transplant recipients. Therefore, we examined the effect of intrapulmonary polyinosinic:polycytidylic acid (poly I:C), a synthetic double-stranded RNA that mimics viral innate immune activation (27, 28), in an established murine HCT model.
3. MATERIALS AND METHODS
3.1. Mice
Male 7-10 week-old C57Bl/6J (H2b) and C3HeB/FeJ (H2k) mice were purchased from Jackson Laboratories (Bar Harbor, ME) and were allowed to acclimate for 7 days prior to use. All animals were housed in a pathogen-free facility on LPS-free bedding (Shepherd Specialty Papers Inc., Kalamazoo, MI), with irradiated food (PicoLab Mouse Diet 20 5058, Purina Mills, Richmond, IN) and antibiotic water (Sulfamethoxazole/Trimethoprim 1.2/0.24 mg/mL). Experimental groups included 3-6 mice. Experiments were approved by the Institutional Animal Care and Use Committees.
3.2. Mixed leukocyte reactions (MLR)
24 hours prior to euthanasia, stimulator C57Bl/6J mice were injected intraperitoneally with 400 μL of DMEM media (Sigma-Aldrich, St. Louis, MO) alone or containing 400 μg of poly I:C. Mice were euthanized using CO2. Splenocytes were isolated via spleen homogenization and filtration and subsequently washed in media, filtered through 70 μm filters (BD, Franklin Lakes, NJ), counted on a hemocytometer, and resuspended in RPMI 1640 (Gibco, Grand Island, NY) containing Penicillin/Streptomycin (Sigma-Aldrich, St. Louis, MO), L-Glutamine (Sigma-Aldrich, St. Louis, MO), and 10% fetal bovine serum (FBS) (Hyclone, Logan, UT). 5×105 C57Bl/6J stimulator splenocytes (irradiated in a Cesium irradiator at 20 Gy) were co-cultured with 1.5×105, 2.5×105, or 5×105 C3HeB/FeJ or C57Bl/6J responder splenocytes. 1 μCi of 3H-thymidine was added for the last 18 hours of culture. 4 days following initial culture, splenocytes were harvested (TomTec harvester 96 Mach III M, Hamden, CT) and radioactivity was measured using a β-counter (Wallac Trilux 1450 Microbeta counter, Waltham, MA). Results are reported as counts per minute (cpm) from wells with 5×105 responder splenocytes. Proliferation was determined by subtracting cpm of responders alone from cpm in wells containing both responders and stimulators.
3.3. Murine hematopoietic cell transplantation (HCT)
C57Bl/6J recipient animals underwent HCT as previously described (23). Briefly, donor mice were euthanized using CO2. Bone marrow was isolated from tibia and femurs and splenocytes were isolated from spleens via homogenization and filtration. All donor cells were washed in media, filtered through 70 μm filters (BD, Franklin Lakes, NJ), counted on a hemocytometer, and resuspended in media containing 10% FBS (Hyclone, Logan, UT), 1% L-glutamine (Sigma-Aldrich, St. Louis, MO) and 1% penicillin/streptomycin (Sigma-Aldrich, St. Louis, MO). Recipient mice were lethally irradiated with 9.5 Gy of Cesium irradiation and intravenously injected via the retro-orbital route with 4×106 bone marrow cells and 1×106 splenocytes from either C57Bl/6J (Syn) or C3HeB/FeJ (Allo). Allogeneic and syngeneic HCT were performed under identical conditions. After HCT, animals were allowed to recover for 4 weeks before further experimental use. In the first 4 weeks, mortality in the allogeneic group was 14% and 3% in the syngeneic group. Following necropsy, gastrointestinal histology was evaluated for inflammation using an established semi-quantitative scoring system (29).
Recipient engraftment was evaluated between 3 and 4 weeks following transplantation. Blood was obtained using maxillary sinus puncture. Cells were stained with anti-H2Kk-FITC, anti-H2Db-PE, anti-CD3-PE-Cy7, and anti-CD11b-APC (BioLegend, San Diego, CA). Flow cytometry was performed as described below. Animals with greater than 90% donor-derived peripheral blood cells were used for experimental exposures.
3.4. Poly I:C Exposures
LPS-free poly I:C (Invivogen, San Diego, CA) was dissolved in LPS-free water. A Limulus amoebocyte lysate assay (Lonza, Basel, Switzerland) performed in-house confirmed absence of LPS contamination. Mice were anesthetized with 1% isoflurane. Intrapulmonary administration of 40 μL of LPS-free water alone or containing 100 μg of poly I:C was performed via oropharyngeal aspiration. Mice were euthanized either 72 hours or 2 weeks after poly I:C exposure.
3.5. Bronchoalveolar lavage (BAL) and analysis
Mice were euthanized using CO2 and their lungs were surgically exposed. The trachea was cannulated and the lungs were lavaged with five 800-μL aliquots of 0.9% saline (VWR International, West Chester, PA). BAL supernatant was analyzed using a mouse 23-plex cytokine assay (Bio-Rad Laboratories, Hercules, CA). BAL cells underwent red blood cell lysis, counting using a hemocytometer, and resuspension in flow cytometry buffer for staining. Flow cytometry was performed as described below.
3.6. Lung tissue extraction and preservation
Lungs were perfused with 0.9% saline. The accessory (smallest) lobe of the right lung was stored in RNAlater (Ambion/Applied Biosystems, Austin, TX) for 24 hours, then stored at −80°C. For flow cytometry, the remaining right lung was placed in a HEPES-based buffer (Sigma-Aldrich, St. Louis, MO), digested with collagenase-A (Roche Diagnostics, Mannheim, Germany) and DNAse-I (Sigma-Aldrich, St. Louis, MO), filtered through a 70 μm filter (BD, Franklin Lakes, NJ), red blood cell-lysed, washed, and resuspended in 500 μL of flow cytometry buffer containing phosphate-buffered sodium solution (Sigma-Aldrich, St. Louis, MO) with 3% FBS (Hyclone, Logan, UT), 0.05% sodium azide (VWR International, West Chester, PA), and 10 mM ethylenediaminetetraacetic acid (EDTA, Sigma-Aldrich, St. Louis, MO). The left lung was gravity-inflated with 10% formalin (VWR International, West Chester, PA), fixed in 10% formalin for 24 hours and then transferred into 70% EtOH.
3.7. Lung Histology
After fixation, the left lung was paraffin-embedded and 5 μm sections were either stained with hematoxylin and eosin (H&E) or deparaffinized for further staining as previously described (30). CD3-immunohistochemistry was performed with rabbit anti-mouse CD3 antibody (Thermo Scientific, Fremont, CA) used at a dilution of 1:150 and developed with horseradish peroxidase-conjugated streptavidin (Vector Laboratory, Burlingame, CA) and then diaminobenzidine (Dako, Carpinteria, CA). Immunofluorescence was performed with mouse anti-mouse β-catenin (BD Transduction Laboratories, BD, Franklin Lakes, NJ) and goat anti-mouse-Alexa-488 (Molecular Probes, Eugene, OR) antibodies, both used at a dilution of 1:400. Stained sections were mounted with Fluoromount-G containing 2 μg/ml 4,6-diamidino-2-phenylindole (DAPI, Sigma-Aldrich, Saint Louis, MO).
3.8. RNA Analysis
RNA was isolated from RNAlater-preserved lung tissue using a commercially available kit (Ambion/Applied Biosystems, Austin, TX). Spectrophotometric analysis was used to assess RNA quantity and Bio-RAD Experion chips (Bio-Rad Laboratories, Hercules, CA) were used to assess RNA quality. cDNA was transcribed using the Applied Biosystems cDNA reverse transcription kit with the addition of a broad-range RNAse inhibitor (Applied Biosystems, Foster City, CA). Transcripts were quantified in triplicate from 2 ng cDNA using the Eppendorf cDNA reverse transcription kit (Eppendorf, Hauppauge, NY) and Taqman probe and primer combinations for β-catenin (Mm00483039_m1), vimentin (Mm01333430_m1), fibronectin (Mm01256734_m1), collagen type I (Mm00801666_g1), E-cadherin (Mm01247357_m1), and the endogenous control β-actin (4352933E) (Applied Biosystems, Foster City, CA). Change in expression was calculated using the 2−ΔΔCt method, normalized to β-actin expression, and expressed as fold-change as compared to commercially-available normal mouse lung total RNA (Ambion/Applied Biosystems, Austin, TX).
3.9. Flow Cytometry
Cells underwent a 10-minute incubation in flow cytometry buffer containing 5% normal mouse serum and 5% normal rat serum (Jackson ImmunoResearch Laboratories Inc., West Grove, PA) and 1% Fc receptor block (Affinity purified anti-mouse CD16/32) (eBioscience, San Diego, CA). Cells were subsequently stained with one of 5 anti-mouse antibody panels (eBioscience, San Diego, CA). The lymphocyte panel included anti-CD3-FITC, anti-H2Kk-PE, anti-CD4-PE-Cy7, anti-DX5-APC, anti-CD8-APC-Cy7, and anti-CD19-PE-Cy5.5; the first myeloid panel included anti-MHCII(IA/IE)-FITC, anti-Ly6G-PE, anti-CD11b-PE-Cy7, anti-CD3-APC, anti-Gr1-APC-Cy7, and anti-CD11c-PE-Cy5.5; the second myeloid panel included anti-MHCII(IA/IE)-FITC, anti-Ly6G-PE, anti-CD11b-PE-Cy7, anti-CD86-APC, anti-Gr1-APC-Cy7, and anti-CD11c-PE-Cy5.5; the regulatory T cell (Treg) panel included anti-CD3-FITC, anti-H2Kk-PE, anti-CD4-PE-Cy7, anti-CD8-APC-Cy7, and anti-CD25-PE-Cy5.5. After surface marker staining, Treg panel cells were fixed and permeabilized with fixation/permeabilization buffer (eBioscience, San Diego, CA) and stained with anti-FOXP3-APC. BAL samples and 72-hour HCT lung samples were analyzed individually. Splenocyte samples and myeloid-stained lung and BAL samples from two-week HCT mice were pooled by group. Reported cell counts consist of individually-analyzed flow cytometry BAL samples. Fluorescence was measured using BD FACS Canto II flow cytometer with BD FACSDiva software (BD, Franklin Lakes, NJ). Flow cytometry analysis was performed using FlowJo software (Tree Star Inc., Ashland, OR). Pulmonary myeloid cell gating was based on a previously published strategy (31). Cells were analyzed using a singlet gate to exclude cell aggregates, followed by an all-cell gate to exclude small debris and dead cells. Absolute numbers were obtained by multiplying the percentage of all cells by the live-cell counts done using Trypan dead-cell exclusion. Lymphocyte subsets were identified within a small-cell gate.
3.10. Statistical analysis
Data are expressed as means ± SEM. For each experiment, the Allo+poly I:C group was compared to the Allo+control and Syn+poly I:C groups using the Student’s t-test. P-values of 0.05 or less were considered significant.
4. RESULTS
4.1. Following allogeneic HCT, poly I:C induces acute and chronic lymphocytic lung inflammation
72 hours after poly I:C exposure, allogeneic HCT mice exposed to poly I:C (Allo+poly I:C) had significantly increased total cell numbers in their bronchoalveolar lavage (BAL) as compared to either allogeneic mice exposed to water (Allo+control) or syngeneic mice exposed to poly I:C (Syn+poly I:C) (Figure 1A). This increase in total cells primarily reflected a marked increase in CD3+ and CD4+ lymphocyte populations with a less prominent increase in CD8+ T cells (Figure 1B-D). Significant elevations in total cells as well as CD3+, CD4+ and CD8+ T lymphocytes persisted at 2 weeks in the BAL of Allo+poly I:C as compared to Allo+control or Syn+poly I:C (Figure 1E-H). The pattern of mononuclear cell recruitment in syngeneic mice resembled that of non-transplanted mice with early lymphocytic inflammation at 72 hours and complete normalization at 2 weeks (data not shown).
Figure 1. Increased and persistent bronchoalveolar lavage (BAL) T cells in allogeneic mice 72 hours and 2 weeks after poly I:C exposure.

BAL cells were evaluated by flow cytometry at both 72 hours (n = 6 mice/group) and two weeks (n = 3 mice/group) following poly I:C exposure in both allogeneic and syngeneic HCT mice. (A) At 72 hours, allogeneic mice exposed to poly I:C (Allo+poly I:C) had significantly more BAL total cells than either unexposed allogeneic mice (Allo+control) (p = 0.001) or poly I:C-exposed syngeneic mice (Syn+poly I:C) (p = 0.04). (B) Closely paralleling the increase in total BAL cells, CD3+ cells in Allo+poly I:C BAL were elevated above both unexposed allogeneic mice (Allo+control) (p = 0.007) and poly I:C-exposed syngeneic mice (Syn+poly I:C) (p = 0.02). (C) CD4+ BAL cells also increased in Allo+poly I:C BAL as compared to both Allo+control (p = 0.04) and Syn+poly I:C (p = 0.02) BAL while (D) CD8+ cells in allogeneic BAL increased following poly I:C exposure (p = 0.01) but did not differ significantly between Allo+poly I:C and Syn+poly I:C mice (p = 0.3). (E) Two weeks following poly I:C exposure, BAL total cells remained elevated in Allo+poly I:C BAL compared to both Allo+control (p = 0.02) and Syn+poly I:C (p = 0.01). (F) Allo+poly I:C BAL also continued to contain more CD3+ cells than both Allo+control (p = 0.005) and Syn+poly I:C (p = 0.003) controls. CD4+ and CD8+ BAL cells followed a similar pattern; Allo+poly I:C BAL contained (G) more CD4+ cells than both Allo+control (p = 0.003) and Syn+poly I:C (p < 0.001) and (H) more CD8+ cells than both Allo+poly I:C (p = 0.02) and Syn+poly I:C (p = 0.02).
On histopathological examination of lung tissue, at both the 72-hour and 2-week time points, poly I:C exposure led to perivascular and peribronchiolar mononuclear inflammation in allogeneic HCT, which was less pronounced in Syn+poly I:C mice and absent in unexposed mice (Figure 2A-D). Lung tissue cellular quantification by flow cytometry showed trends in T cell recruitment that were similar, but less striking, compared to the BAL (data not shown). Immunohistochemistry with anti-CD3 antibody staining confirmed the presence of T cells within the perivascular and peribronchiolar mononuclear infiltrates (Figure 2F, H).
Figure 2. Prominent peribronchiolar lymphocytic inflammation, epithelium-infiltrating CD3+ cells, and epithelial dysplasia in allogeneic HCT mice exposed to poly I:C (Allo+poly I:C).

Lungs were inflated and preserved two-weeks following poly I:C exposure. Representative sections photographed at 400x magnification are shown. H&E staining demonstrated minimal peribronchiolar cellular inflammation in both (A) unexposed syngeneic (Syn+control) and (B) unexposed allogeneic (Allo+control) lungs. Small, mononuclear cells in the peribronchiolar region were visible in sections from both (C) Syn+poly I:C and (D&E) Allo+poly I:C lungs, with more prominent cellular inflammation in Allo+poly I:C samples. (F) Additional immunohistochemistry revealed these peribronchiolar cells to be CD3-positive. Only Allo+poly I:C animals demonstrated epithelium-infiltrating CD3+ cells (black arrows) and epithelial dysplasia with several superimposed disorganized epithelial cell layers (white arrows) on both (E) H&E-stained and (F) CD3-stained sections. (G&H) Further digital enlargement of E&F (inset boxes) are representative images of infiltrating CD3+ cells (black arrows) and epithelial dysplasia (white arrows) seen throughout Allo+poly I:C lung tissue.
4.2. Poly I:C exposure induces epithelial T cell infiltration and epithelial injury in allogeneic HCT animals
Microscopic analysis of lung tissue showed that peribronchiolar T cells in Allo+poly I:C animals were infiltrating the epithelial cell layer, a phenomenon not observed in any of the control animals (Figure 2E-H). Immunohistochemistry with anti-CD3 antibody staining identified the epithelium-infiltrating mononuclear cells as CD3-positive (Figure 2F, H). In addition, Allo+poly I:C lung sections also demonstrated marked epithelial dysplasia with epithelial cell crowding and disorganization, changes not observed in lung sections from Allo+control or Syn+poly I:C mice (Figure 2A-E).
Given the epithelial changes observed in the Allo+poly I:C lungs, in order to further evaluate the extent of epithelial injury and assess the possibility of early epithelial to mesenchymal transition (EMT), we measured pulmonary expression of multiple markers of EMT in our model, including vimentin, fibronectin, collagen type I, E-cadherin, and β-catenin. At two weeks after treatment, β-catenin, an epithelial cell junctional protein, was significantly reduced in the lung tissue of Allo+poly I:C mice as compared to Allo+control lung tissue, while expression in Syn+poly I:C compared to Syn+control lung tissue was not significantly different after poly I:C treatment (Figure 3A). Immunofluorescent staining for β-catenin demonstrated large sections of diminished and disordered β-catenin in Allo+poly I:C lungs that aligned with sections of dysplastic epithelium and peribronchiolar mononuclear inflammation observed on the H&E and CD3-positive stained sections (Figure 3B-E), suggesting active epithelial injury.
Figure 3. Decreased and disorganized β-catenin in allogeneic HCT mice exposed to poly I:C (Allo+poly I:C).

β-catenin transcripts were measured using RT-PCR in lung tissue. In addition, sections of preserved and paraffin-embedded lungs were deparaffinized and stained for β-catenin (red) and DAPI (white). Representative sections photographed at 400x magnification are shown. (A) Allo+poly I:C lungs contained fewer transcripts for β-catenin compared to unexposed allogeneic lungs (Allo+control) (p < 0.001) while β-catenin expression in syngeneic lungs was unaffected by poly I:C exposure (p = 0.94). In comparison to (B-D) control groups, (E) only Allo+poly I:C lungs displayed sections of decreased β-catenin staining (white brackets) and irregular β-catenin staining (blue bracket).
4.3. Poly I:C increases monocyte recruitment and antigen presenting cell (APC) activation in allogeneic HCT
To understand whether the increased lymphocytic inflammation in Allo+poly I:C was associated with increased acute inflammation and antigen presentation, we used flow cytometry to identify neutrophils (defined as CD11c+, MHCII−, CD11b+, Ly6G+), monocytes (defined as CD11c−, MHCII−, CD11b+, Ly6G−), and APCs (defined as CD11c+, MHCII+, markers common to both macrophages and dendritic cells) (31). At 72 hours post poly I:C exposure, BAL neutrophils increased only modestly and to the same degree in both allogeneic and syngeneic mice (data not shown). In contrast, monocytes increased to a significantly higher degree in allogeneic compared to syngeneic HCT mice (Figure 4A, C). This increase was associated with increased levels of the monocyte chemotactic protein-1 (MCP-1/CCL2) in the BAL of Allo+poly I:C mice as compared to controls (Figure 4B). Furthermore, monocyte expression of the activation markers Gr1 and CD86 was significantly increased in Allo+poly I:C as compared to controls (Figure 4D-G), along with an increase in the percentage of monocytes that co-expressed both activation markers (Figure 4D). Although absolute numbers of APCs were not significantly increased, significant increases in APC expression of Gr1, CD86, as well as CD11b, were also found in Allo+poly I:C mice compared to controls (data not shown).
Figure 4. Increased BAL monocytes in allogeneic mice post poly I:C (Allo+poly I:C) with increased expression of activation markers.

BAL myeloid cells were evaluated by flow cytometry 72 hours following poly I:C exposure in both allogeneic and syngeneic HCT mice (n = 6 mice/group). (A) The total numbers of monocytes increased in Allo+poly I:C BAL above both unexposed allogeneic recipients (Allo+control) (p = 0.001) and poly I:C-exposed syngeneic recipients (Syn+poly I:C) (p = 0.04). (B) The monocyte chemotactic protein-1 (MCP-1/CCL2) chemokine was significantly elevated in the BAL of Allo+poly I:C mice as compared to Allo+control (p = 0.0007) and Syn+poly I:C (p = 0.007). (C) Representative flow cytometric plots show the CD11b+ monocyte population as a percentage of all IAIE-negative Ly6G-negative cells for each experimental group. (D-G) In addition, markers of monocyte activation were increased: (D) Representative flow cytometric plots show the Gr1+ CD86+ double-positive activated population as a percentage of monocytes for each experimental group. This activated monocyte population was increased in the Allo+poly I:C mice as compared to controls. (E) the mean fluorescence intensity (MFI) of the fluorochrome conjugated to Gr1 was elevated on Allo+poly I:C monocytes compared to Allo+control (p < 0.001) and Syn+poly I:C (p < 0.001) monocytes, and (F) the MFI of the fluorochrome conjugated to CD86 was elevated on Allo+poly I:C monocytes compared to Allo+control (p = 0.002) and Syn+poly I:C (p < 0.001) monocytes. (G) Representative MFI histograms of Gr1 and CD86 on monocytes are shown (solid medium gray curve = unstained control; solid light gray curve = Syn+poly I:C; thick dark gray curve = Allo+poly I:C sample).
4.4. APCs exposed to poly I:C in vivo potentiate allogeneic proliferation in vitro
In order to test the idea that poly I:C augments chronic lung inflammation through an effect on APCs, we harvested splenocytes 24 hours after intraperitoneal poly I:C injection, for use as stimulators in a mixed leukocyte reaction. Poly I:C pre-treated stimulators significantly enhanced allospecific proliferation of MHC-mismatched responders as compared to untreated stimulators. In contrast, syngeneic splenocyte proliferation under identical conditions with or without poly I:C was modest (Figure 5).
Figure 5. Stimulator cells from animals pre-treated with poly I:C in vivo exacerbate allogeneic proliferation in an MLR.
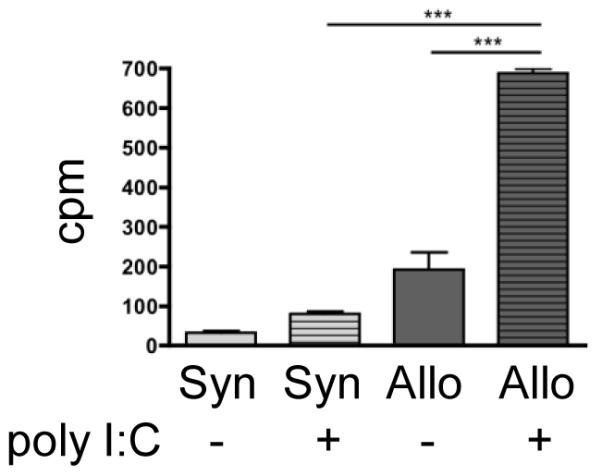
Stimulator C57Bl/6J donors were injected intraperitoneally with either control media or 400 μg of poly I:C. On day 0, 5×105 irradiated stimulator splenocytes were co-cultured with 5×105 splenocytes from either allogeneic C3HB/FeJ or syngeneic C57Bl/6J mice. Following the addition of 3H-thymidine for the last 18 hours of culture, proliferation was measured on day 4 in counts per minute (cpm). Allogeneic splenocytes co-cultured with stimulators pretreated with poly I:C proliferated significantly more than both allogeneic splenocytes co-cultured with control stimulators (p < 0.001) and syngeneic splenocytes co-cultured with poly I:C pre-treated stimulators (p < 0.001). Error bars represent differences between triplicate wells.
4.5. Regulatory T cell (Treg) response after poly I:C is diminished in allogeneic HCT mice
Next, we investigated whether the increased lymphocyte inflammation and increased epithelial disruption in Allo+poly I:C mice were associated with lower levels of Tregs. At 72 hours, poly I:C significantly increased the percentage of CD4+FOXP3+ Tregs in both allogeneic (from 1.6% to 5.0%) and syngeneic mice (from 6.3% to 13.4%) (Figure 6A-B). This poly I:C effect on Tregs disappeared by two weeks in all mice (Figure 6C). More notably, at both time points and in both exposed and unexposed groups, allogeneic animals had significantly fewer Tregs as compared to syngeneic animals (Figure 6A-C).
Figure 6. Decreased regulatory T cells (Tregs) in allogeneic HCT animals compared to syngeneic controls at both 72-hours and two-weeks post poly I:C.

CD3+ BAL cells were evaluated for CD4− and FOXP3-positivity (Tregs) using flow cytometry at both 72-hours (n = 6 mice/group) and two weeks (n = 3 mice/group) following poly I:C exposure in both allogeneic and syngeneic HCT animals. Both (A) representative samples and (B) collective data at 72 hours show the percentage of Tregs increased following poly I:C exposure in both allogeneic HCT animals (from 1.6% to 5.0%) (p = 0.02) and syngeneic HCT animals (6.3% to 13.4%) (p = 0.002). However, allogeneic BAL contained a significantly smaller percentage of Tregs than syngeneic BAL both at baseline (p = 0.005) and following poly I:C exposure (p < 0.001); (C) A similar deficit in allogeneic Tregs was observed at two weeks following poly I:C exposure both at baseline (p = 0.02) and following poly I:C exposure (p = 0.03).
4.6. Intrapulmonary poly I:C does not exacerbate systemic GVHD
In order to understand to what extent increased pulmonary inflammation in Allo+poly I:C mice reflected systemic GVHD, we evaluated the effect of poly I:C on mouse weights and gastrointestinal pathology. Intrapulmonary poly I:C had no effect on total, CD3+, CD4+, or CD8+ splenocyte numbers in either allogeneically- or syngeneically-transplanted animals at 72 hours or at two weeks (data not shown). Intrapulmonary poly I:C had also no significant effect on overall weight loss, even though allogeneic animals lost more weight than syngeneic (data not shown). In addition, while allogeneic animals developed mild gastrointestinal GVHD that was significantly greater than that observed in syngeneic animals, intrapulmonary poly I:C exposure did not alter the extent of gastrointestinal pathology (Figure 7). Thus, poly I:C exacerbates pulmonary lymphocytic inflammation in allogeneic HCT in the absence of a systemic effect.
Figure 7. Poly I:C does not exacerbate allogeneic GVHD.
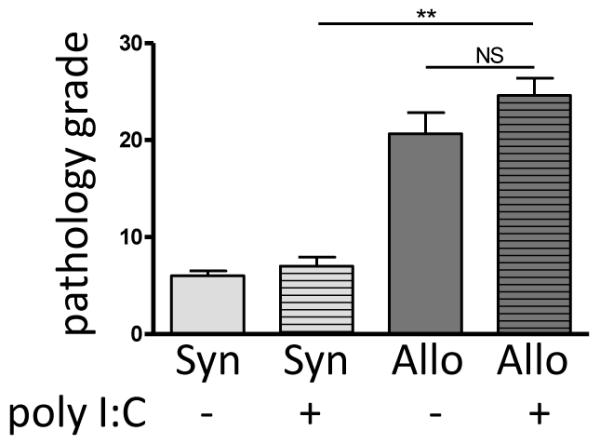
Two weeks following poly I:C exposure, samples from the gastroduodenal junction, ileum, cecum, colon, rectum, and liver were obtained. Sections of preserved and paraffin-embedded tissue were stained with H&E and scored on a semi-quantitative grading scale for signs of gastrointestinal GVHD by an experienced and blinded pathologist. Allogeneic animals developed significantly more gastrointestinal GVHD than their syngeneic counterparts (p < 0.001) but the severity was unaffected by poly I:C exposure (p =0.4).
5. DISCUSSION
Intrapulmonary poly I:C induces prominent lymphocyte recruitment into the lung fluid and T cell infiltration into the bronchiolar epithelium in the setting of allogeneic but not syngeneic HCT. The persistence of lymphocytic inflammation in Allo+poly I:C mice at two-weeks after exposure is striking given the near-complete resolution of lung inflammation at the same time point in syngeneic animals. To our knowledge, this is the first study to demonstrate that the innate antiviral response to a single viral PAMP is sufficient to potentiate transplant related lung disease in the allogeneic HCT setting.
Although the precise antigenic targets of the recruited lymphocytes are uncertain at this time, several lines of evidence suggest that potentiation of a cellular immune response to lung allo-antigens may be taking place in response to innate immune activation. First, we demonstrate in vitro that poly I:C can enhance allogeneic lymphocyte proliferation using strain combinations identical to those employed in the in vivo experiments. Second, T-cell epithelial infiltration occurred only in Allo+poly I:C mice, suggesting an immune response had developed against allo-antigens present on bronchiolar epithelium. Similarly, in human GVHD and lung allograft rejection, the pulmonary epithelium has been found to represent a major target of allorecognition (32). In addition, K-alpha-1 tubulin, a lung epithelial gap-junction protein, has recently been identified as a self-antigen target recognized by antibodies in the setting of lung transplant-related allograft rejection (33). This finding is particularly intriguing in light of the decreased β-catenin, an epithelial gap-junctional protein, in Allo+poly I:C lungs in this study.
Although poly I:C resulted in the development of LB and epithelial injury in allogeneic HCT mice, we did not visualize BO, a characteristic lesion of chronic pulmonary alloimmune disease. Nevertheless, epithelial changes have been described as part of the spectrum of pathologic findings that accompany chronic pulmonary GVHD and have been brought forth as a likely precursor to bronchiolar obliteration (13). In fact, epithelial injury accompanies the process of epithelial to mesenchymal transition (EMT), which is thought to contribute to airway remodeling and fibrosis in BO (34, 35). In our model, poly I:C exposure in allogeneic HCT mice was associated with reduced expression and disordered appearance of epithelial β-catenin, a marker of EMT (36).
Our current study extends the idea developed in our previous work that local pulmonary innate immune activation by PAMPs is an important mechanism that contributes to alloimmune lung disease (20-23). Studies by other groups have also demonstrated the loss of transplant tolerance following systemic poly I:C administration leading to skin allograft loss (26, 37). Furthermore, systemic administration of TLR agonists has been shown to lead to increased systemic GVHD (38) or idiopathic pneumonia syndrome after HCT (39). A unique observation of the current study, however, is the demonstration that local poly I:C administration can exacerbate localized pulmonary GVHD, in the absence of systemic GVHD. This organ-specificity of the GVHD process in our model is demonstrated by the lack of significant increase in weight loss or gastrointestinal disease in poly I:C-treated, as compared to untreated, mice. A similar idea of a local effect on alloimmunity has been introduced in the setting of skin GVHD with the use of cutaneous application of a TLR7 agonist to induce localized skin disease; however, that study involved a mixed chimera model rather than fully mismatched allogeneic HCT (24). To our knowledge, our study constitutes the first clear evidence that interactions between the lung and innate environmental stimuli can contribute to local pulmonary GVHD in the absence of systemic disease.
A potential mechanism, by which intrapulmonary poly I:C leads to the development of increased lymphocytic lung inflammation and GVHD after allogeneic HCT, may involve increased antigen presentation with decreased opposition by regulatory T cells. In our studies, increased recruitment of APCs, particularly monocytes, was seen in allogeneic mice exposed to poly I:C, with increased expression of activation markers Gr1 and CD86. Furthermore, mouse APCs stimulated with poly I:C in vivo were able to elicit a stronger alloimmune response in vitro. The in vivo APC recruitment and activation was associated with increased levels of the monocyte chemotactic protein-1, a recruiter of monocytes and monocyte-derived dendritic cells, as well as T cells (31). This strong APC activation in poly I:C-induced pulmonary GVHD may favor an increased effector T cell response over a regulatory T cell differentiation. While systemic APC activation in HCT has been described and attributed to the conditioning regimen and associated tissue damage (40, 41), our data proposes that pulmonary innate immune stimulation has a stronger activating effect on APCs, particularly monocytes, in the allogeneic, compared to the syngeneic, environment. This is consistent with recent murine orthotopic lung transplant data showing that recruitment of activated monocyte-derived APCs to the lung is associated with increased allorecognition (42). Furthermore, our data is consistent with human HCT studies, in which allogeneic recipients with a paucity of Tregs are at greater risk for GVHD (43, 44). While we show clear changes in Tregs and APCs, further studies are needed to fully elucidate their precise roles in poly I:C-induced exacerbation of pulmonary GVHD.
Collectively, these results are consistent with clinical observations that link RVIs in both HCT (5) and lung transplant (17, 18) recipients to increased rates of pulmonary GVHD or rejection. Furthermore, this data demonstrates a novel mechanism by which RVIs could contribute to transplant-related lung disease. Namely, viral innate immune activation alone, in the absence of an established anti-viral cellular response, can exacerbate features of chronic pulmonary GVHD in the setting of allogeneic HCT, along with early excessive APC, particularly monocyte, recruitment and activation. In contrast to existing ideas regarding mechanisms of viral exacerbation of transplant rejection based on molecular mimicry or epitope spreading occurring as part of an ongoing adaptive immune response, our results suggest that strategies that focus on limiting early viral-associated innate immune activation could prove clinically useful in reducing the burden of viral-associated transplant lung disease.
ACKNOWLEDGEMENTS
The authors would like to thank Barry Stripp and Brian Brockway for kindly providing reagents, equipment, as well as their assistance for immunofluorescence staining and interpretation. The authors are also grateful to Dr. Diana Cardona for her generous help with gastrointestinal pathology grading.
FUNDING SOURCES
This work was supported by National Institutes of Health Grants 1P50-HL084917-011 (project 3 to SMP), 1 K24 HL091140 (to SMP), and RR024 127-03 (to TM). Additional support was also provided by research awards from the Duke University School of Medicine Stead Scholarship (to CVK) and Continuing Research Scholarship (to CVK).
Abbreviations
- PAMP
Pathogen-associated molecular pattern
- Poly I:C
polyinosinic:polycytidylic acid
- GVHD
graft-versus-host disease
- HCT
hematopoietic cell transplant
- RVI
respiratory viral infection
- LB
lymphocytic bronchiolitis
- BO
bronchiolitis obliterans
- LPS
lipopolysaccharide
- TLR
Toll-like receptor
- BAL
bronchoalveolar lavage
- Allo+poly I:C
allogeneic HCT mice exposed to intrapulmonary poly I:C
- Allo+control
allogeneic HCT mice exposed to intrapulmonary water
- Syn+poly I:C
syngeneic HCT mice exposed to intrapulmonary poly I:C
- Syn+control
syngeneic HCT mice exposed to intrapulmonary water
- EMT
epithelial to mesenchymal transition
- APC
antigen presenting cell
- MHC
major histocompatibility complex
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Ferrara JL, Deeg HJ. Graft-versus-host disease. N Engl J Med. 1991;324(10):667. doi: 10.1056/NEJM199103073241005. [DOI] [PubMed] [Google Scholar]
- 2.Socie G, Stone JV, Wingard JR, et al. Long-term survival and late deaths after allogeneic bone marrow transplantation. Late Effects Working Committee of the International Bone Marrow Transplant Registry. N Engl J Med. 1999;341(1):14. doi: 10.1056/NEJM199907013410103. [DOI] [PubMed] [Google Scholar]
- 3.Chao NJ, Schmidt GM, Niland JC, et al. Cyclosporine, methotrexate, and prednisone compared with cyclosporine and prednisone for prophylaxis of acute graft-versus-host disease. N Engl J Med. 1993;329(17):1225. doi: 10.1056/NEJM199310213291703. [DOI] [PubMed] [Google Scholar]
- 4.Afessa B, Litzow MR, Tefferi A. Bronchiolitis obliterans and other late onset non-infectious pulmonary complications in hematopoietic stem cell transplantation. Bone Marrow Transplant. 2001;28(5):425. doi: 10.1038/sj.bmt.1703142. [DOI] [PubMed] [Google Scholar]
- 5.Versluys AB, Rossen JW, van Ewijk B, Schuurman R, Bierings MB, Boelens JJ. Strong association between respiratory viral infection early after hematopoietic stem cell transplantation and the development of life-threatening acute and chronic alloimmune lung syndromes. Biol Blood Marrow Transplant. 2010;16(6):782. doi: 10.1016/j.bbmt.2009.12.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Afessa B, Peters SG. Chronic lung disease after hematopoietic stem cell transplantation. Clin Chest Med. 2005;26(4):571. doi: 10.1016/j.ccm.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 7.Soubani AO, Miller KB, Hassoun PM. Pulmonary complications of bone marrow transplantation. Chest. 1996;109(4):1066. doi: 10.1378/chest.109.4.1066. [DOI] [PubMed] [Google Scholar]
- 8.Griese M, Rampf U, Hofmann D, Fuhrer M, Reinhardt D, Bender-Gotze C. Pulmonary complications after bone marrow transplantation in children: twenty-four years of experience in a single pediatric center. Pediatr Pulmonol. 2000;30(5):393. doi: 10.1002/1099-0496(200011)30:5<393::aid-ppul5>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 9.Sharma S, Nadrous HF, Peters SG, et al. Pulmonary complications in adult blood and marrow transplant recipients: autopsy findings. Chest. 2005;128(3):1385. doi: 10.1378/chest.128.3.1385. [DOI] [PubMed] [Google Scholar]
- 10.Yokoi T, Hirabayashi N, Ito M, et al. Nagoya BMT Group Broncho-bronchiolitis obliterans as a complication of bone marrow transplantation: a clinicopathological study of eight autopsy cases. Virchows Arch. 1997;431(4):275. doi: 10.1007/s004280050099. [DOI] [PubMed] [Google Scholar]
- 11.Estenne M, Maurer JR, Boehler A, et al. Bronchiolitis obliterans syndrome 2001: an update of the diagnostic criteria. J Heart Lung Transplant. 2002;21(3):297. doi: 10.1016/s1053-2498(02)00398-4. [DOI] [PubMed] [Google Scholar]
- 12.Tazelaar HD, Yousem SA. The pathology of combined heart-lung transplantation: an autopsy study. Hum Pathol. 1988;19(12):1403. doi: 10.1016/s0046-8177(88)80233-8. [DOI] [PubMed] [Google Scholar]
- 13.Martinu T, Howell DN, Davis RD, Steele MP, Palmer SM. Pathologic correlates of bronchiolitis obliterans syndrome in pulmonary retransplant recipients. Chest. 2006;129(4):1016. doi: 10.1378/chest.129.4.1016. [DOI] [PubMed] [Google Scholar]
- 14.Taylor DO, Stehlik J, Edwards LB, et al. Registry of the International Society for Heart and Lung Transplantation: Twenty-sixth Official Adult Heart Transplant Report-2009. J Heart Lung Transplant. 2009;28(10):1007. doi: 10.1016/j.healun.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 15.Erard V, Chien JW, Kim HW, et al. Airflow decline after myeloablative allogeneic hematopoietic cell transplantation: the role of community respiratory viruses. J Infect Dis. 2006;193(12):1619. doi: 10.1086/504268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan CK, Hyland RH, Hutcheon MA, et al. Small-airways disease in recipients of allogeneic bone marrow transplants. An analysis of 11 cases and a review of the literature. Medicine (Baltimore) 1987;66(5):327. doi: 10.1097/00005792-198709000-00001. [DOI] [PubMed] [Google Scholar]
- 17.Vilchez RA, Dauber J, McCurry K, Iacono A, Kusne S. Parainfluenza virus infection in adult lung transplant recipients: an emergent clinical syndrome with implications on allograft function. Am J Transplant. 2003;3(2):116. doi: 10.1034/j.1600-6143.2003.00024.x. [DOI] [PubMed] [Google Scholar]
- 18.Kumar D, Erdman D, Keshavjee S, et al. Clinical impact of community-acquired respiratory viruses on bronchiolitis obliterans after lung transplant. Am J Transplant. 2005;5(8):2031. doi: 10.1111/j.1600-6143.2005.00971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chien JW, Zhao LP, Hansen JA, Fan WH, Parimon T, Clark JG. Genetic variation in bactericidal/permeability-increasing protein influences the risk of developing rapid airflow decline after hematopoietic cell transplantation. Blood. 2006;107(5):2200. doi: 10.1182/blood-2005-06-2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palmer SM, Burch LH, Davis RD, et al. The role of innate immunity in acute allograft rejection after lung transplantation. Am J Respir Crit Care Med. 2003;168(6):628. doi: 10.1164/rccm.200303-447OC. [DOI] [PubMed] [Google Scholar]
- 21.Palmer SM, Burch LH, Trindade AJ, et al. Innate immunity influences long-term outcomes after human lung transplant. Am J Respir Crit Care Med. 2005;171(7):780. doi: 10.1164/rccm.200408-1129OC. [DOI] [PubMed] [Google Scholar]
- 22.Palmer SM, Klimecki W, Yu L, et al. Genetic regulation of rejection and survival following human lung transplantation by the innate immune receptor CD14. Am J Transplant. 2007;7(3):693. doi: 10.1111/j.1600-6143.2007.01669.x. [DOI] [PubMed] [Google Scholar]
- 23.Garantziotis S, Palmer SM, Snyder LD, et al. Alloimmune lung injury induced by local innate immune activation through inhaled lipopolysaccharide. Transplantation. 2007;84(8):1012. doi: 10.1097/01.tp.0000286040.85007.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chakraverty R, Cote D, Buchli J, et al. An inflammatory checkpoint regulates recruitment of graft-versus-host reactive T cells to peripheral tissues. J Exp Med. 2006;203(8):2021. doi: 10.1084/jem.20060376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen L, Wang T, Zhou P, et al. TLR engagement prevents transplantation tolerance. Am J Transplant. 2006;6(10):2282. doi: 10.1111/j.1600-6143.2006.01489.x. [DOI] [PubMed] [Google Scholar]
- 26.Thornley TB, Phillips NE, Beaudette-Zlatanova BC, et al. Type 1 IFN mediates cross-talk between innate and adaptive immunity that abrogates transplantation tolerance. J Immunol. 2007;179(10):6620. doi: 10.4049/jimmunol.179.10.6620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsumoto M, Seya T. TLR3: interferon induction by double-stranded RNA including poly(I:C) Adv Drug Deliv Rev. 2008;60(7):805. doi: 10.1016/j.addr.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 28.Stowell NC, Seideman J, Raymond HA, et al. Long-term activation of TLR3 by poly(I:C) induces inflammation and impairs lung function in mice. Respir Res. 2009;10:43. doi: 10.1186/1465-9921-10-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cooke KR, Hill GR, Crawford JM, et al. Tumor necrosis factor-alpha production to lipopolysaccharide stimulation by donor cells predicts the severity of experimental acute graft-versus-host disease. J Clin Invest. 1998;102(10):1882. doi: 10.1172/JCI4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reynolds SD, Giangreco A, Hong KU, McGrath KE, Ortiz LA, Stripp BR. Airway injury in lung disease pathophysiology: selective depletion of airway stem and progenitor cell pools potentiates lung inflammation and alveolar dysfunction. Am J Physiol Lung Cell Mol Physiol. 2004;287(6):L1256. doi: 10.1152/ajplung.00203.2004. [DOI] [PubMed] [Google Scholar]
- 31.Lin KL, Suzuki Y, Nakano H, Ramsburg E, Gunn MD. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J Immunol. 2008;180(4):2562. doi: 10.4049/jimmunol.180.4.2562. [DOI] [PubMed] [Google Scholar]
- 32.Devouassoux G, Pison C, Drouet C, Pin I, Brambilla C, Brambilla E. Early lung leukocyte infiltration, HLA and adhesion molecule expression predict chronic rejection. Transpl Immunol. 2001;8(4):229. doi: 10.1016/s0966-3274(00)00029-0. [DOI] [PubMed] [Google Scholar]
- 33.Goers TA, Ramachandran S, Aloush A, Trulock E, Patterson GA, Mohanakumar T. De novo production of K-alpha1 tubulin-specific antibodies: role in chronic lung allograft rejection. J Immunol. 2008;180(7):4487. doi: 10.4049/jimmunol.180.7.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Borthwick LA, Parker SM, Brougham KA, et al. Epithelial to mesenchymal transition (EMT) and airway remodelling after human lung transplantation. Thorax. 2009;64(9):770. doi: 10.1136/thx.2008.104133. [DOI] [PubMed] [Google Scholar]
- 35.Borthwick LA, McIlroy EI, Gorowiec MR, et al. Inflammation and epithelial to mesenchymal transition in lung transplant recipients: role in dysregulated epithelial wound repair. Am J Transplant. 2010;10(3):498. doi: 10.1111/j.1600-6143.2009.02953.x. [DOI] [PubMed] [Google Scholar]
- 36.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112(12):1776. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller DM, Thornley TB, Pearson T, et al. TLR agonists prevent the establishment of allogeneic hematopoietic chimerism in mice treated with costimulation blockade. J Immunol. 2009;182(9):5547. doi: 10.4049/jimmunol.0802077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nestel FP, Price KS, Seemayer TA, Lapp WS. Macrophage priming and lipopolysaccharide-triggered release of tumor necrosis factor alpha during graft-versus-host disease. J Exp Med. 1992;175(2):405. doi: 10.1084/jem.175.2.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cooke KR, Kobzik L, Martin TR, et al. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood. 1996;88(8):3230. [PubMed] [Google Scholar]
- 40.Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet. 2009;373(9674):1550. doi: 10.1016/S0140-6736(09)60237-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lakkis FG. Where is the alloimmune response initiated? Am J Transplant. 2003;3(3):241. doi: 10.1034/j.1600-6143.2003.00054.x. [DOI] [PubMed] [Google Scholar]
- 42.Gelman AE, Okazaki M, Sugimoto S, et al. CCR2 regulates monocyte recruitment as well as CD4 T1 allorecognition after lung transplantation. Am J Transplant. 2010;10(5):1189. doi: 10.1111/j.1600-6143.2010.03101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zorn E, Kim HT, Lee SJ, et al. Reduced frequency of FOXP3+ CD4+CD25+ regulatory T cells in patients with chronic graft-versus-host disease. Blood. 2005;106(8):2903. doi: 10.1182/blood-2005-03-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fondi C, Nozzoli C, Benemei S, et al. Increase in FOXP3+ regulatory T cells in GVHD skin biopsies is associated with lower disease severity and treatment response. Biol Blood Marrow Transplant. 2009;15(8):938. doi: 10.1016/j.bbmt.2009.04.009. [DOI] [PubMed] [Google Scholar]