

Abstract
Phosphorylated derivatives of the membrane lipid phosphatidylinositol (PtdIns), known as phosphoinositides (PIs), regulate membrane-proximal cellular processes by recruiting specific protein effectors involved in cell signalling, membrane trafficking and cytoskeletal dynamics. Two PIs that are generated through the activities of distinct PI 3-kinases (PI3Ks) are of special interest in cancer research. PtdIns(3,4,5)P3, generated by class I PI3Ks, functions as tumour promotor by recruiting effectors involved in cell survival, proliferation, growth and motility. Conversely, there is evidence that PtdIns3P, generated by class III PI3K, functions in tumour suppression. Three subunits of the class III PI3K complex (Beclin 1, UVRAG and BIF-1) have been independently identified as tumour suppressors in mice and humans, and their mechanism of action in this context has been proposed to entail activation of autophagy, a catabolic pathway that is considered to mediate tumour suppression by scavenging damaged organelles that would otherwise cause DNA instability through the production of reactive oxygen species. Recent studies have revealed two additional functions of PtdIns3P that might contribute to its tumour suppressor activity. The first involves endosomal sorting and lysosomal downregulation of mitogenic receptors. The second involves regulation of cytokinesis, which is the final stage of cell division. Further elucidation of the mechanisms of tumour suppression mediated by class III PI3K and PtdIns3P will identify novel Achilles’ heels of the cell’s defence against tumourigenesis and will be useful in the search for prognostic and diagnostic biomarkers in cancer.
Keywords: autophagy, cancer, cell division, cytokinesis, endocytosis, PI 3-kinase, tumour suppressor
Introduction
Eukaryotic cells contain very extensive intracellular membrane systems, and many vital cellular processes, such as metabolic reactions, signal transduction, cytoskeletal rearrangements, protein sorting and regulation of membrane dynamics, occur partially or entirely at membrane–cytosol interfaces. The main advantages of executing biochemical reactions on membranes include the limitation of substrate diffusion (i.e. limited to two dimensions instead of three) and the possibility of confining biochemical processes to restricted subcellular locations.
The containment of cellular processes to intracellular membranes requires the reversible assembly of protein complexes onto specific membranes. A group of phosphorylated derivatives of phosphatidylinositol (PtdIns), collectively known as phosphoinositides (PIs), are ideally suited for this task [1]. The PIs are generated and metabolized through the activities of a number of substrate-specific PI kinases and phosphatases, the dysfunctions of which are associated with various human diseases [2]. Of special interest in cancer research are two PI 3-kinases (i.e. kinases that phosphorylate the inositol headgroup in the 3-position) that have opposing roles in tumourigenesis. Class I PI 3-kinase (PI3K-I), on the one hand, is a well-known tumour promoting enzyme (or to be more precise, a small group of related enzymes) whose hyperactivity is strongly associated with carcinogenesis in humans [3]. Consistent with this, PTEN, a phosphatase that essentially reverses the 3-phosphorylation mediated by PI3K-I, is an important tumour suppressor [4]. The catalytic product of PI3K-I, PtdIns(3,4,5)P3, recruits several proteins involved in cell signalling to the plasma membrane, the most important one being the protein kinase AKT, which orchestrates various signalling cascades that promote cell growth and survival. On the other hand, class III PI 3-kinase (PI3K-III) is considered to be a tumour suppressor based on the findings that three of its accessory subunits, Beclin 1, UVRAG and BIF-1, have been independently identified as tumour suppressors whose partial or complete inactivation causes the increased occurrence of spontaneous tumours in mice and (probably) humans [5–7]. Recently identified molecular and cellular mechanisms that may serve to explain the tumour suppressor functions of PI3K-III are the topic of the present review.
PI3K-III: a conserved lipid kinase complex
The catalytic subunit of PI3K-III was first identified as Vps34, an enzyme that mediates vacuolar protein sorting in the yeast Saccharomyces cerevisiae [8]. By contrast to PI3K-I, PI3K-III is conserved between yeast, plants and humans, and the human homologue of Vps34 is referred to as hVps34, PIK3C3 or VPS34. In the present review, the latter term is used. Subsequent work soon revealed that Vps34 is associated with a regulatory subunit, Vps15, a putative protein kinase [9]. More recently, Vps34 was found to engage in two functionally distinct complexes in yeast. One complex, consisting of Vps34, Vps15, Vps30 and Vps38, regulates endosomal sorting, whereas another complex, in which Vps38 is replaced by Atg14, is required for autophagy [10].
The two PI3K-III complexes in yeast have their human counterparts: one consisting of VPS34, VPS15, the Vps30 homologue Beclin 1 and the Vps38 homologue UVRAG, and the other containing ATG14 (also called hAtg14, Atg14L or Barkor) instead of UVRAG [11–14]. Thus, we can consider VPS34-VPS15-Beclin 1 as a core complex onto which the accessory subunits UVRAG and ATG14 can assemble in a competitive manner (Fig. 1A). In addition, the UVRAG containing complex can associate with Rubicon, a protein that negatively regulates the function of this complex in endosomal and autophagic trafficking [11,12], and with BIF-1 (also known as endophilin B1 or SH3GLB1), identified as a positive regulator of autophagy [5] (Fig. 1B). Although Rubicon can be isolated as a major constituent of UVRAG-containing PI3K-III complexes, this is not the case with BIF-1, which appears to be more transiently associated [12,15]. Nevertheless, the fact that BIF-1 contains an amino-terminal BAR domain predicted to have membrane-bending ability, as well as the finding that knockdown of BIF-1 inhibits the catalytic activity of PI3K-III, suggests that this protein is an important accessory subunit of mammalian PI3K-III [5]. BAR domain proteins have been found to associate with membranes of high curvature [16], and it is tempting to speculate that BIF-1 might direct PI3K-III activity to such membranes.
Fig. 1.
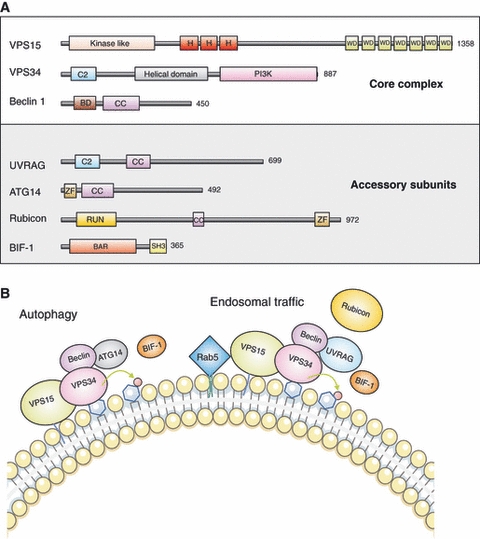
The human PI3K-III complex. (A) The various subunits of PI3K-III are indicated. VPS34, VPS15 and Beclin 1 are considered to form a core complex onto which accessory subunits can assemble. HEAT repeats (H), WD40 repeats (WD), Bcl-2 binding domain (BD), zinc fingers (ZF) and coiled-coil domains (CC) are indicated. Note that there exist alternative sequence variants for most of the subunits (not indicated). (B) Distinct PI3K-III subcomplexes regulate autophagosome formation and endosomal traffic.
The recently solved crystal structure of Drosophila melanogaster Vps34 revealed that this PI3K has a considerably smaller ATP-binding pocket than class I PI3Ks [17]. This explains why several inhibitors of class I PI3Ks fail to inhibit class III PI3K (i.e. they are too bulky to fit into the ATP-binding pocket). More importantly, the structural insight offers a rationale for designing specific PI3K-III inhibitors in the future.
Even though considerable knowledge has been gained about the biochemical composition of PI3K-III, we still know little about the regulation of its catalytic activity. The catalytic activity appears to be stimulated by BIF-1, whereas Rubicon inhibits the overall function of PI3K-III [5,11,12]. However, the exact mechanisms of these regulations remain unsolved. An important clue to the regulation of VPS34 has emerged recently with the finding that VPS34 is phosphorylated on threonine 159 by the cyclin-dependent kinase Ckd1 during mitosis, and that this causes its dissociation from Beclin 1 and an inhibition of autophagy [18].
Recognition of PtdIns3P by FYVE and phox homology (PX) domains
A breakthrough in our understanding of how PI3K-III and its catalytic product control cellular functions came with the identification of the FYVE (conserved in Fab1, YOTB, Vac1 and EEA1) domain, and the demonstration that this domain binds PtdIns3P. The FYVE domain was originally identified as a zinc finger required for localization of the early endosomal autoantigen 1 (EEA1) to early endosomes [19]. The finding that wortmannin, a general PI3K inhibitor, prevents the localization of EEA1 to endosomes, provided a clue that EEA1 might be recruited by a 3-phosphorylated PI [20], and biochemical studies showed that the FYVE domains from yeast and mammalian proteins bind to PtdIns3P with high specificity [21–23]. Further progress in deciphering the downstream functions of PtdIns3P was obtained when the PX domain was identified as a second PtdIns3P binding domain [24–27]. Although a few mammalian PX domains bind to other 3-PIs than PtdIns3P, most of the PX domains bind specifically to PtdIns3P with affinities comparable to those of FYVE domains [28]. The human genome encodes approximately 30 FYVE domain-containing proteins and some 45 PX domain-containing proteins that presumably mediate most (but not all) of the downstream functions of PtdIns3P [29]. Additional PtdIns3P-binding proteins that do not contain FYVE or PX domains include the Proppin/WIPI proteins, which bind PtdIns3P [and to some extent the related PtdIns(3,5)P2] through a WD40-repeat-containing β-propeller structure [30,31], and certain variant pleckstrin homology domains such as the GLUE (GRAM-like ubiquitin-binding in EAP45) domain [32–34].
Intracellular localization of PtdIns3P
The identification of PtdIns3P-binding domains offered the possibility to design probes that reveal the intracellular distribution of this lipid. Early work revealed that a single FYVE domain from the endosomal protein HRS binds PtdIns3P with too low affinity to be useful as a probe. Remarkably, however, when two FYVE domains from HRS were fused in tandem (2xFYVE), the resulting construct could be used for imaging of cellular PtdIns3P with high sensitivity and specificity, presumably as a result of an avidity effect [35]. The fact that 2xFYVE can be either transfected into cells as a fusion with green fluorescent protein or another tag, or expressed in bacteria and purified as a recombinant probe that can be used directly on fixed specimens, makes this probe very versatile for monitoring the distribution of PtdIns3P. Subsequently, other probes have been used for monitoring PtdIns3P, including the FYVE domain of SARA, which has an intrinsic ability to dimerize and therefore does not need to be expressed as tandem fusion [36], and certain PX domains [37]. In general, the various probes have yielded comparable results, although the 2xFYVE probe has been most rigorously tested for ligand specificity. The original studies using 2xFYVE by fluorescence and electron microscopy showed that the bulk of PtdIns3P is associated with the limiting and intraluminal membranes of endosomes at steady-state [35]. Subsequent studies have revealed that PtdIns3P can also be detected at the plasma membrane of cells stimulated with insulin or lysophosphatidic acid [38,39]. Because this pool of PtdIns3P is generated through the activity of PI3K-II [40], which has so far not been implicated in tumour suppression, it will not be considered further in the present review. Recent analyses of starved yeast cells have revealed a strong localization of PtdIns3P on autophagosomes, especially on the inner membranes [41], and, during induction of autophagy in mammalian cells, PtdIns3P is formed on membranes of the endoplasmic reticulum (ER) [42]. The importance of this is discussed below.
PI3K-III and PtdIns3P binding proteins in endosomal trafficking
Because Vps34 and Vps15 were originally identified as mediators of vacular protein sorting in yeast, the first characterized functions of PI3K-III and PtdIns3P were those associated with endosomal trafficking (Fig. 2A).
Fig. 2.
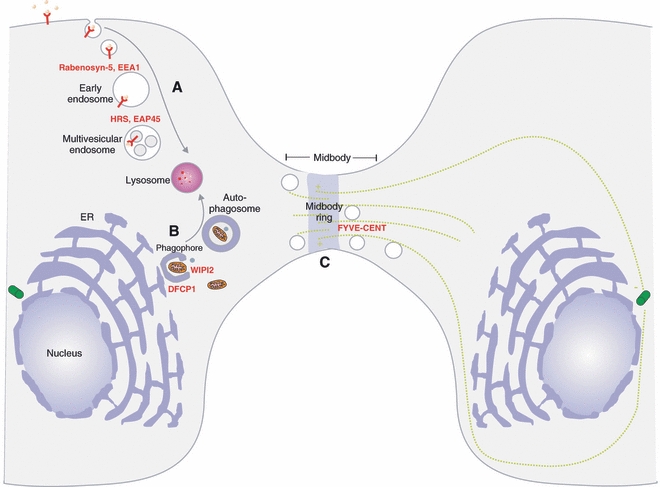
PtdIns3P effectors in cell regulation. Endocytic downregulation of a mitogenic receptor (A), autophagy (B) and cytokinesis (C). PtdIns3P effectors involved in the various processes are shown in red. Rabenosyn-5 and EEA1 mediate membrane fusion in the early endocytic pathway. HRS and EAP45 are subunits of the ESCRT machinery that sorts ubiquitinated mitogenic receptors into the ILVs of multivesicular endosomes. DFCP1 mediates phagophore biogenesis, whereas WIPI2 mediates autophagosome biogenesis. FYVE-CENT facilitates cytokinesis. Microtubules are indicated by dashed green lines.
PtdIns3P and endosomal fusion
Yeast cells devoid of functional Vps34 or Vps15 secrete several hydrolases that normally are transported to the lysosome-like vacuole [8,43], suggesting that these proteins control trafficking between the secretory and endosomal pathways and/or between endosomes. A key effector of PtdIns3P in endocytic trafficking is the FYVE-domain-containing protein Vac1, which interacts genetically and physically with the small GTPase Vps21, and with Vps45, a member of the Sec1/Munc18 family of proteins regulating the formation of SNARE complexes that mediate membrane fusion [44,45]. Indeed, studies of the mammalian homologues of Vac1, Vps21 and Vps45, termed Rabenosyn-5, RAB5 and VPS45, respectively, have revealed that these proteins function in tethering and fusion reactions in the early endocytic pathway [46]. Moreover, studies in Drosophila have revealed that Rabenosyn functions to bridge Rab5 with Vps45, thereby regulating the function of the SNARE protein Avalanche in endosomal fusion [47]. Interestingly, functional interference with Rabenosyn-5 and its interacting partners causes a loss of both epithelial and planar polarity [47,48]. The loss of epithelial polarity is a prevailing characteristic of carcinomas, and mutation of Rabenosyn indeed causes epithelial tumours in Drosophila. To date, the mechanisms by which Rabenosyn controls epithelial polarity have not been elucidated, whereas more detailed insight is available in the case of planar cell polarity. One consequence of interferring with Rabenosyn function is that Flamingo, a determinant of planar cell polarity through the noncanonical Wnt signalling pathway, accumulates in the cytoplasm instead of translocating to polarized membrane domains [48]. Accumulating evidence suggests a link between improper development of planar cell polarity and cancer [49], and even though it is still not clear whether Rabenosyn-5 is a tumour suppressor in mammals, the epithelial and planar cell polarity maintained by this RAB5 and PtdIns3P effector has to be considered when dissecting the tumour suppressor activities of PI3K-III.
Early studies in mammalian cells have also revealed another important PtdIns3P effector in endosomal tethering and fusion, namely EEA1, a protein that contains a Rab5 binding domain and a FYVE domain at its C-terminus, and a distinct Rab5 binding domain at its N-terminus [50]. EEA1 forms rod-shaped dimers through parallel coiled-coil interactions, and is well suited for tethering two opposing Rab5-containing membranes [51]. The exquisite localization of EEA1 to early endosomes is probably conferred by the coincident detection of PtdIns3P and GTP-bound Rab5 [50]. EEA1 is structurally related to Rabenosyn-5, and also interacts with SNARE molecules [52,53]. In a remarkable reconstitution of Rab5-mediated fusion using liposomes and recombinant SNAREs and Rab5 effectors, the inclusion of PtdIns3P could bypass the requirement for PI3K-III, as expected based on previous studies [54]. Importantly, the omission of either EEA1 or Rabenosyn-5 was sufficient to inhibit fusion strongly, indicating that these proteins, despite their structural relatedness, have nonredundant functions in endocytic membrane fusion.
PtdIns3P and endosomal sorting to the degradative pathway
Consistent with the fact that Vps34 was originally identified as a mediator of protein sorting, studies of both yeast and mammalian cells have shown that PI3K-III is required for proper sorting of certain membrane proteins from endosomes to vacuoles/lysosomes [8,55]. Moreover, interference with the function of VPS34 in mammalian cells results in dilated late endosomes devoid of intraluminal vesicles (ILVs) [56,57]. Recent studies have revealed that not only VPS34, but also VPS15, Beclin 1, UVRAG and BIF-1 are required for proper degradation of endocytosed epidermal growth factor receptors in lysosomes, highlighting the involvement of an entire PI3K-III complex in endosomal sorting [15].
A mechanistic explanation for these findings has emerged with the discovery of the endosomal sorting complex required for transport (ESCRT) machinery [58,59]. This molecular machinery, which consists of at least four subcomplexes (ESCRT-0, -I, -II and -III), is recruited to endosome membranes where it recognizes ubiquitinated membrane proteins (e.g. activated growth factor receptors and membrane-anchored hydrolases) and sorts these into ILVs. Recent reconstitution studies employing giant unilamellar vesicles have revealed that ESCRT-0, which contains as many as five ubiquitin-binding domains, serves to sequester ubiquitinated cargoes, whereas ESCRT-I and -II, which also contain ubiquitin-binding domains, serve to form invaginations of the endosomal membrane [60]. Finally, ESCRT-III is recruited, cargo is deubiquitinated by deubiquitinating enzymes recruited by ESCRT-III [61], and ESCRT-III serves to sever the neck of the forming invagination, thereby securing the inclusion of cargo proteins within ILVs [60]. The main link between PI3K-III and the ESCRT pathway is the fact that Vps27/HRS, a core component of ESCRT-0, contains a FYVE domain that mediates its recruitment to endosomal membranes through binding PtdIns3P [62]. Vps27/HRS in turn recruits ESCRT-I through interaction with the Vps23/TSG101 subunit, so the initial recruitment of ESCRT-0 to endosomal membranes via FYVE-PtdIns3P interactions is crucial for the function of the ESCRT machinery. In addition, a subunit of ESCRT-II, Vps36/EAP45, contains a PtdIns3P-binding GLUE domain that is predicted to contribute to the membrane recruitment of ESCRT-II [32,34]. The involvement of the ESCRT machinery in protein sorting and ILV biogenesis, as well as the notion that key subunits of this machinery require PtdIns3P for their membrane recruitment, readily explains why interference with PI3K-III functions results in impaired protein sorting and causes endosomes devoid of ILVs.
PI3K-III and PtdIns3P binding proteins in autophagy
Macroautophagy (referred to here as autophagy) is a process that involves the sequestration of cytoplasm by a double membrane called phagophore or isolation membrane, followed by fusion of the resulting autophagosome with endosomes and lysosomes [63] (Fig. 2B). The degradation of the sequestered cytosolic material in autolysosomes is beneficial for the cell, both under starvation conditions (when it is crucial to recycle free amino acids by degrading cytosolic proteins that are not housekeeping), during infection with cytosolic parasites, and under various stress conditions (e.g. those that result in the accumulation of cytosolic protein aggregates that are not readily degraded by proteasomes). The exact origin of the phagophore membrane is still a matter of debate, although there are strong arguments in favour of a contribution from both ER and mitochondrial membranes [42,64,65]. At least in the case of ER membranes, there is evidence for the production of PtdIns3P during the early phase of autophagosome formation [42,66]. Several lines of evidence point to a crucial role for PI3K-III in autophagy [66], and immunoelectron microscopy of starved yeast cells using the 2xFYVE probe has revealed that PtdIns3P is enriched on inner side of the phagophore during autophagosome formation [41].
Studies in yeast have revealed that a complex consisting of Vps34, Vps15, Vps30 and Atg14 is required for autophagy [10], and subsequent work in mammalian cells has shown a similar requirement for the mammalian homologues of these proteins [11,12]. In addition, an involvement of the Vps38 homologue UVRAG has been reported [7], which is surprising in light of the strong evidence that Vps38 mediates endosomal trafficking and not autophagy in yeast. A possible role of UVRAG in autophagy is supported by the independent identification of BIF-1, an interactor of UVRAG, as a regulator of autophagy [5]. On the other hand, monoallelic UVRAG mutations associated with microsatellite unstable colon cancer have no effect on autophagy, and the depletion of UVRAG has an undetectable effect on autophagy in HEK293 cells, whereas endosomal sorting is affected [67]. One explanation for the conflicting findings on UVRAG may stem from the involvement of UVRAG in fusion between autophagosomes (and late endosomes) and lysosomes through its interactions with the HOPS complex [68]. Except for the catalytic activity of VPS34, little is known about the specific functions of the various PI3K-III subunits in autophagy. However, recent evidence suggests that the specific function of ATG14 in autophagy may reflect the ability of this protein to target PI3K-III to ER membranes [69].
How does PtdIns3P mediate autophagy? The only known PtdIns3P effector in autophagy in yeast is the Proppin protein Atg18 [70], whose binding to PtdIns3P is required for autophagy [71]. Although the exact function of Atg18 in autophagy remains to be clarified, current evidence suggests that this protein, in complex with Atg2, facilitates the recruitment of lipidated Atg8, a key effector in autophagosome formation to phagophore membranes [71]. A mammalian Atg18 homologue, WIPI2, is recruited to phagophore membranes along with ULK1, a protein kinase that positively regulates autophagy [72]. Interestingly, the depletion of WIPI2 leads to a strong accumulation of omegasomes, comprising ER-localized PtdIns3P-containing structures positive for DFCP1 (double FYVE domain-containing protein 1) that are considered to act as platforms for autophagosome formation. This suggests a role for WIPI2 in the progression from omegasomes into autophagosomes.
PI3K-III and PtdIns3P binding proteins in cytokinesis
A surprising finding when using a green fluorescent protein-tagged version of the 2xFYVE probe was that PtdIns3P accumulates in the bridge separating two dividing cells, the so-called midbody. PtdIns3P is frequently associated with the central, electron-dense part of the midbody, referred to as the midbody ring or the Flemming body, but can also be observed on small vesicles throughout the midbody region [73]. This localization of PtdIns3P raises the question of whether its formation is required during cytokinesis, the final stage of cell division (Fig. 2C). Indeed, small interfering RNA-mediated knockdown of VPS34, as well as of the accessory PI3K-III components VPS15, Beclin 1, UVRAG and BIF-1, causes an increased proportion of cells undergoing cytokinesis, suggesting a role for PI3K-III in the completion of cytokinesis [15,73]. Small interfering RNA screening identified the large FYVE domain-containing protein FYVE-CENT (FYVE protein on centrosomes) as a PtdIns3P effector in cytokinesis. FYVE-CENT localizes to the centrosome in interphase cells and translocates to the midbody during telophase. This translocation appears to be mediated by the microtubule-based motor protein KIF13A. The precise function of FYVE-CENT during cytokinesis remains to be characterized, although one clue arises from the finding that TTC19, a protein that associates with FYVE-CENT and accompanies it on its translocation from the centrosome to the midbody, interacts with the ESCRT-III subunit CHMP4B [73]. This is interesting because ESCRT-III has been proposed to mediate the final membrane abscission step during cytokinesis [74,75], and it is possible that TTC19, brought to the midbody by FYVE-CENT and KIF13A, may be a positive regulator of CHMP4B in cytokinesis. By analogy with its yeast counterpart Vps32/Snf7, CHMP4B is predicted to form spiral-shaped oligomers that constrict to mediate membrane severing [76], and TTC19 might serve to control this oligomerization.
PI3K-III and PtdIns3P effectors in tumour suppression
The PI3K-III subunit Beclin 1 is monoallelically deleted in a large proportion of breast and ovarian cancers, and heterozygous beclin 1 knockout mice develop spontaneous mammary tumours [6]. These findings, combined with the observation that both Beclin 1 and its yeast homologue Vps30 mediate autophagy, suggest that Beclin 1 acts as a tumour suppressor because of its function in autophagy. In support of this idea, there is evidence that autophagy functions as tumour suppressor by scavenging damaged mitochondria and peroxisomes that would otherwise cause genomic instability through oxygen radical-induced DNA damage [77]. Further supporting the notion that PI3K-III acts as a tumour suppressor through its function in autophagy is the observation that two other PI3K-III accessory proteins identified as positive regulators of autophagy, UVRAG and BIF-1, are also tumour suppressors [5,7]. Even though these are compelling data, one obvious question arises regarding the role of PI3K-III in autophagy-mediated tumour suppression: are other autophagy regulators also tumour suppressors? One would expect that this would be the case but, so far, only one of the many other proteins implicated in autophagy regulation has been identified as a putative tumour suppressor, the protease ATG4C [78]. Because, among the more than 30 positive regulators of autophagy, three out of four identified tumour suppressors belong to the PI3K-III complex, the possibility exists that PI3K-III may mediate tumour suppression not by autophagy but by an alternative means.
Given the importance of endocytosis and lysosomal downregulation of growth factor receptors in attenuation of mitogenic cell signalling [79], one distinct possibility is that PI3K-III could (at least in part) exert its tumour suppressor function through its role in endosome fusion and endosomal receptor sorting. Although there is no direct evidence that this is the case, it is interesting to note that the PtdIns3P-binding endosomal fusion regulator, Rabenosyn, is a tumour suppressor in flies [47], and the same is the case with multiple components of the ESCRT machinery that acts downstream of PtdIns3P in the endosomal sorting of mitogenic receptors [80–83]. Arguing against this idea is the fact that Hrs, the PtdIns3P binding ESCRT-0 protein that initiates further ESCRT recruitment to membranes, is not a tumour suppressor in Drosophila.
The recent discovery that PI3K-III regulates cytokinesis has offered a third possible explanation for the tumour suppressor activity of this enzyme complex [73]. Inhibition of PI3K-III activity not only causes an increased proportion of cells undergoing cytokinesis, but also an increase in bi- and multinucleate cells. Under certain conditions, tetraploidy may develop into aneuploidy, which is strongly associated with cancer. It is therefore likely that incomplete cytokinesis, as experienced when PI3K-III or the PtdIns3P effector FYVE-CENT is functionally ablated, may represent a step in oncogenesis [84]. It is interesting to note that FYVE-CENT is frequently mutated in breast cancer [85,86], although it remains to be established whether this PtdIns3P effector is a genuine tumour suppressor.
Conclusions and perspectives
As discussed in the present review, PI3K-III may theoretically function as a tumour suppressor via at least three different mechanisms (Fig. 3). The involvement of PI3K-III in autophagy maintains genome stability by eliminating damaged organelles that produce reactive oxygen species; the role of PI3K-III in endosomal fusion and sorting ensures correct downregulation of mitogenic signalling; and the correct function of PI3K-III in cytokinesis prevents aneuploidy. Further work is required to establish which of these three processes is most important in PI3K-III-mediated tumour suppression. It can also not be excluded that PI3K-III may act as tumour suppressor by additional means. For example, SARA, a mediator of transforming growth factor-β signalling, requires PtdIns3P binding for its function [87], and the transforming growth factor-β signalling pathway does have a tumour suppressor role under most conditions [88]. Furthermore, PtdIns3P binding subunits mediate membrane association of the retromer complex [89], which sorts cargoes such as mannose 6-phosphate receptors and Wntless (an accessory factor in Wnt secretion) for trafficking from endosomes to the biosynthetic pathway [89,90]. Even though there is no evidence so far that implicates the retromer in tumour suppression, this possibility cannot at present be discarded.
Fig. 3.
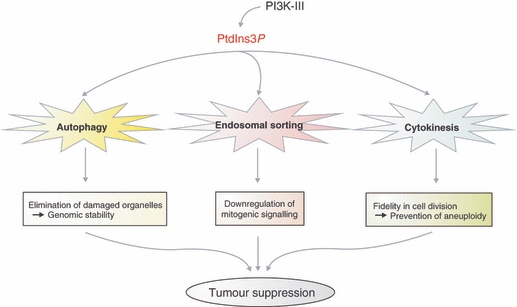
Alternative tumour suppressor mechanisms of PI3K-III. Three alternative (hypothetic) tumour suppressor mechanisms downstream of PtdIns3P formation are shown. The relative importance of these mechanisms in the tumour suppressor activity of PI3K-III subunits remains to be established.
The involvement of PI3K-III in diverse cellular processes raises the question of how this complex is recruited to the correct membranes at the right time. There is evidence that PI3K-III is recruited to early and late endosomal membranes through interactions with the small GTPases RAB5 and RAB7, respectively [91–93]. Less is known about how PI3K-III is recruited to midbodies and autophagic membranes, although the latter is likely to be mediated by the autophagy-specific subunit ATG14 [69]. In addition, the finding that phosphorylation of VPS34 during mitosis causes its dissociation from Beclin 1 [18] provides a hint that post-translational modifications of PI3K-III may contribute to regulate its activity and specificity.
Although considerable efforts have been made to understand how PtdIns3P is formed by PI3K-III, we are also beginning to learn about the metabolism of this lipid. Three known routes for PtdIns3P metabolism have been described: degradation by lysosomal lipases, phosphorylation by the PtdIns3P 5-kinase Fab1/PIKfyve, and dephosphorylation by 3-phosphatases [94]. It is worth noting that Fab1/PIKfyve is itself a PtdIns3P binding protein [95], and that MTM1 and MTMR2, two phosphatases capable of dephosphorylating PtdIns3P, can associate with PI3K-III on endosomal membranes [96,97]. This suggests a tight regulation of PtdIns3P formation and turnover, and it will be interesting to determine whether PIKfyve and MTM1/MTMR2 may play any role in tumourigenesis.
Even though it is assumed that PI3K-III functions as tumour suppressor through its ability to produce PtdIns3P at the correct intracellular membranes, this has not been formally demonstrated and, to date, we do not know whether the catalytic subunit of PI3K-III, VPS34, is a tumour suppressor. Further studies should clarify this, and it will also be important to establish whether the catalytic function of VPS34 is required for its eventual tumour suppressor function.
If we nevertheless accept that PI3K-III acts as tumour suppressor through PtdIns3P generation, can this be exploited in cancer diagnosis and therapy? Because it is much easier to inhibit an enzyme pharmacologically than to boost its function, the tumour promotor PI3K-I is a more attractive drug target than the tumour suppressor PI3K-III. On the other hand, we know that PtdIns3P can be dephosphorylated and that PI3K-III undergoes negative regulation [11,12], and alleviation of these inhibitory mechanisms might provide a viable strategy towards increasing the tumour suppressor activity of PI3K-III and its catalytic product in cancer treatment. More straightforwardly, knowing that PI3K-III subunits such as Beclin 1 and UVRAG are frequently downregulated or mutated in cancers [6,7,67,98,99], it will be interesting to perform systematic analyses of PI3K-III subunits and key PtdIns3P effectors in various cancers. Such studies should reveal mutational and expression-based signatures that can be used to predict the outcome of disease, and to guide the choice of therapeutic regimens.
Acknowledgments
I thank my mentors Sjur Olsnes and Marino Zerial, and my excellent co-workers at the Institute for Cancer Research. Research in my laboratory is generously sponsored by the Norwegian Cancer Society, the Research Council of Norway, the South-Eastern Norway Regional Health Authority, the European Research Foundation, and by an Advanced Grant from the European Research Council.
Glossary
Abbreviations
- EEA1
early endosomal autoantigen 1
- ER
endoplasmic reticulum
- ESCRT
endosomal sorting complex required for transport
- ILV
intraluminal vesicles
- PI
phosphoinositide
- PI3K
phosphoinositide 3-kinase
- PtdIns
phosphatidylinositol
- PX
phox homology
References
- 1.Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- 2.McCrea HJ, De Camilli P. Mutations in phosphoinositide metabolizing enzymes and human disease. Physiology. 2009;24:8–16. doi: 10.1152/physiol.00035.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev. 2010;20:87–90. doi: 10.1016/j.gde.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li L, Ross AH. Why is PTEN an important tumor suppressor? J Cell Biochem. 2007;102:1368–1374. doi: 10.1002/jcb.21593. [DOI] [PubMed] [Google Scholar]
- 5.Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mul JJ, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9:1142–1151. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 7.Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–699. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- 8.Schu PV, Takegawa K, Fry MJ, Stack JH, Waterfield MD, Emr SD. Phosphatidylinositol 3-kinase encoded by yeast VPS34 gene essential for protein sorting. Science. 1993;260:88–91. doi: 10.1126/science.8385367. [DOI] [PubMed] [Google Scholar]
- 9.Stack JH, DeWald DB, Takegawa K, Emr SD. Vesicle-mediated protein transport: regulatory interactions between the Vps15 protein kinase and the Vps34 PtdIns 3-kinase essential for protein sorting to the vacuole in yeast. J Cell Biol. 1995;129:321–334. doi: 10.1083/jcb.129.2.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kihara A, Noda T, Ishihara N, Ohsumi Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol. 2001;152:519–530. doi: 10.1083/jcb.152.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11:468–476. doi: 10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11:385–396. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- 13.Sun Q, Fan W, Chen K, Ding X, Chen S, Zhong Q. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA. 2008;105:19211–19216. doi: 10.1073/pnas.0810452105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19:5360–5372. doi: 10.1091/mbc.E08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thoresen SB, Pedersen NM, Liestol K, Stenmark H. A phosphatidylinositol 3-kinase class III sub-complex containing VPS15, VPS34, Beclin 1, UVRAG and BIF-1 regulates cytokinesis and degradative endocytic traffic. Exp Cell Res. 2010 doi: 10.1016/j.yexcr.2010.07.008. doi: 10.1016/j.yexcr.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 16.Gallop JL, McMahon HT. BAR domains and membrane curvature: bringing your curves to the BAR. Biochem Soc Symp. 2005;72:223–231. doi: 10.1042/bss0720223. [DOI] [PubMed] [Google Scholar]
- 17.Miller S, Tavshanjian B, Oleksy A, Perisic O, Houseman BT, Shokat KM, Williams RL. Shaping development of autophagy inhibitors with the structure of the lipid kinase Vps34. Science. 2010;327:1638–1642. doi: 10.1126/science.1184429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furuya T, Kim M, Lipinski M, Li J, Kim D, Lu T, Shen Y, Rameh L, Yankner B, Tsai L-H, et al. Negative regulation of Vps34 by Cdk mediated phosphorylation. Mol Cell. 2010;38:500–511. doi: 10.1016/j.molcel.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stenmark H, Aasland R, Toh BH, D’Arrigo A. Endosomal localization of the autoantigen EEA1 is mediated by a zinc-binding FYVE finger. J Biol Chem. 1996;271:24048–24054. doi: 10.1074/jbc.271.39.24048. [DOI] [PubMed] [Google Scholar]
- 20.Patki V, Virbasius J, Lane WS, Toh BH, Shpetner HS, Corvera S. Identification of an early endosomal protein regulated by phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA. 1997;94:7326–7330. doi: 10.1073/pnas.94.14.7326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gaullier J-M, Simonsen A, D’Arrigo A, Bremnes B, Aasland R, Stenmark H. FYVE fingers bind PtdIns(3)P. Nature. 1998;394:432–433. doi: 10.1038/28767. [DOI] [PubMed] [Google Scholar]
- 22.Burd CG, Emr SD. Phosphatidylinositol(3)-phosphate signaling mediated by specific binding to RING FYVE domains. Mol Cell. 1998;2:157–162. doi: 10.1016/s1097-2765(00)80125-2. [DOI] [PubMed] [Google Scholar]
- 23.Patki V, Lawe DC, Corvera S, Virbasius JV, Chawla A. A functional PtdIns(3)P-binding motif. Nature. 1998;394:433–434. doi: 10.1038/28771. [DOI] [PubMed] [Google Scholar]
- 24.Ellson CD, Gobert-Gosse S, Anderson KE, Davidson K, Erdjument-Bromage H, Tempst P, Thuring JW, Cooper MA, Lim Z-Y, Holmes AB, et al. Phosphatidylinositol 3-phosphate regulates the neutrophil oxidase complex by binding to the PX domain of p40phox. Nature Cell Biol. 2001;3:679–682. doi: 10.1038/35083076. [DOI] [PubMed] [Google Scholar]
- 25.Xu YHH, Seet L, Wong SH, Hong W. Sorting nexin 3 (SNX3) regulates endosomal function via its PX domain-mediated interaction with phosphatidylinositol 3-phosphate. Nature Cell Biol. 2001;3:658–666. doi: 10.1038/35083051. [DOI] [PubMed] [Google Scholar]
- 26.Kanai F, Liu H, Akbary H, Field S, Matsuo T, Brown G, Cantley LC, Yaffe MB. The PX domains of p47phox and p40phox bind to lipid products of phosphoinositide 3-kinase. Nature Cell Biol. 2001;3:675–678. doi: 10.1038/35083070. [DOI] [PubMed] [Google Scholar]
- 27.Cheever ML, Sato TK, de Beer T, Kutateladze T, Emr SD, Overduin M. Phox domain interaction with PtdIns(3)P targets Vam7 t-SNARE to vacuole membranes. Nature Cell Biol. 2001;3:613–618. doi: 10.1038/35083000. [DOI] [PubMed] [Google Scholar]
- 28.Lemmon MA. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol. 2008;9:99–111. doi: 10.1038/nrm2328. [DOI] [PubMed] [Google Scholar]
- 29.Birkeland HC, Stenmark H. Protein targeting to endosomes and phagosomes via FYVE and PX domains. Curr Top Microbiol Immunol. 2004;282:89–115. doi: 10.1007/978-3-642-18805-3_4. [DOI] [PubMed] [Google Scholar]
- 30.Proikas-Cezanne T, Waddell S, Gaugel A, Frickey T, Lupas A, Nordheim A. WIPI-1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene. 2004;23:9314–9325. doi: 10.1038/sj.onc.1208331. [DOI] [PubMed] [Google Scholar]
- 31.Dove SK, Piper RC, McEwen RK, Yu JW, King MC, Hughes DC, Thuring J, Holmes AB, Cooke FT, Michell RH, et al. Svp1p defines a family of phosphatidylinositol 3,5-bisphosphate effectors. EMBO J. 2004;23:1922–1933. doi: 10.1038/sj.emboj.7600203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Slagsvold T, Aasland R, Hirano S, Bache KG, Raiborg C, Trambaiolo D, Wakatsuki S, Stenmark H. Eap45 in mammalian ESCRT-II binds ubiquitin via a phosphoinositide-interacting GLUE domain. J Biol Chem. 2005;280:19600–19606. doi: 10.1074/jbc.M501510200. [DOI] [PubMed] [Google Scholar]
- 33.Hirano S, Suzuki N, Slagsvold T, Kawasaki M, Trambaiolo D, Kato R, Stenmark H, Wakatsuki S. Structural basis of ubiquitin recognition by mammalian Eap45 GLUE domain. Nat Struct Mol Biol. 2006;13:1031–1032. doi: 10.1038/nsmb1163. [DOI] [PubMed] [Google Scholar]
- 34.Teo H, Gill DJ, Sun J, Perisic O, Veprintsev DB, Vallis Y, Emr SD, Williams RL. ESCRT-I core and ESCRT-II GLUE domain structures reveal central role for GLUE domain in linking to ESCRT-I and membranes. Cell. 2006;125:99–111. doi: 10.1016/j.cell.2006.01.047. [DOI] [PubMed] [Google Scholar]
- 35.Gillooly DJ, Morrow IC, Lindsay M, Gould R, Bryant NJ, Gaullier J-M, Parton RG, Stenmark H. Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J. 2000;19:4577–4588. doi: 10.1093/emboj/19.17.4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hayakawa A, Hayes SJ, Lawe DC, Sudharshan E, Tuft R, Fogarty K, Lambright D, Corvera S. Structural basis for endosomal targeting by FYVE domains. J Biol Chem. 2004;279:5958–5966. doi: 10.1074/jbc.M310503200. [DOI] [PubMed] [Google Scholar]
- 37.Scott CC, Cuellar-Mata P, Matsuo T, Davidson HW, Grinstein S. Role of 3-phosphoinositides in the maturation of Salmonella-containing vacuoles within host cells. J Biol Chem. 2002;277:12770–12776. doi: 10.1074/jbc.M110399200. [DOI] [PubMed] [Google Scholar]
- 38.Maffucci T, Brancaccio A, Piccolo E, Stein RC, Falasca M. Insulin induces phosphatidylinositol-3-phosphate formation through TC10 activation. EMBO J. 2003;22:4178–4189. doi: 10.1093/emboj/cdg402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maffucci T, Cooke FT, Foster FM, Traer CJ, Fry MJ, Falasca M. Class II phosphoinositide 3-kinase defines a novel signaling pathway in cell migration. J Cell Biol. 2005;169:789–799. doi: 10.1083/jcb.200408005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11:329–341. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 41.Obara K, Noda T, Niimi K, Ohsumi Y. Transport of phosphatidylinositol 3-phosphate into the vacuole via autophagic membranes in Saccharomyces cerevisiae. Genes Cells. 2008;13:537–547. doi: 10.1111/j.1365-2443.2008.01188.x. [DOI] [PubMed] [Google Scholar]
- 42.Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Herman PK, Stack JH, DeModena JA, Emr SD. A novel protein kinase homolog essential for protein sorting to the yeast lysosome-like vacuole. Cell. 1991;64:425–437. doi: 10.1016/0092-8674(91)90650-n. [DOI] [PubMed] [Google Scholar]
- 44.Burd CG, Peterson M, Cowles CR, Emr SD. A novel Sec18p/NSF-dependent complex required for Golgi-to- endosome transport in yeast. Mol Biol Cell. 1997;8:1089–1104. doi: 10.1091/mbc.8.6.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tall GG, Hama H, DeWald DB, Horazdovsky BF. The phosphatidylinositol 3-phosphate binding protein Vac1p interacts with a Rab GTPase and a Sec1p homologue to facilitate vesicle-mediated vacuolar protein sorting. Mol Biol Cell. 1999;10:1873–1889. doi: 10.1091/mbc.10.6.1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nielsen E, Christoforidis S, Uttenweiler-Joseph S, Miaczynska M, Dewitte F, Wilm M, Hoflack B, Zerial M. Rabenosyn-5, a novel Rab5 effector, is complexed with hVPS45 and recruited to endosomes through a FYVE finger domain. J Cell Biol. 2000;151:601–612. doi: 10.1083/jcb.151.3.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morrison HA, Dionne H, Rusten TE, Brech A, Fisher WW, Pfeiffer BD, Celniker SE, Stenmark H, Bilder D. Regulation of early endosomal entry by the Drosophila tumor suppressors Rabenosyn and Vps45. Mol Biol Cell. 2008;19:4167–4176. doi: 10.1091/mbc.E08-07-0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mottola G, Classen AK, Gonzalez-Gaitan M, Eaton S, Zerial M. A novel function for the Rab5 effector Rabenosyn-5 in planar cell polarity. Development. 2010;137:2353–2364. doi: 10.1242/dev.048413. [DOI] [PubMed] [Google Scholar]
- 49.Wang Y. Wnt/Planar cell polarity signaling: a new paradigm for cancer therapy. Mol Cancer Ther. 2009;8:2103–2109. doi: 10.1158/1535-7163.MCT-09-0282. [DOI] [PubMed] [Google Scholar]
- 50.Simonsen A, Lippé R, Christoforidis S, Gaullier J-M, Brech A, Callaghan J, Toh B-H, Murphy C, Zerial M, Stenmark H. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature. 1998;394:494–498. doi: 10.1038/28879. [DOI] [PubMed] [Google Scholar]
- 51.Callaghan J, Simonsen A, Gaullier J-M, Toh B-H, Stenmark H. The endosome fusion regulator EEA1 is a dimer. Biochem J. 1999;338:539–543. [PMC free article] [PubMed] [Google Scholar]
- 52.McBride HM, Rybin V, Murphy C, Giner A, Teasdale R, Zerial M. Oligomeric complexes link Rab5 effectors with NSF and drive membrane fusion via interactions between EEA1 and syntaxin 13. Cell. 1999;98:377–386. doi: 10.1016/s0092-8674(00)81966-2. [DOI] [PubMed] [Google Scholar]
- 53.Simonsen A, Gaullier J-M, D’Arrigo A, Stenmark H. The Rab5 effector EEA1 interacts directly with syntaxin-6. J Biol Chem. 1999;274:28857–28860. doi: 10.1074/jbc.274.41.28857. [DOI] [PubMed] [Google Scholar]
- 54.Ohya T, Miaczynska M, Coskun U, Lommer B, Runge A, Drechsel D, Kalaidzidis Y, Zerial M. Reconstitution of Rab- and SNARE-dependent membrane fusion by synthetic endosomes. Nature. 2009;459:1091–1097. doi: 10.1038/nature08107. [DOI] [PubMed] [Google Scholar]
- 55.Siddhanta U, McIlroy J, Shah A, Zhang YT, Backer JM. Distinct roles for the p110α and hVPS34 phosphatidylinositol 3′- kinases in vesicular trafficking, regulation of the actin cytoskeleton, and mitogenesis. J Cell Biol. 1998;143:1647–1659. doi: 10.1083/jcb.143.6.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fernandez-Borja M, Wubbolts R, Calafat J, Janssen H, Divecha N, Dusseljee S, Neefjes J. Multivesicular body morphogenesis requires phosphatidylinositol 3-kinase activity. Curr Biol. 1999;14:55–58. doi: 10.1016/s0960-9822(99)80048-7. [DOI] [PubMed] [Google Scholar]
- 57.Futter CE, Collinson LM, Backer JM, Hopkins CR. Human VPS34 is required for internal vesicle formation within multivesicular bodies. J Cell Biol. 2001;155:1251–1263. doi: 10.1083/jcb.200108152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Katzmann DJ, Odorizzi G, Emr SD. Receptor downregulation and multivesicular-body sorting. Nat Rev Mol Cell Biol. 2002;3:893–905. doi: 10.1038/nrm973. [DOI] [PubMed] [Google Scholar]
- 59.Raiborg C, Stenmark H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature. 2009;458:445–452. doi: 10.1038/nature07961. [DOI] [PubMed] [Google Scholar]
- 60.Wollert T, Hurley JH. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature. 2010;464:864–869. doi: 10.1038/nature08849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Williams RL, Urbe S. The emerging shape of the ESCRT machinery. Nat Rev Mol Cell Biol. 2007;8:355–368. doi: 10.1038/nrm2162. [DOI] [PubMed] [Google Scholar]
- 62.Raiborg C, Bremnes B, Mehlum A, Gillooly DJ, Stang E, Stenmark H. FYVE and coiled-coil domains determine the specific localisation of Hrs to early endosomes. J Cell Sci. 2001;114:2255–2263. doi: 10.1242/jcs.114.12.2255. [DOI] [PubMed] [Google Scholar]
- 63.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11:1433–1437. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- 65.Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–667. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol. 2009;186:773–782. doi: 10.1083/jcb.200907014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Knaevelsrud H, Ahlquist T, Merok MA, Nesbakken A, Stenmark H, Lothe RA, Simonsen A. UVRAG mutations associated with microsatellite unstable colon cancer do not affect autophagy. Autophagy. 2010;6:863–870. doi: 10.4161/auto.6.7.13033. [DOI] [PubMed] [Google Scholar]
- 68.Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, Vergne I, Deretic V, Feng P, Akazawa C, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol. 2008;10:776–787. doi: 10.1038/ncb1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Matsunaga K, Morita E, Saitoh T, Akira S, Ktistakis NT, Izumi T, Noda T, Yoshimori T. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol. 2010;190:511–521. doi: 10.1083/jcb.200911141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Obara K, Sekito T, Niimi K, Ohsumi Y. The ATG18-ATG2 complex is recruited to autophagic membranes via PtdIns(3)P and exerts an essential function. J Biol Chem. 2008;283:23972–23980. doi: 10.1074/jbc.M803180200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nair U, Cao Y, Xie Z, Klionsky DJ. Roles of the lipid-binding motifs of Atg18 and Atg21 in the cytoplasm to vacuole targeting pathway and autophagy. J Biol Chem. 2010;285:11476–11488. doi: 10.1074/jbc.M109.080374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Polson HE, de LJ, Rigden DJ, Reedijk M, Urbe S, Clague MJ, Tooze SA. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. 2010;6:506–522. doi: 10.4161/auto.6.4.11863. [DOI] [PubMed] [Google Scholar]
- 73.Sagona AP, Nezis IP, Pedersen NM, Liestol K, Poulton J, Rusten TE, Skotheim RI, Raiborg C, Stenmark H. PtdIns(3)P controls cytokinesis through KIF13A-mediated recruitment of FYVE-CENT to the midbody. Nat Cell Biol. 2010;12:362–371. doi: 10.1038/ncb2036. [DOI] [PubMed] [Google Scholar]
- 74.Morita E, Sandrin V, Chung HY, Morham SG, Gygi SP, Rodesch CK, Sundquist WI. Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. EMBO J. 2007;26:4215–4227. doi: 10.1038/sj.emboj.7601850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Carlton JG, Martin-Serrano J. Parallels between cytokinesis and retroviral budding: a role for the ESCRT machinery. Science. 2007;316:1908–1912. doi: 10.1126/science.1143422. [DOI] [PubMed] [Google Scholar]
- 76.Teis D, Saksena S, Emr SD. Ordered assembly of the ESCRT-III complex on endosomes is required to sequester cargo during MVB formation. Dev Cell. 2008;15:578–589. doi: 10.1016/j.devcel.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 77.Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367–1381. doi: 10.1101/gad.1545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marino G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, Lopez-Otin C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem. 2007;282:18573–18583. doi: 10.1074/jbc.M701194200. [DOI] [PubMed] [Google Scholar]
- 79.Bache KG, Slagsvold T, Stenmark H. Defective downregulation of receptor tyrosine kinases in cancer. EMBO J. 2004;23:2707–2712. doi: 10.1038/sj.emboj.7600292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vaccari T, Bilder D. The Drosophila tumor suppressor vps25 prevents nonautonomous overproliferation by regulating notch trafficking. Dev Cell. 2005;9:687–698. doi: 10.1016/j.devcel.2005.09.019. [DOI] [PubMed] [Google Scholar]
- 81.Vaccari T, Rusten TE, Menut L, Nezis IP, Brech A, Stenmark H, Bilder D. Comparative analysis of ESCRT-I, ESCRT-II and ESCRT-III function in Drosophila by efficient isolation of ESCRT mutants. J Cell Sci. 2009;122:2413–2423. doi: 10.1242/jcs.046391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Thompson BJ, Mathieu J, Sung HH, Loeser E, Rorth P, Cohen SM. Tumor suppressor properties of the ESCRT-II complex component Vps25 in Drosophila. Dev Cell. 2005;9:711–720. doi: 10.1016/j.devcel.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 83.Moberg KH, Schelble S, Burdick SK, Hariharan IK. Mutations in erupted, the Drosophila ortholog of mammalian tumor susceptibility gene 101, elicit non-cell-autonomous overgrowth. Dev Cell. 2005;9:699–710. doi: 10.1016/j.devcel.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 84.Sagona AP, Stenmark H. Cytokinesis and cancer. FEBS Lett. 2010;584:2652–2661. doi: 10.1016/j.febslet.2010.03.044. [DOI] [PubMed] [Google Scholar]
- 85.Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 86.Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 87.Tsukazaki T, Chiang TA, Davison AF, Attisano L, Wrana JL. SARA, a FYVE domain protein that recruits Smad2 to the TGFβ receptor. Cell. 1998;95:779–791. doi: 10.1016/s0092-8674(00)81701-8. [DOI] [PubMed] [Google Scholar]
- 88.Heldin CH, Landstrom M, Moustakas A. Mechanism of TGF-beta signaling to growth arrest, apoptosis, and epithelial-mesenchymal transition. Curr Opin Cell Biol. 2009;21:166–176. doi: 10.1016/j.ceb.2009.01.021. [DOI] [PubMed] [Google Scholar]
- 89.Bonifacino JS, Hurley JH. Retromer. Curr Opin Cell Biol. 2008;20:427–436. doi: 10.1016/j.ceb.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lorenowicz MJ, Korswagen HC. Sailing with the Wnt: charting the Wnt processing and secretion route. Exp Cell Res. 2009;315:2683–2689. doi: 10.1016/j.yexcr.2009.06.015. [DOI] [PubMed] [Google Scholar]
- 91.Christoforidis S, Miaczynska M, Ashman K, Wilm M, Zhao L, Yip S-C, Waterfield MD, Backer JM, Zerial M. Phosphatidylinositol-3-OH kinases are Rab5 effectors. Nature Cell Biol. 1999;1:249–252. doi: 10.1038/12075. [DOI] [PubMed] [Google Scholar]
- 92.Murray JT, Panaretou C, Stenmark H, Miaczynska M, Backer JM. Role of Rab5 in the recruitment of hVps34/p150 to the early endosome. Traffic. 2002;3:416–427. doi: 10.1034/j.1600-0854.2002.30605.x. [DOI] [PubMed] [Google Scholar]
- 93.Stein MP, Feng Y, Cooper KL, Welford AM, Wandinger-Ness A. Human VPS34 and p150 are Rab7 interacting partners. Traffic. 2003;4:754–771. doi: 10.1034/j.1600-0854.2003.00133.x. [DOI] [PubMed] [Google Scholar]
- 94.Stenmark H, Gillooly DJ. Intracellular trafficking and turnover of phosphatidylinositol 3-phosphate. Semin Cell Dev Biol. 2001;12:193–199. doi: 10.1006/scdb.2000.0236. [DOI] [PubMed] [Google Scholar]
- 95.Odorizzi G, Babst M, Emr SD. Fab1p PtdIns(3)P 5-kinase function essential for protein sorting in the multivesicular body. Cell. 1998;95:847–858. doi: 10.1016/s0092-8674(00)81707-9. [DOI] [PubMed] [Google Scholar]
- 96.Cao C, Laporte J, Backer JM, Wandinger-Ness A, Stein MP. Myotubularin lipid phosphatase binds the hVPS15/hVPS34 lipid kinase complex on endosomes. Traffic. 2007;8:1052–1067. doi: 10.1111/j.1600-0854.2007.00586.x. [DOI] [PubMed] [Google Scholar]
- 97.Cao C, Backer JM, Laporte J, Bedrick EJ, Wandinger-Ness A. Sequential actions of myotubularin lipid phosphatases regulate endosomal PI(3)P and growth factor receptor trafficking. Mol Biol Cell. 2008;19:3334–3346. doi: 10.1091/mbc.E08-04-0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Morselli E, Galluzzi L, Kepp O, Vicencio JM, Criollo A, Maiuri MC, Kroemer G. Anti- and pro-tumor functions of autophagy. Biochim Biophys Acta. 2009;1793:1524–1532. doi: 10.1016/j.bbamcr.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 99.Brech A, Ahlquist T, Lothe RA, Stenmark H. Autophagy in tumour suppression and promotion. Mol Oncol. 2009;3:366–375. doi: 10.1016/j.molonc.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]